
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalpol attenuates oxidative stress and promotes autophagy in TNF-α-exposed HAECs by up-regulating AMPK†
Yukun Zhang,
Changyuan Wang ,
Qining Yang,
Yue Jin,
Qiang Meng,
Qi Liu,
Yongguo Dai,
Zhihao Liu,
Kexin Liu and
Huijun Sun*
,
Qining Yang,
Yue Jin,
Qiang Meng,
Qi Liu,
Yongguo Dai,
Zhihao Liu,
Kexin Liu and
Huijun Sun*
Department of Clinical Pharmacology, College of Pharmacy, Dalian Medical University, 9 West Section, Lvshun South Road, Lvshunkou District, Dalian 116044, China. E-mail: sunhuijun@dlmedu.edu.cn; Fax: +86 411 86110413; Tel: +86 411 86110413
First published on 15th November 2017
Abstract
Oxidative stress and autophagy dysfunction are critical factors in the pathogenesis of atherosclerosis. Adenosine 5′ monophosphate-activated protein kinase (AMPK) plays an important role in inhibiting oxidative stress and promoting autophagy. Catalpol, an iridoid glucoside extracted from the root of Rehmanniae glutinosa L, was reported to produce a potent antioxidant effect. However, the mechanism of catalpol inhibiting oxidative stress and its effect on AMPK remain unclear. This study aims to investigate the potential role of catalpol in regulating oxidative stress and autophagy in tumor necrosis factor-α (TNF-α)-treated human aorta epithelial cells (HAECs), and the important role of AMPK involved in catalpol's effects. In the present study, effects of catalpol on inhibiting oxidative stress and the related adhesion, apoptosis and promotion of autophagy were demonstrated. Catalpol also produces a potent effect on activating AMPK in TNF-α-treated HAECs. Mechanistically, the effects of catalpol on inhibiting oxidative stress and increasing the autophagy level were partly blocked by a pharmacological AMPK inhibitor, compound C or AMPK small interfering RNA, indicating that catalpol performed such protective effects by activating AMPK. In summary, this study demonstrated that AMPK is a key treatment target for endothelial cell injury and catalpol confers protection on TNF-α-treated HAECs by up-regulating AMPK activity. Catalpol has highly favorable characteristics for the treatment of atherosclerosis.
1. Introduction
Aging is the greatest risk factor for the development of atherosclerosis (AS). One important mechanism that links aging and the development of vascular pathology is endothelial cell chronic inflammation. In the early stage of atherosclerosis, chronic inflammation leads to stress, metabolic abnormalities associated with reactive oxygen species (ROS) overproduction, monocyte adhesion to endothelial cells and dysfunction of autophagy as well as cell apoptosis.1,2Oxidative stress has been considered as an important factor in the pathophysiology of AS.3 Membrane lipids, organelles, and even DNA are believed to be the main targets of oxidative stress, which disturbs endothelial homeostasis and induces apoptosis.4 The NADPH oxidase family including NOX1–5 is the most important source of ROS. NOX4 is the major source of ROS in endothelial cells.5,6 In the early progression of atherosclerotic plaque formation, increased NOX4 expression has been detected.7 Increased expression of NOX4 can promote oxidative stress and inflammation related transcription factor nuclear factor-kappa B (NF-κB) activation.8 Additionally, excess ROS production and NF-κB activation can promote endothelial cells expressing cell adhesion molecules, which play key roles in the formation of AS plaques, including intercellular cell adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1) and monocyte chemotactic protein-1 (MCP-1).9 In addition, autophagy is also a process in the cells to maintain intracellular homeostasis during various cell stresses.10 It has been reported that autophagy level is suppressed in injured endothelial cells, and autophagy deficiency promoted AS.11 Therefore, enhancing suppressed autophagy level implicates a protective effect and the precise targets responsible for such effects might be an ideal treatment target for AS. Hence, these indexes which are characteristic in oxidative stress, cell adhesion, apoptosis and autophagy are often employed to measure the treating possible effects of drugs on AS.
Adenosine 5′ monophosphate-activated protein kinase (AMPK) is a key enzyme participating in cellular energy metabolism and serves as a cellular energy sensor. The role of AMPK in preventing endothelial dysfunction has been proposed in many investigations, which include anti-inflammation via SIRT1-related pathway, inducing cell autophagy via mTOR-related pathway12 and protecting cells from oxidative stress13 and so on. Studies also provide that activation of AMPK protects endothelial cells from AS through decreasing adhesion molecule expression, attenuating endothelial inflammation13,14 and reducing oxidative stress. Thus, augmentation of AMPK activity is posited to be a potential target for therapeutic interventions in AS.15,16
Catalpol, whose structure is shown in Fig. S1,† is an iridoid glucoside and has been found to be present in large quantities in the root of Romania glutinosa L.12 As a traditional medicine, catalpol demonstrates a variety of biological activities including anticancer, neuro-protective, anti-inflammatory, diuretic, hypoglycemic and anti-hepatitis virus effects.17,18 Previous studies have also provided some clues that catalpol can affect energy metabolism through increasing mitochondrial biogenesis, enhancing endogenous antioxidant enzymatic activities and inhibiting free radical generation.19,20 What's more, recent studies provided that catalpol could up-regulate the decreased AMPK phosphorylation in db/db mice and ameliorate high-fat diet-induced adipose tissue inflammation by suppressing NF-κB.12 Therefore, we presumed that catalpol could protect against endothelial dysfunction via an AMPK dependent way.
In the present study, human aortic epithelium cells (HAECs) were cultured and treated with TNF-α to test this hypothesis. The results truly showed that phosphorylated AMPK expression was decreased in cultured HAECs exposed to TNF-α and the suppressed phosphorylated AMPK could be reversed by catalpol. The results also indicated that TNF-α induced ROS over-production, apoptosis and inflammation as well as the decreased autophagy level could be restored by catalpol in an AMPK-dependent way through using pharmacological inhibitor of AMPK, compound C or AMPK small interfere RNA (siRNA). We conclude that targeting AMPK is an attractive strategy for the treatment of AS and catalpol might be a promising agent.
