
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comparison of macrocyclic and acyclic chelators for gallium-68 radiolabelling†
Maria Iris Tsionoua,
Caroline E. Knapp b,
Calum A. Foleya,
Catherine R. Munteanua,
Andrew Cakebreadc,
Cinzia Imbertia,
Thomas R. Eykyna,
Jennifer D. Younga,
Brett M. Patersond,
Philip J. Blowera and
Michelle T. Ma*a
b,
Calum A. Foleya,
Catherine R. Munteanua,
Andrew Cakebreadc,
Cinzia Imbertia,
Thomas R. Eykyna,
Jennifer D. Younga,
Brett M. Patersond,
Philip J. Blowera and
Michelle T. Ma*a
aKing's College London, Division of Imaging Sciences and Biomedical Engineering, St Thomas' Hospital, London SE1 7EH, UK. E-mail: michelle.ma@kcl.ac.uk
bDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK
cDivision of Analytical and Environmental Sciences, King's College London, Franklin Wilkin's Building, London SE1 9NH, UK
dSchool of Chemistry, Bio21 Molecular Science and Biotechnology Institute, The University of Melbourne, 3010, Victoria, Australia
First published on 25th October 2017
Abstract
Gallium-68 (68Ga) is a positron-emitting isotope used for clinical PET imaging of peptide receptor expression. 68Ga radiopharmaceuticals used in molecular PET imaging consist of disease-targeting biomolecules tethered to chelators that complex 68Ga3+. Ideally, the chelator will rapidly, quantitatively and stably coordinate 68Ga3+ at room temperature, near neutral pH and low chelator concentration, allowing for simple routine radiopharmaceutical formulation. Identification of chelators that fulfil these requirements will facilitate development of kit-based 68Ga radiopharmaceuticals. Herein the reaction of a range of widely used macrocyclic and acyclic chelators with 68Ga3+ is reported. Radiochemical yields have been measured under conditions of varying chelator concentrations, pH (3.5 and 6.5) and temperature (25 and 90 °C). These chelators are: 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA), 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA), 1,4,7-triazacyclononane macrocycles substituted with phosphonic (NOTP) and phosphinic (TRAP) groups at the amine, bis(2-hydroxybenzyl)ethylenediaminediacetic acid (HBED), a tris(hydroxypyridinone) containing three 1,6-dimethyl-3-hydroxypyridin-4-one groups (THP) and the hexadentate tris(hydroxamate) siderophore desferrioxamine-B (DFO). Competition studies have also been undertaken to assess relative complexation efficiencies of each chelator for 68Ga3+ under different pH and temperature conditions. Performing radiolabelling reactions at pH 6.5, 25 °C and 5–50 μM chelator concentration resulted in near quantitative radiochemical yields for all chelators, except DOTA. Radiochemical yields either decreased or were not substantially improved when the reactions were undertaken at lower pH or at higher temperature, except in the case of DOTA. THP and DFO were the most effective 68Ga3+ chelators at near-neutral pH and 25 °C, rapidly providing near-quantitative radiochemical yields at very low chelator concentrations. NOTP and HBED were only slightly less effective under these conditions. In competition studies with all other chelators, THP demonstrated highest reactivity for 68Ga3+ complexation under all conditions. These data point to THP possessing ideal properties for rapid, one-step kit-based syntheses of 68Ga-biomolecules for molecular PET imaging. LC-MS and 1H, 13C{1H} and 71Ga NMR studies of HBED complexes of Ga3+ showed that under the analytical conditions employed in this study, multiple HBED-bound Ga complexes exist. X-ray diffraction data indicated that crystals isolated from these solutions contained octahedral [Ga(HBED)(H2O)], with HBED coordinated in a pentadentate N2O3 mode, with only one phenolic group coordinated to Ga3+, and the remaining coordination site occupied by a water molecule.
Introduction
Gallium-68 (68Ga) is a positron-emitting isotope with emission properties (t1/2 = 68 min, β+ 90%, Emax = 1880 keV) that make it suitable for diagnostic imaging with positron emission tomography (PET). A pharmaceutical grade 68Ge/68Ga generator has recently become commercially available,1 providing hospitals with on-site access to a GMP-grade diagnostic PET radionuclide without the need for local cyclotron facilities. The most widely utilised 68Ga radiopharmaceuticals consist of 68Ga coordinated to a chelator that is attached to a peptide for targeting cell-surface receptors of tumours. Numerous centres already routinely produce diagnostic 68Ga–HBED–PSMA2,3 and 68Ga–DOTA–TATE4–6 for whole-body PET imaging of prostate and neuroendocrine cancers respectively. These radiotracers have had a significant impact on patient management in centres where they are available, but the complexity of their radiosynthesis in hospitals is a barrier to widespread implementation.Radiosynthesis of 68Ga–DOTA–TATE requires heating at 80–100 °C in order for the DOTA chelator to chelate radiopharmaceutical concentrations of 68Ga3+ with yields greater than 80%.7–9 On the other hand, 68Ga–HBED–PSMA can be prepared at ambient temperatures, but HBED forms multiple species when complexed to Ga3+.10 This is undesirable as it is possible that the different species have different pharmacological profiles. Heating is employed to increase formation of the most thermodynamically favoured compound, although the structure of this complex has not been defined. Even with heating, populations of other isomers are observed.10 The radiosyntheses of 68Ga–DOTA–TATE and 68Ga–HBED–PSMA are undertaken at pH 3–5.
As a result of heating requirements at acidic pH, clinical radiosyntheses of both 68Ga–DOTA–TATE and 68Ga–HBED–PSMA require multiple manipulations or complex automated equipment.9,10 Typical 68Ga radiopharmaceutical syntheses involve (i) elution of 68Ga from a generator, (ii) pretreatment of eluate to remove contaminating metal impurities that interfere with radiolabelling, as well as 68Ge “breakthrough”, (iii) addition of 68Ga to aqueous solutions of peptide–chelator precursor at pH 3–5, (iv) heating for 5–10 min (followed by cooling) (v) removal of unreacted 68Ga and buffering salts (using solid phase extraction cartridges) and (vi) reconstitution in physiologically compatible solutions for patient administration. In centres that are equipped for more complex preparations of 18F radiopharmaceuticals, this is not a barrier to routine radiosynthesis but it is time-consuming and costly. However, in regional healthcare centres, or hospitals in countries with developing healthcare systems, such complexity will be a barrier to widespread implementation.
Chelators that quantitatively coordinate 68Ga3+ at near-neutral pH, room temperature and low concentrations of chelator-bioconjugate will enable one-step, kit-based radiolabelling protocols, with concomitant widespread patient benefit. Such radiosyntheses would ideally only require a kit vial containing bioconjugate and buffer components, 68Ga generator eluate, a syringe and appropriate radiation shielding. Chelators that fulfil these requirements would also be useful for radiolabelling of small proteins that are susceptible to unfolding or degradation at extremes of pH and temperature. Over the past decade, several chelators have been evaluated and/or developed for 68Ga radiolabelling of biomolecules, to overcome the limitations of DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid). These include chelators based on 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA and its derivative NODAGA),11–15 1,4,7-triazacyclononane (tacn) macrocycles substituted with phosphonic (NOTP16,17) and phosphinic (TRAP18,19) groups at the amine, hexaazamacrobicycles,20 a pyridyl-substituted DOTA macrocycle (PCTA),15,21 bis(2-hydroxybenzyl)ethylenediaminediacetic acid (HBED) and related compounds possessing phenol, amine and carboxyl donor groups,3,22 6-amino-1,4-diazepanes with acetate substituents at the amines (DATA),23–26 a siderophore-derived macrocyclic chelator with hydroxamate groups (FSC),27 the acyclic siderophore desferrioxamine-B (which also contains hydroxamates),28,29 and an acyclic chelator based on a substituted pyridine carboxylate with an N4O2 binding mode (DEDPA) (Chart 1).30–32 Our research group has recently developed tris(hydroxypyridinone) (THP) derivatives based on 1,6-dimethyl-3-hydroxypyridin-4-one units.33–39 Of these chelators, NOTA/NODAGA, TRAP/NOPO, HBED, FSC, DATA, DFO, DEDPA and THP can reportedly be radiolabelled with 68Ga3+ at ambient temperature. Only DATA derivatives24 and THP derivatives33–35,37,38 have been reported to complex 68Ga3+ above pH 5 at ambient temperature. Many of these ligands provide highly rigid and inert Ga3+–chelator complexes. Rigidity is imparted by both selection of appropriate “hard” donor atoms with high affinity for Ga3+, which has a relatively high charge density, and the geometry or topology of the chelator itself. Chelators with pre-arranged conformations18,26,32 that accommodate octahedral binding of Ga3+ favour very high complex rigidity, contributing to kinetic stability of the resulting Ga3+ complex.