2. Materials and methods
2.1 Reagents
Catalpol (Fig. S1,† 98%) was obtained from Nanjing Jingzhu biotech Ltd (Nanjing, China). Co. Recombinant human TNF-α was purchased from Peprotech Inc (Rocky Hill, USA). Dorsomorphin (compound C, 6-[4-(2-piperidin-1-ylethoxy)phenyl]-3-pyridin-4-ylpyrazolo[1,5-]pyrimidine) was obtained from Selleck company (Shanghai, China). DMEM medium was purchased from Gibco-BRL Company (Gaithersburg, MD, USA). DCFH-DA fluorescent probe and ECL Plus were obtained from Biotool (Shanghai, China). Monodansylcadaverine (MDC) was purchased from KeyGEN Biotech (Nanjing, China). Antibodies specific for Bcl-2, caspase-3, caspase-9, NF-κB p65, phosphorylated NF-κB p65, ICAM-1, VCAM-1, AMPK, phosphorylated-AMPK and SIRT1 were obtained from Proteintech Group (Wuhan, China). Antibody specific for β-actin was purchased from Beyotime (Jiangsu, China). Antibody specific for acetyl-lysine was purchased from Abcam Ltd. (Cambridge, UK). | ||
| Fig. 1 Effect of catalpol on biochemical parameters of TNF-α-induced oxidative in HAECs. (A) The level of GSH. (B) The release of LDH. (C) The level of SOD. (D) Intracellular level of MDA. Data are expressed as mean ± SD from three independent experiments. **p < 0.05 vs. control group, ##p < 0.05 vs. TNF-α group. | ||
2.2 Cell culture and treatment
HAECs were purchased from ScienCell Company (CA, USA) and cultured in Modified Eagle's Medium that was supplemented with 10% (v/v) fetal bovine serum (Gibco, CA, USA). The cells were kept at 37 °C in a humidified incubator with 5% CO2. The in vitro model was established by treating HAECs with TNF-α (50 ng mL−1) for 24 h.82.3 SOD, MDA, GSH and LDH assay
Levels of superoxide dismutase (SOD), malonaldehyde (MDA), glutathione (GSH) and lactate dehydrogenase (LDH) were measured to detect the level of endothelial cell damage with colorimetric assay kit (Nanjing Jiancheng Bioengineering, China) according to the manufacturer's instructions.2.4 Measurement of intracellular ROS
2′,7′-Dichlorodihydrofluorescein diacetate (H2DCFDA) probe was employed to measure ROS level as previously described. HAECs were incubated with TNF-α and different concentrations of catalpol for 24 h at 37 °C and then collected and incubated with H2DCFHDA for 30 min at 37 °C. The fluorescence intensity was immediately measured using FACS Calibur (BD, USA) equipped with an argon ion laser (488 nm excitation) and 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per sample were measured.
000 cells per sample were measured.
2.5 Isolation of cytoplasmic and nuclear proteins
Cytoplasmic and nuclear protein fractions from cultured HAECs were prepared using a commercial protein isolation kit (KeyGEN Biotech, Nanjing, China) according to the manufacturer's instructions.2.6 Western blot analysis
Equal amounts of proteins were separated by 10–20% SDS-PAGE and transferred onto a PVDF membrane (Millipore, Bedford, MA, USA). The blocked membranes were then immunoblotted with the indicated primary antibodies at 4 °C overnight. After washing, the membranes were then incubated with the appropriate secondary antibodies. The membranes were exposed to enhance chemiluminescence-plus reagents (Beyotime Institute of Biotechnology, Hangzhou, China). The emitted light was captured by a Bio-rad imaging system with a Chemi HR camera 410 and analyzed with a Gel-Pro Analyzer Version 4.0 (Media Cybernetics, MD, USA).2.7 Analysis of acetylated NF-κB p65 by immunoprecipitation
A sufficient amount of NF-κB p65 antibody (Proteintech group, Wuhan, China) was added into 200 μg protein and gently rotated at 4 °C overnight. The immunocomplex was captured by adding 25 μL of protein A + G agarose beads (Biotool, Shanghai, China) and gently rotating at 4 °C for 3 h. Then, the mixture was centrifuged at 1500g for 5 min at 4 °C. The precipitate was washed three times with ice-cold phosphate buffer saline, resuspended in 1× sample buffer and boiled for 5 min to dissociate the immune-complex from the beads. The supernatant was collected by centrifugation and subjected to Western blotting. Results were expressed relative to the control on the same blot, and the values were expressed as fold increase after normalization with total NF-κB.2.8 Down-regulation of gene expression by small interfering RNA
2 × 105 HAECs were seeded in 6-well plates and were transfected with specific siRNA (50 nM) using Lipofectamine 2000 (Invitrogen, Karlsruhe, Germany) according to the manufacturer's instructions. Cells transfected with control plasmids were pooled and used as negative control. The sequence of AMPK siRNA: sense 5′-UGCCUACCAUCUCAUAAUAdTdT-3′; antisense 5′-UAUUAUGAGAUGGUAGGCAdTdT-3′ (Genepharma, Shanghai, China). The transfected cells were incubated at 37 °C in serum free MEM and fetal bovine serum was added to 10% 6 h after transfection. After growing for additional 24 h, cells were treated with TNF-α (50 ng mL−1), catalpol (20 μM) or both for additional 24 h and then collected for Western blot analysis to determine the levels of the indicated proteins.2.9 MDC staining
Cultures were stained with MDC (50 μmol L−1) at 37 °C for 40 min. After incubation, cells were washed three times with PBS, fixed with 5% paraformaldehyde, and observed under a fluorescence microscope (Leica, Germany) immediately.2.10 Transmission electron microscopy
Cells were treated as indicated in ref. 21. At first, 2 × 106 cells were seeded in culture dishes. After treatment with TNF-α (50 ng mL−1), catalpol (20 μM) or both for 24 h, cells were fixed for 2 h at 4 °C in 1.6% glutaraldehyde in 0.1 M Sörensen phosphate buffer (pH 7.3) and washed once with PBS and then re-fixed in aqueous 2% osmium tetroxide and finally embedded in Epon® epoxy resin, until imaging. The examination was performed at 80–100 kV under a transmission electron microscope, on ultrathin sections (80 nm) stained with 0.1% lead citrate and 10% uranyl acetate.2.11 Fluorescence microscopy
HAECs were seeded in 6-well plates at a density of 1 × 105 per well. The cells were treated with catalpol (10, 20 and 40 μM) and TNF-α (50 ng mL−1) at 37 °C for 24 h. At the end of treatment, cells were fixed with 4% paraformaldehyde for 15 min, rinsed with PBS and mounted in mounting medium with DAPI. The samples were observed by inverted fluorescence microscope.2.12 Transfection of HAECs and luciferase reporter assay
On day 0, HAECs cells were plated at a density of 5 × 104 cells per well in 24-well plates in DMEM supplemented with 10% FBS and incubated at 37 °C in a 5% CO2 incubator. On day 1, cells were washed with 0.5 mL PBS and 0.5 mL fresh DMEM was added to each well before transfection. Cells were cotransfected with 2 μg plasmid containing wild-type AMPK promoter linked to the firefly luciferase reporter gene and 2 μg control plasmid containing the Renilla luciferase reporter gene by using lipo2000 transfection reagent. 6 h after transfection, cells were washed with 0.5 mL PBS, switched to DMEM supplemented with 10% dilapidated FBS and catalpol (10, 20, 40 μM) for 24 h at 37 °C and 5% CO2. On day 3, cells were washed with 0.5 mL PBS and firefly and Renilla luciferase activities were measured using the Dual Luciferase Reporter Assay System and Turner Designs TD-20/20 Luminometer. Firefly luciferase activities in the transfected lysates were normalized by Renilla luciferase activity from the same tube. Each transfection was done in triplicate independent experiments.2.13 Statistical analysis
All analyses were performed with the SPSS software package, version 19.0. Data are presented as means ± SD. Comparison of quantitative variables was performed by either Student's t test or ANOVA. p values <0.05 (two-tailed) were considered statistically significant.3. Results
3.1 Catalpol enhanced SOD activity, intracellular GSH level and reduced the level of MDA as well as LDH release
GSH, a cofactor for glutathione peroxidase and the redox enzyme, can catalyze the reduction of lipid peroxide.22 SOD is an important enzyme in cellular defense against oxidative stress and MDA is an essential index positively correlated with lipid peroxidation. The production of LDH in the culture supernatant is regarded as an early indicator of cell death.15 Thus, we examined the effects of catalpol on levels of SOD, MDA, GSH and LDH. As shown in Fig. 1, TNF-α treatment significantly decreased GSH and SOD activity and elevated MDA and LDH release in HAECs. On the contrary, catalpol apparently reversed the effects of TNF-α in a concentration dependent manner. The results indicated that catalpol could produce anti-oxidant effects in TNF-α-treated HAECs.3.2 Catalpol inhibited TNF-α-induced ROS production and NOX4 expression in HAECs
NADPH oxidase is a key determinant in ROS generation, while NOX4 is the most abundant NOX isoform in the vasculature.6 Therefore, the effects of catalpol on the expression of NOX4 protein level and ROS generation in TNF-α-treated HAECs were measured. As shown in Fig. 2A and B, TNF-α significantly increased ROS production and NOX4 protein expression by 1.94 and 2.99-fold compared with control group, respectively. In addition, compared with cells exposed to TNF-α, catalpol obviously alleviated the increased NOX4 expression and abundance of ROS generation. These results suggested a potent effect of catalpol on inhibiting oxidative stress. | ||
| Fig. 2 Catalpol inhibited TNF-α-induced ROS over production and NOX4 over expression in HAECs. (A) Effect of catalpol on ROS generation induced by TNF-α. (B) Effect of catalpol on NOX4 protein expression induced by TNF-α. (C) I-κBα protein expression. (D) NF-κB/p65 protein expression. (E) The phosphorylation level of p65. Data illustrated on the graph bar represents the mean ± SD from three independent experiments. **p < 0.05 vs. control group, ##p < 0.05 vs. TNF-α group. | ||
3.3 Catalpol inhibited TNF-α-induced IκBα degradation and NF-κB translocation in HAECs
The pro-inflammatory mediator, NF-κB is a nuclear transcription factor that plays a central role in inflammatory responses during early AS, and inhibiting NF-κB activity is beneficial to reduce cell adhesion molecules expression including ICAM-1, VCAM-1 and MCP-1.16,23 To examine the influence of catalpol on NF-κB signal pathway, IκBα degradation, NF-κB/p65 nuclear translocation and phosphorylation were measured by western blotting. As shown in Fig. 2C, TNF-α alone significantly induced IκBα degradation in cytosol. However, catalpol inhibited TNF-α-induced IκBα decrease in a concentration-dependent manner. We then investigated the translocation of NF-κB/p65 in the nucleus as well as phosphorylation of p65. TNF-α significantly induced the translocation of NF-κB/p65 and phosphorylation of p65, and treatment with catalpol partially blocked TNF-α-induced NF-κB/p65 translocation and p65 phosphorylation (Fig. 2D and E). These results suggested that catalpol inhibited the nuclear translocation of the NF-κB/p65 subunit in to the nucleus.3.4 Catalpol inhibited TNF-α-induced cell adhesion and apoptosis in HAECs
Leukocyte adhesion and trans-endothelial migration into the surrounding tissues are critical steps in vascular inflammation and AS.24 Endothelial cells play an imperative role in this process via expressing cell adhesion molecules, such as ICAM-1, VCAM-1, which augment their adhesiveness to leukocytes.25 MCP-1, a chemokine attracting white blood cells migrating to the injured area, is mainly secreted by endothelial cells and monocytes in response to an inflammatory stimulus.26 Therefore, effects of catalpol on TNF-a-induced over expression of ICAM-1, VCAM-1 as well as intracellular MCP-1 and secreted MCP-1 were detected. As shown in Fig. 3A–D, the expression of ICAM-1, VCAM-1 and MCP-1 in cells exposed to TNF-α was significantly increased compared with the control group, and the released MCP-1 in the culture medium was also obviously increased compared with the control group. However, as found, catalpol inhibited the over-expressions in a concentration dependent manner. These results demonstrated that catalpol could inhibit cell adhesion and chemotaxis in TNF-α-exposed HAECs. | ||
| Fig. 3 Catalpol inhibited TNF-α-induced adhesion molecule expression and apoptosis. (A) Catalpol inhibited TNF-α induced ICAM-1 protein expression. (B) Catalpol inhibited TNF-α induced VCAM-1 protein expression. (C) Catalpol inhibited TNF-α induced MCP-1 protein expression. (D) Catalpol inhibited TNF-α induced MCP-1 release in the culture medium. (E) Anti-apoptotic protein Bcl-2 level. (F) Pro-apoptotic protein caspase-3 level. (G) Pro-apoptotic protein caspase-9 protein level. (H) DAPI staining in TNF-α-treated HAECs. Data illustrated on the graph bar represent the mean ± SD from three independent experiments. **p < 0.05 vs. control group, ##p < 0.05 vs. TNF-α group. | ||
Cell apoptosis plays a crucial role in the pathogenesis of cardiovascular disease. Thus, to evaluate the effect of catalpol on cell apoptosis, Bcl-2, caspase-3 and caspase-9 expressions were determined by western blotting. As shown in Fig. 3E, catalpol obviously upgraded the decreased Bcl-2 level induced by TNF-α. Furthermore, a significantly increased expression of caspase-3 and caspase-9 induced by TNF-α was also down-regulated after catalpol administration (Fig. 3F and G). Additionally, as shown in Fig. 3H, in DAPI staining, TNF-α-treated cells appeared as blue colored uniform oval, and increased karyopycnosis, chromatin marginalized and nuclear fragmentation showing typical apoptosis. Whereas catalpol obviously reduced the number of apoptotic cells, indicating the intense anti-apoptotic effect of catalpol. The above results demonstrated the effects of catalpol on inhibiting cell adhesion and apoptosis.
3.5 Catalpol up-regulated autophagy in TNF-α induced HAECs
Autophagy modulates endothelial inflammation and retards the development of AS.27,28 LC3-2/1 is required for the formation of the autophagosome and decreased autophagy level inhibits the degradation of p62. Thus, LC3-2/1 and p62 protein expressions were measured to investigate the effect of catalpol on autophagy. As shown in Fig. 4A, decreased conversion of LC3-1 to LC3-2 and increased p62 level were observed after TNF-α treatment compared with that in the control group. Conversely, increased conversion of LC3-1 to LC3-2 and decreased p62 protein expression were shown after catalpol treatment compared with TNF-α group. Monodansylcadaverine (MDC) staining, which was able to stain autophagosomes specifically, was employed to further evaluate the effect of catalpol on autophagy. We could found that TNF-α caused significantly decreased fluorescence intensity compared with the control group, and the fluorescence intensity was significantly increased in a concentration dependent way after treated with catalpol (Fig. 4B). Additionally, typical autophagosome and autophagolysosome were detected by transmission electron microscopy. Ultrastructural image analysis also showed the decreased double-membrane autophagic vesicles after TNF-α treatment compared with that in control group. In contrast, treatment with catalpol obviously increased the number of double-membrane autophagic vesicles (Fig. 4C). Collectively, these results demonstrated that catalpol could promote autophagy and autophagic flux in TNF-α-treated HAECs. | ||
| Fig. 4 Catalpol increased TNF-α-induced decrease autophagy level in HAECs. (A) Effect of catalpol on LC3-II/I and p62/SQSTM expression. (B) Catalpol treatment for 24 h increased the number of autophagosomes in HAECs, determined by MDC staining. (C) Catalpol treatment for 24 h increased the number of autophagosomes in HAECs, determined by TEM analysis. Data illustrated on the graph bar represent the mean ± SD from three independent experiments. **p < 0.05 vs. control group, ##p < 0.05 vs. TNF-α group. | ||
3.6 Catalpol increased AMPK, SIRT1 expression and decreased NF-κB p65 acetylation in TNF-α-treated HAECs
AMPK is a heterotrimer consisting α, β, γ three subunits, and α-subunit is a catalytic subunit. Since activation of AMPK is benefit to inhibit oxidative stress and promoting autophagy, thus, AMPK expression in TNF-α-treated HAECs and effect of catalpol on AMPK activity were investigated. AMPK activation is characterized as phosphorylation at αThr172, so we evaluated AMPK activity by measuring the abundance of phosphorylated-AMPK protein at αThr. As shown in Fig. 5A, the phosphorylated-AMPK protein level was remarkably reduced in TNF-α treated HAECs, but catalpol could reverse the loss of phosphorylated-AMPK level in a concentration-dependent manner. SIRT1 is known as a mammalian ortholog of Sir2 (silent information regulator 2) that acts as a master metabolic sensor of NAD+ and modulates cellular metabolism and life span.29 SIRT1 protein expression can be positively regulated by AMPK indicating a coordination of their effects on energy metabolism.30,31 SIRT1 also modulates the acetylation status of p65. To validate the activation effect of catalpol on AMPK, effects of catalpol on SIRT1 and acetylated-p65 (Ac-p65) were detected. Compared with the control group, the expression of SIRT1 was down-regulated in the TNF-α-treated cells and after treatment with catalpol expression of SIRT1 was up-regulated markedly in a concentration-dependent manner (Fig. 5B). Notably, the level of Ac-p65 was shown as predicted in an opposite way, that the Ac-p65 was significantly increased after treated with TNF-α compared with the control group, then after treated with catalpol the up-regulated Ac-p65 was decreased obviously in a concentration dependently manner (Fig. 5C). These results indicated the potential effect of catalpol on promoting AMPK activation.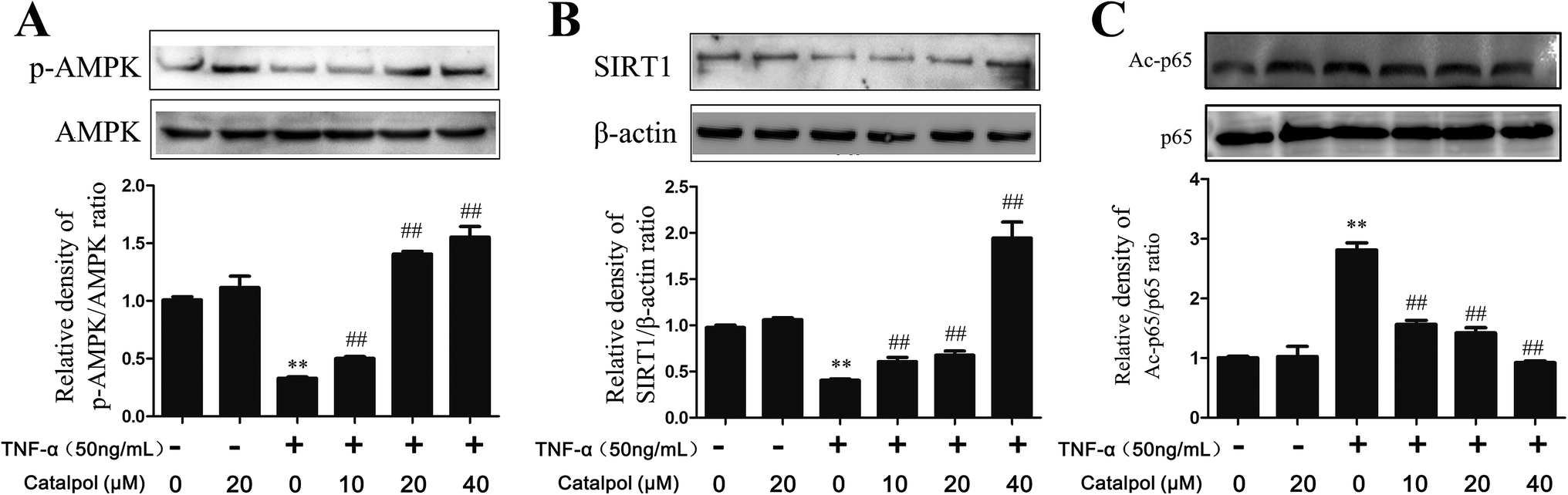 | ||
| Fig. 5 Catalpol increased TNF-α-induced decrease of AMPK, SIRT1 protein expression and enhanced p65 deacetylation. (A) Catalpol reversed TNF-α induced down-regulation of phosphorylated AMPK. (B) Catalpol reversed TNF-α induced down-regulation of SIRT1 protein expression. (C) Catalpol reversed TNF-α induced p65 acetylation. Data illustrated on the graph bar represent the mean ± SD from three independent experiments. **p < 0.05 vs. control group, ##p < 0.05 vs. TNF-α group. | ||
3.7 Catalpol increased SIRT1 expression and decreased NF-κB p65 acetylation in TNF-α-treated HAECs via AMPK activation
Then, whether the effects of catalpol on inhibiting oxidative stress and promoting autophagy were based on activating AMPK was considered in the present study. Thus pharmacological AMPK inhibitor compound C and AMPK siRNA were employed. First, the effects of catalpol on AMPK activity with or without compound C or AMPK siRNA treatment were detected. As shown in Fig. 6A, AMPK siRNA significantly inhibited AMPK and phorsphorylated-AMPK protein level. The content of SIRT1 was also detected after transfection with AMPK siRNA. In normal cells, catalpol significantly increased SIRT1 protein expression compared to TNF-α-treated group. But in cells transfected with AMPK siRNA, the increased effect of catalpol on SIRT1 protein expression was abrogated (Fig. 6B). Similarly, transfection with AMPK siRNA also significantly attenuated the inhibitory effect of catalpol on Ac-p65 expression from 5.26 to 1.26 fold compared with that in normal cells (Fig. 6C). Treatment with compound C significantly decreased phosphorylated AMPK protein expression compared to that in untreated cells and the stimulating effect of catalpol on AMPK protein expression was abrogated by compound C (Fig. S2A†). In addition, the up-regulating effect of catalpol on SIRT1 protein expression in TNF-α-treated HAECs was also partly blocked by compound C (Fig. S2B†). Moreover, the inhibitory effect of catalpol on Ac-p65 expression was also attenuated by compound C from 3.34 to 1.26 fold (Fig. S2C†). Collectively, the above results demonstrated that catalpol could increase SIRT1 protein expression and promote p65 deacetylation through up-regulating AMPK activity. | ||
| Fig. 6 Catalpol up-regulated SIRT1 expression and enhanced p65 deacetylation through activating AMPK. (A) Effect of catalpol on AMPK phosphorylation after treated with AMPK siRNA. (B) Effect of catalpol on SIRT1 protein expression after treated with AMPK siRNA. (C) Effect of catalpol on p65 acetylation after treated with AMPK siRNA. Data illustrated on the graph bar represent the mean ± SD from three independent experiments. **p < 0.05 vs. control group, ##p < 0.05 vs. TNF-α group. &&p < 0.05 vs. AMPK siRNA or compound C, $$p < 0.05 vs. AMPK siRNA or compound C with TNF-α treatment. | ||
3.8 Catalpol inhibited ROS production and NOX4 expression via AMPK activation
We hypothesized that the effect of catalpol attenuating ROS production and NOX4 expression was also mediated by increasing AMPK activity. Thus, we detected NOX4 expression and ROS production after treated with compound C or AMPK siRNA. In mock cells, ROS production and NOX4 protein expression were increased in TNF-α-treated group compared to the control group and catalpol could reverse the effects of TNF-α significantly. However, transfection with AMPK siRNA partly blocked the inhibitory effects of catalpol on ROS over-generation from 1.64-fold to 1.32-fold (Fig. 7A). The inhibiting effect of catalpol on NOX4 expression was also synchronously attenuated from 4.55-fold to 1.64-fold compared with that in mock cells (Fig. 7B). Similarly, compound C significantly attenuated the inhibitory effect of catalpol from 1.75-fold to 1.47-fold (Fig. S3A†) and partly blocked the inhibitory effect of catalpol from 2.78-fold to 1.67-fold compared to that without compound C treatment (Fig. S3B†). All these results suggested that the inhibitory effects of catalpol on oxidative stress were partly mediated by activating AMPK. | ||
| Fig. 7 Catalpol inhibited TNF-α-induced oxidative stress, cell apoptosis and autophagy deficiency through activating AMPK. (A) Effect of catalpol on ROS production after treated with AMPK siRNA. (B) Effect of catalpol on NOX4 expression after treated with AMPK siRNA. (C) Effect of catalpol on LC3-II/I and p62/SQSTM expression after treated with AMPK siRNA. (D) Effect of catalpol on ICAM-1 protein expression after treated with AMPK siRNA. (E) Effect of catalpol on VCAM-1 protein expression after treated with AMPK siRNA. (F) Effect of catalpol on Bcl-2 protein expression after treated with AMPK siRNA. (G) Effect of catalpol on caspase-3 protein expression after treated with AMPK siRNA. Data illustrated on the graph bar represent the mean ± SD from three independent experiments. **p < 0.05 vs. control group, ##p < 0.05 vs. TNF-α group. &&p < 0.05 vs. AMPK siRNA treatment, $$p < 0.05 vs. AMPK siRNA with TNF-α treatment. | ||
3.9 Catalpol inhibited cell adhesion and apoptosis via AMPK activation
To evaluate whether the AMPK pathway contributed to the inhibiting effects of catalpol on endothelial cell adhesion and apoptosis, the expressions of adhesion and apoptosis related proteins were detected after transfection with AMPK siRNA or administration with AMPK inhibitor compound C. In normal cells, ICAM-1 and VCAM-1 protein expression was increased in TNF-α treated group compared to the control group and catalpol significantly reduced the over-expression of ICAM-1 and VCAM-1 protein expression. Whereas transfection with AMPK siRNA partly blocked the inhibitory effect of catalpol on ICAM-1 (2.04-fold to 1.18-fold) and VCAM-1 (2.27-fold to 1.02-fold) expressions compared with that in normal cells (Fig. 7D and E) (in cells co-treated with TNF-α and compound C ICAM-1 and VCAM-1 protein level was inhibited significantly compared with TNF-α treated alone, as shown in Fig. S3D and E.† The inhibitory effect might contribute to the direct inhibitory effect of compound C on ICAM-1 and VCAM-1 (ref. 32)). As shown in Fig. 7F, G, S3F and G,† in normal cells, Bcl-2 protein expression was decreased and caspase-3 protein expression was increased in TNF-α treated group compared to the control group, and catalpol could reverse the effects of TNF-α significantly. But transfection with AMPK siRNA or treatment by compound C partly blocked the stimulating effect of catalpol on Bcl-2 expression (2.02-fold to 1.28-fold, 1.52-fold to 1.27-fold) compared with that in normal cells. The inhibitory effect of catalpol on caspase-3 expression was also attenuated by compound C or AMPK siRNA (2.56 to 1.22-fold, 4.00 to 1.20-fold). These results suggested that the inhibitory effects of catalpol on TNF-α-induced adhesion and apoptosis were possibly mediated by AMPK activation.3.10 Catalpol promoted endothelial cell autophagy via AMPK activation
Then of course, whether the effects of catalpol on promoting cellular autophagy was related to AMPK activation aroused our interest. Thus, AMPK siRNA and AMPK inhibitor compound C were also employed in the subsequent study. As shown in Fig. 7C, in normal cells, LC3-2/1 ratio was decreased and p62 protein expression level was increased in TNF-α group compared to control group. And catalpol resulted in a significant increase in the LC3-2/1 ratio and decline in p62 protein expression level compared to TNF-α-group, respectively. Moreover, in cells transfected with AMPK siRNA, the increased effect of catalpol on LC3-2/1 ratio was lower than that in normal cells (2.32 to 1.75-fold). However, p62 expression level had almost no change. As shown in Fig. S3C,† the increased effects of catalpol on LC3-2/1 ratio was attenuated from 2.74-fold to 1.83-fold in cells treated by compound C, however, the expression of p62 didn't change much. Collectively, these results demonstrated that catalpol could increase autophagy and autophagic flux through up-regulating AMPK activity but independent of p62.3.11 Catalpol enhanced AMPK activity without changing AMPK promoter activity
On the bases of above observations, we proposed that catalpol might have the capacity to enhance AMPK activity. Thus, effect of catalpol on AMPK activity was first studied in a concentration dependent manner. As shown in Fig. 8A, catalpol triggered a concentration-dependent increase in phosphorylated AMPK expression. In addition, dual-luciferase reporter assay was employed to investigate the effect of catalpol on AMPK promoter activity. Luciferase reporter plasmid that contains the wild-type human AMPK promoter was established (Fig. 8B). As shown in Fig. 8C, catalpol (10, 20, 40 μM) didn't promote AMPK promoter activity compared with control group. Thus, the results suggested that the increase effect of catalpol on AMPK activity was not depending on enhancing AMPK promoter activity. | ||
| Fig. 8 Catalpol up-regulated AMPK activity without affecting AMPK promoter activity. (A) Catalpol enhanced AMPK phosphorylation level in a concentration dependent manner. (B) AMPK promoter was inserted into the PGL4.18-AMPK vector. (C) Effect of catalpol on AMPK promoter activity. Data illustrated on the graph bar represent the mean ± SD from three independent experiments. **p < 0.05 vs. control group. | ||
4. Discussion
AS is the major cause of death in the world and chronic inflammation is a shared pathophysiological feature of AS. Chronic inflammation induced endothelial oxidative stress, and apoptosis as well as decreased autophagy which largely interfered with the normal function of endothelial cells, and promoted the development of AS.Catalpol, an iridoid glucoside, is present in larger quantities in several plants in genus Rehmannia (Orobanchaceae). Biological activities of catalpol including anti-cancer, neuroprotective, anti-inflammatory, diuretic, hypoglycemic and anti-hepatitis virus effects have been reported.33,34 In the present study, we firstly attempted to demonstrate (1) catalpol inhibited oxidative stress and related cell adhesion, apoptosis as well as increased cellular autophagy in TNF-α-treated endothelial cells; (2) catalpol prevented against TNF-α induced oxidative stress, apoptosis and autophagy deficiency through activating AMPK.
TNF-α is a multifunctional cytokine that plays important role in inducing inflammatory response in AS. Thus, TNF-α induced HAECs was employed as a in vitro AS model in the present study. In the pre-experiments concentration of catalpol was determined by cell viability assay (Fig. S4†) based on our previous study. AMPK is a key regulator of energy metabolism in molecular biology and is expressed in variety of metabolism-related organs. Loss of AMPK activity was reported in diabetes and other metabolic disorders.35,36 In AS, inhibition of AMPK activity has been reported in multiple cell types. Decreased AMPK activity promotes smooth muscle cell proliferation, monocyte-to-macrophage differentiation,37 accelerates endothelium inflammation14,38 and so on. As catalpol has been reported to play a relieving role in diabetes through regulating glucose and lipid metabolism,39 and AMPK is a critical enzyme in energy metabolism, we posited that the protective effect of catalpol on TNF-α-treated HAECs was depending on activating AMPK. Previous studies indicated that activated AMPK could promote the accumulation of NAD+, thereby affecting SIRT1 deacetylase activity and performing variety of effects.40 Our study demonstrated that catalpol induced activation of AMPK accompanied with increased SIRT1 protein expression and decreased deacetylation of p65. Further, by using chemical synthesis inhibitor of AMPK, compound C and AMPK siRNA, we verified that catalpol increased SIRT1 protein expression and decreased deacetylation of p65 via activating AMPK.
Oxidative stress refers to the oxidation and antioxidant imbalance, resulting in releasing a large number of oxidation intermediates mainly ROS in endothelial cells. The excess production of ROS has been proved to be related to vascular diseases, such as AS, hypertension, diabetes vasculopathy and restenosis.41 As the first line of defense against ROS, antioxidant enzymes metabolize ROS innocuous byproducts.42 In the present study, catalpol up-regulated the expression of superoxide scavenger SOD and down-regulated the level of MDA indicating a potent antioxidative effect of catalpol. NADPH oxidase family consisting NOX1–5 is the most important source of ROS42 and NOX4 is considered to be the major source of ROS in endothelial cells.43,44 The increased NOX4 expression was found to be related to the formation of atherosclerotic plaque in its early progression.45 The present study demonstrated that catalpol significantly attenuated ROS generation and reduced NOX4 expression in TNF-α induced HAECs. Previous studies indicated that regulation of AMPK activity could inhibit oxidative stress through inhibiting NADPH oxidase expression in normal or palmitate-treated HUVECs.46,47 It was also reported that AMPK was involved in modulating endothelial cell injury in diabetes induced by NADPH oxidase overexpression.39,48,49 By using AMPK inhibitor compound C and AMPK siRNA, we proved that catalpol enhanced AMPK activity to reduce NOX4 expression and ROS production. These findings revealed that catalpol attenuated TNF-α induced oxidative stress partially via enhancing AMPK activity.
NF-κB, a transcription factor that can be activated by oxidative stress and mediates cell inflammation, adhesion and survival, plays a vital role in the process of AS.50 Inhibition of NF-κB activity is one of the reliable measures in treating AS. Our data demonstrated that catalpol could inhibit NF-κB pathway activation, including inhibiting IκB degradation, NF-κB p65 translocation to nuclear, p65 phosphorylation as well as inhibiting NF-κB deacetylation. Leukocyte adhesion and trans-endothelial migration into the surrounding tissues are critical steps in vascular inflammation and AS, followed by NF-κB activation.23 The depressing effects of catalpol on VCAM-1, ICAM-1 and MCP-1 expression showed its potent effects on vascular endothelial cell adhesion. Much evidence has shown that ROS are the mediators inducing adhesion molecules,51,52 and AMPK is effective in mediating ROS production. By using compound C and AMPK siRNA, we proved that catalpol could decrease TNF-α induced adhesion molecule expression via activating AMPK.
Endothelial cell apoptosis, which can be triggered by oxidative stress,53 plays a paramount role in the pathogenesis of atherosclerosis. The apoptosis of endothelial cells contributes to instability of atherosclerotic plaques and thrombosis leading to the acute symptoms in patients with cardiovascular diseases.54 Thereby, blocking endothelial cell apoptosis in AS is an important treatment strategy. Caspase family is an integral part of the apoptotic pathway in which caspase-3 and caspase-9 are the most important.55 B-cell lymphoma 2 (Bcl-2) is the founding member of the Bcl-2 family of regulator proteins that inhibits apoptosis.56 In our study, anti-apoptotic effect of catalpol was proved by increased expression of Bcl-2 and decreased expression of caspase-3, and 9. According to the reports, activation of AMPK is benefit to inhibit cell apoptosis.57 By using AMPK inhibitor compound C and AMPK siRNA, we verified the anti-apoptotic effect of catalpol was partly mediated by AMPK activation.
Autophagy is a catabolic process involving in physiological turnover of long-lived proteins and damaged organelles in autophagosome.58 Proper autophagy is an important response to the extrinsic and intrinsic cellular environments, and positively regulates cellular processes for chronic inflammation.59 In endothelial cells, suppressed autophagy level may contribute to further injury in human atherosclerotic lesions. Previous studies indicated that decreased cell autophagy level was detected in ox-LDL or TNF-α treated HUVECs,2,60 and inducing cell autophagy contributed to ameliorating vascular endothelial inflammation and lipid accumulation in vascular smooth muscle cells.61 In the present study, we found that catalpol restored inhibited autophagic flux in TNF-α-treated HAECs. AMPK is known to regulate cell autophagy through mTOR related pathway. AMPK activation can prevent hydrogen peroxide-induced impairment of the autophagic flux in senescent cells.62 By using AMPK inhibitor compound C or transfection of AMPK siRNA, we demonstrated that the up-regulating effect of catalpol on autophagy was mediated by AMPK activation. All above results indicated that catalpol attenuated oxidative stress and promotes autophagy in TNF-α-treated HAECs via activating AMPK. However, dual-luciferase reporter assay showed hardly any stimulatory effect of catalpol on AMPK promoter. Thus, we suggest that the effect of catalpol on AMPK may be associated with SIRT1, cAMP signaling, LKB1, CAMKK or other factors.63,64 Consequently, more experiments are needed to explore the precise regulatory mechanism of catalpol-mediated AMPK activation.
5. Conclusions
The present study is the first to reveal that catalpol, a poorly investigated natural product from Rehmannia has protective effects against TNF-α induced epithelial dysfunction in HAECs through reducing oxidative stress and enhancing autophagy level. And the protective effects of catalpol are associated with increased AMPK activity or at least partly through AMPK-mediated pathway, which is concluded in Fig. S5.† Thus, AMPK may represent an attractive pharmacological target to arrest endothelial dysfunction and AS progression. Importantly, our research provides a novel active ingredient for AS. Catalpol has highly favorable characteristics for the treatment of AS via activating AMPK to inhibit endothelial inflammation, oxidative stress and enhancing autophagy level.Conflicts of interest
The authors declare no conflicts of interest.Abbreviations
| AMPK | Adenosine 5′ monophosphate-activated protein kinase |
| TNF-α | Tumor necrosis factor-α |
| siRNA | Small interfere RNA |
| HAECs | Human aortic epithelial cells |
| AS | Atherosclerosis |
| ROS | Reactive oxygen species |
| NF-κB | Nuclear factor-kappa B |
| ICAM-1 | Intercellular cell adhesion molecule-1 |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| MCP-1 | Monocyte chemotactic protein-1 |
| MDC | Monodansylcadaverine |
| SOD | Superoxide dismutase |
| MDA | Malonaldehyde |
| GSH | Glutathione |
| LDH | Lactate dehydrogenase |
| H2DCFDA | 2′,7′-Dichlorodihydrofluorescein diacetate |
| Ac-p65 | Acetylated-p65 |
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 81273508).References
- X. Fan, J. Wang, J. Hou, C. Lin, A. Bensoussan, D. Chang, J. Liu and B. Wang, J. Transl. Med., 2015, 13, 92 CrossRef PubMed.
- M. L. Chen, L. Yi, X. Jin, X. Y. Liang, Y. Zhou, T. Zhang, Q. Xie, X. Zhou, H. Chang, Y. J. Fu, J. D. Zhu, Q. Y. Zhang and M. T. Mi, Autophagy, 2013, 9, 2033–2045 CrossRef CAS PubMed.
- P. Libby, P. M. Ridker and G. K. Hansson, Nature, 2011, 473, 317–325 CrossRef CAS PubMed.
- B. Wu, W. Qiu, P. Wang, H. Yu, T. Cheng, G. P. Zambetti, L. Zhang and J. Yu, Gut, 2007, 56, 645–654 CrossRef CAS PubMed.
- M. Yao, F. Gao, X. Wang, Y. Shi, S. Liu and H. Duan, Mol. Med. Rep., 2017, 15, 4319–4325 CrossRef CAS PubMed.
- E. Crosas-Molist, E. Bertran, I. Rodriguez-Hernandez, C. Herraiz, G. Cantelli, A. Fabra and V. Sanz-Moreno, Oncogene, 2017, 36, 3002–3014 CrossRef CAS PubMed.
- L. Xiao, L. Liu, X. Guo, S. Zhang, J. Wang, F. Zhou, L. Liu, Y. Tang and P. Yao, Food Chem. Toxicol., 2017, 105, 22–33 CrossRef CAS PubMed.
- W. Li, C. Wang, J. Peng, J. Liang, Y. Jin, Q. Liu, Q. Meng, K. Liu and H. Sun, Curr. Pharm. Biotechnol., 2014, 15, 1173–1182 CAS.
- C. K. Glass and J. L. Witztum, Cell, 2001, 104, 503–516 CrossRef CAS PubMed.
- G. Kroemer, G. Marino and B. Levine, Mol. Cell, 2010, 40, 280–293 CrossRef CAS PubMed.
- B. Razani, C. Feng, T. Coleman, R. Emanuel, H. Wen, S. Hwang, J. P. Ting, H. W. Virgin, M. B. Kastan and C. F. Semenkovich, Cell Metab., 2012, 15, 534–544 CrossRef CAS PubMed.
- Q. Bao, X. Shen, L. Qian, C. Gong, M. Nie and Y. Dong, Korean J. Physiol. Pharmacol., 2016, 20, 153–160 CrossRef CAS PubMed.
- P. B. Ham 3rd and R. Raju, Prog. Neurobiol., 2017, 157, 92–116 CrossRef PubMed.
- H. Yang, P. Zhao and S. Tian, J. Diabetes Res., 2016, 2016, 9128050 Search PubMed.
- A. M. D'Cruz and V. Pathiyil, South Asian Journal of Cancer, 2015, 4, 58–60 CrossRef PubMed.
- Q. Pan, X. H. Yang and Y. X. Cheng, Braz. J. Med. Biol. Res., 2009, 42, 531–536 CrossRef CAS PubMed.
- Z. Xia, F. Wang, S. Zhou, R. Zhang, F. Wang, J. H. Huang, E. Wu, Y. Zhang and Y. Hu, Oncotarget, 2017, 8, 69303–69315 Search PubMed.
- Y. Xiong, L. Shi, L. Wang, Z. Zhou, C. Wang, Y. Lin, D. Luo, J. Qiu and D. Chen, Pharmacol. Res., 2017, 123, 73–82 CrossRef CAS PubMed.
- X. Li, Z. Xu, Z. Jiang, L. Sun, J. Ji, J. Miao, X. Zhang, X. Li, S. Huang, T. Wang and L. Zhang, Acta Biochim. Biophys. Sin., 2014, 46, 738–748 CrossRef CAS PubMed.
- X. L. Zhang, B. Jiang, Z. B. Li, S. Hao and L. J. An, Pharmacol. Biochem. Behav., 2007, 88, 64–72 CrossRef CAS PubMed.
- X. Li, S. He, X. Zhou, Y. Ye, S. Tan, S. Zhang, R. Li, M. Yu, M. C. Jundt, A. Hidebrand, Y. Wang, G. Li, C. Huang and M. Wu, PLoS Pathog., 2016, 12, e1005363 Search PubMed.
- L. J. Niedernhofer, J. S. Daniels, C. A. Rouzer, R. E. Greene and L. J. Marnett, J. Biol. Chem., 2003, 278, 31426–31433 CrossRef CAS PubMed.
- S. R. Kim, Y. H. Bae, S. K. Bae, K. S. Choi, K. H. Yoon, T. H. Koo, H. O. Jang, I. Yun, K. W. Kim, Y. G. Kwon, M. A. Yoo and M. K. Bae, Biochim. Biophys. Acta, 2008, 1783, 886–895 CrossRef CAS PubMed.
- X. Zhang, W. Liu, X. Niu and L. An, Neurosci. Lett., 2010, 473, 224–228 CrossRef CAS PubMed.
- H. Yamada, M. Yoshida, Y. Nakano, T. Suganami, N. Satoh, T. Mita, K. Azuma, M. Itoh, Y. Yamamoto, Y. Kamei, M. Horie, H. Watada and Y. Ogawa, Arterioscler., Thromb., Vasc. Biol., 2008, 28, 2173–2179 CrossRef CAS PubMed.
- L. C. Rempel, A. B. Finco, R. A. Maciel, B. Bosquetti, L. M. Alvarenga, W. M. Souza, R. Pecoits-Filho and A. E. Stinghen, Toxins, 2015, 7, 1722–1737 CrossRef CAS PubMed.
- M. M. Mihaylova and R. J. Shaw, Nat. Cell Biol., 2011, 13, 1016–1023 CrossRef CAS PubMed.
- U. Sachdev and M. T. Lotze, J. Leukocyte Biol., 2017, 102, 221–235 CrossRef CAS PubMed.
- B. L. Tang, Mol. Cells, 2016, 39, 87–95 CrossRef CAS PubMed.
- S. A. Shah, G. H. Yoon, S. S. Chung, M. N. Abid, T. H. Kim, H. Y. Lee and M. O. Kim, Mol. Psychiatry, 2017, 22, 407–416 CrossRef CAS PubMed.
- N. B. Ruderman, X. J. Xu, L. Nelson, J. M. Cacicedo, A. K. Saha, F. Lan and Y. Ido, Am. J. Physiol.: Endocrinol. Metab., 2010, 298, E751–E760 CrossRef CAS PubMed.
- Y. M. Kim, M. Y. Kim, H. J. Kim, G. S. Roh, G. H. Ko, H. G. Seo, J. H. Lee and K. C. Chang, Atherosclerosis, 2011, 219, 57–64 CrossRef CAS PubMed.
- A. Zhang, S. Hao, J. Bi, Y. Bao, X. Zhang, L. An and B. Jiang, Exp. Toxicol. Pathol., 2009, 61, 461–469 CrossRef CAS PubMed.
- K. Fu, T. Piao, M. Wang, J. Zhang, J. Jiang, X. Wang and H. Liu, Int. Immunopharmacol., 2014, 23, 400–406 CrossRef CAS PubMed.
- G. de Moraes and C. J. Layton, Clin. Exp. Ophthalmol., 2016, 44, 838–852 Search PubMed.
- C. Liu, A. G. Rajapakse, E. Riedo, B. Fellay, M. C. Bernhard, J. P. Montani, Z. Yang and X. F. Ming, Sci. Rep., 2016, 6, 20405 CrossRef CAS PubMed.
- S. B. Vasamsetti, S. Karnewar, R. Gopoju, P. N. Gollavilli, S. R. Narra, J. M. Kumar and S. Kotamraju, Free Radical Biol. Med., 2016, 96, 392–405 CrossRef CAS PubMed.
- L. Buldak, K. Labuzek, R. J. Buldak, G. Machnik, A. Boldys, M. Basiak and O. Boguslaw, Exp. Ther. Med., 2017, 13, 794 CrossRef PubMed.
- H. Zhu, Y. Wang, Z. Liu, J. Wang, D. Wan, S. Feng, X. Yang and T. Wang, Chin. Med., 2016, 11, 25 CrossRef PubMed.
- R. Velagapudi, A. El-Bakoush, I. Lepiarz, F. Ogunrinade and O. A. Olajide, Mol. Cell. Biochem., 2017 DOI:10.1007/s11010-017-3064-3.
- Y. Taniyama and K. K. Griendling, Hypertension, 2003, 42, 1075–1081 CrossRef CAS PubMed.
- A. Ugusman, Z. Zakaria, C. K. Hui and N. A. Nordin, BMC Complementary Altern. Med., 2011, 11, 31 CrossRef PubMed.
- H. Xu, C. Goettsch, N. Xia, S. Horke, H. Morawietz, U. Forstermann and H. Li, Free Radical Biol. Med., 2008, 44, 1656–1667 CrossRef CAS PubMed.
- R. Ray, C. E. Murdoch, M. Wang, C. X. Santos, M. Zhang, S. Alom-Ruiz, N. Anilkumar, A. Ouattara, A. C. Cave, S. J. Walker, D. J. Grieve, R. L. Charles, P. Eaton, A. C. Brewer and A. M. Shah, Arterioscler., Thromb., Vasc. Biol., 2011, 31, 1368–1376 CrossRef CAS PubMed.
- D. Sorescu, D. Weiss, B. Lassegue, R. E. Clempus, K. Szocs, G. P. Sorescu, L. Valppu, M. T. Quinn, J. D. Lambeth, J. D. Vega, W. R. Taylor and K. K. Griendling, Circulation, 2002, 105, 1429–1435 CrossRef CAS PubMed.
- M. Zhang, C. M. Wang, J. Li, Z. J. Meng, S. N. Wei, J. Li, R. Bucala, Y. L. Li and L. Chen, Mediators Inflammation, 2013, 2013, 260464 Search PubMed.
- G. Spanier, H. Xu, N. Xia, S. Tobias, S. Deng, L. Wojnowski, U. Forstermann and H. Li, J. Physiol. Pharmacol., 2009, 60(suppl. 4), 111–116 Search PubMed.
- A. A. Eid, B. M. Ford, K. Block, B. S. Kasinath, Y. Gorin, G. Ghosh-Choudhury, J. L. Barnes and H. E. Abboud, J. Biol. Chem., 2010, 285, 37503–37512 CrossRef CAS PubMed.
- G. H. Li, X. L. Lin, H. Zhang, S. Li, X. L. He, K. Zhang, J. Peng, Y. L. Tang, J. F. Zeng, Y. Zhao, X. F. Ma, J. J. Lei, R. Wang, D. H. Wei, Z. S. Jiang and Z. Wang, Atherosclerosis, 2015, 243, 223–235 CrossRef CAS PubMed.
- Y. L. Tang, J. H. Jiang, S. Wang, Z. Liu, X. Q. Tang, J. Peng, Y. Z. Yang and H. F. Gu, PLoS One, 2015, 10, e0123685 Search PubMed.
- C. J. Liang, S. H. Wang, Y. H. Chen, S. S. Chang, T. L. Hwang, Y. L. Leu, Y. C. Tseng, C. Y. Li and Y. L. Chen, Free Radical Biol. Med., 2011, 51, 1337–1346 CrossRef CAS PubMed.
- X. Liu, L. Pan, X. Wang, Q. Gong and Y. Z. Zhu, Atherosclerosis, 2012, 222, 34–42 CrossRef CAS PubMed.
- B. Lu, B. Wang, S. Zhong, Y. Zhang, F. Gao, Y. Chen, F. Zheng and G. Shi, Oncotarget, 2016, 7, 34800–34810 Search PubMed.
- F. Chen, P. Eriksson, T. Kimura, I. Herzfeld and G. Valen, Coron. Artery Dis., 2005, 16, 191–197 CrossRef PubMed.
- J. F. Zhao, A. H. Sun, P. Ruan, X. H. Zhao, M. Q. Lu and J. Yan, Microb. Pathog., 2009, 46, 194–200 CrossRef CAS PubMed.
- Y. Tsujimoto, L. R. Finger, J. Yunis, P. C. Nowell and C. M. Croce, Science, 1984, 226, 1097–1099 CAS.
- G. Y. Wang, Y. G. Bi, X. D. Liu, J. F. Han, M. Wei and Q. Y. Zhang, Mol. Med. Rep., 2017, 16, 3262–3268 Search PubMed.
- A. Kumar, U. K. Singh and A. Chaudhary, Future Med. Chem., 2015, 7, 1535–1542 CrossRef CAS PubMed.
- T. Shen, O. Alvarez-Garcia, Y. Li, M. Olmer and M. K. Lotz, Osteoarthritis Cartilage, 2017, 25, 287–296 CrossRef CAS PubMed.
- X. Jin, M. Chen, L. Yi, H. Chang, T. Zhang, L. Wang, W. Ma, X. Peng, Y. Zhou and M. Mi, Mol. Nutr. Food Res., 2014, 58, 1941–1951 CAS.
- B. H. Li, S. Q. Liao, Y. W. Yin, C. Y. Long, L. Guo, X. J. Cao, Y. Liu, Y. Zhou, C. Y. Gao, L. L. Zhang and J. C. Li, Mol. Biol. Rep., 2015, 42, 179–186 CrossRef CAS PubMed.
- X. Han, H. Tai, X. Wang, Z. Wang, J. Zhou, X. Wei, Y. Ding, H. Gong, C. Mo, J. Zhang, J. Qin, Y. Ma, N. Huang, R. Xiang and H. Xiao, Aging Cell, 2016, 15, 416–427 CrossRef CAS PubMed.
- K. Inoki, J. Kim and K. L. Guan, Annu. Rev. Pharmacol. Toxicol., 2012, 52, 381–400 CrossRef CAS PubMed.
- E. Y. Chan, Antioxid. Redox Signaling, 2012, 17, 775–785 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09085d |
| This journal is © The Royal Society of Chemistry 2017 |