 | ||
| Chart 1 | ||
Most radiosyntheses of 68Ga–chelator complexes have been undertaken in acidic solution, below pH 5. This is because hydrated Ga3+ species such as [Ga(H2O)6]3+ predominate in solution below pH 4 but as the pH is raised above 4, the poorly soluble hydroxide species Ga(OH)3 is formed, until the pH exceeds 6.3, where tetracoordinate [Ga(OH)4]− predominates,40–43 although this is strongly dependant on temperature and is influenced by concentrations of other metal ions and coordinating molecules in solution.44,45 For efficient 68Ga3+ radiolabelling of chelate–peptide conjugates above pH 4, chelate complex formation must effectively compete with 68Ga-colloid formation. Preferably, the rate of chelation will be diffusion-controlled, so that complex formation outcompetes 68Ga3+ colloid formation. The amounts of 68Ga eluted from clinical generators are in the range of 400–2000 MBq, approximately equivalent to 4–20 pmol of 68Ga3+ in 1–5 mL of solution, i.e. nanomolar concentrations. Highly efficient chelators are required to quantitatively coordinate such low concentrations of metal ion without excessively high chelator concentrations.
For most of the chelators mentioned above, their Ga3+ complexes and their complexes with some other metal ions, metal stability constants and protonation constants have been reported (Table 1). These data are very useful in predicting the ability of a chelator to coordinate Ga3+, the selectivity of a chelator for Ga3+ over other metal ions, and the ability of other ligands such as hydroxide ions, to compete for Ga3+ binding under physiological conditions. These data do not, however, predict the kinetics of complexation, and without very detailed speciation studies, they do not describe the complexity of the reaction matrix in 68Ga radiolabelling solutions where other adventitious metal ions are present, as well as buffer components.
| Chelator | log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Ka Ka |
logK1 |
|---|---|---|
| a In the absence of published stability constants for THP, we have included logβ3 values for the related [Ga(deferiprone)3] complex (deferiprone = 1,2-dimethyl-3-hydroxypyridin-4-one). |
||
| DOTA48,49 | 11.74, 9.76, 4.68, 4.11, 2.37 | 26.05 |
| NOTA18,50 | 13.17, 5.74, 3.22, 1.96 | 29.63 |
| NOTP51 | 11.7, 9.1, 7.5, 5.8, 3.1, 0.9 | — |
| TRAP19 | 11.48, 5.44, 4.84, 4.23, 3.45, 1.66 | 26.24 |
| HBED52 | 12.60, 11.00, 8.44, 4.72, 2.53, 1.74 | 39.57 |
| DFO53 | 10.79, 9.55, 8.96, 8.32 | 28.65 |
| Deferipronea,54,55 | 9.86, 3.70;54 9.78, 3.6155 |
(logβ3) 38.42;54 37.3555 |
To the best of our knowledge, there have been no comprehensive side-by-side comparisons of Ga3+ chelators to evaluate relative radiolabelling efficiencies. Several prior studies have compared 68Ga radiolabelling for a limited number of chelators.16,21,24,33,35,46,47 Some of these studies have compared different chelator concentrations or amounts, and all of these studies only explore one or two specific reaction conditions. In some of these studies including our own, the 68Ga radiolabelling conditions used for each chelator are not identical.33,35,46 Therefore, to identify the most suitable chelators to take forward for kit-based 68Ga radiolabelling, we have compared the efficiency of 68Ga radiolabelling of commercially available DOTA, NOTA, NOTP, TRAP, HBED, THP and DFO, under four different reaction conditions (high and ambient temperatures, and neutral and low pH conditions), and across five orders of magnitude of chelator concentration (50 nM to 500 mM). We also report data that reveal the complexity of Ga3+–HBED coordination.
Experimental
Materials and instrumentation
All solvents and reagents were obtained from Sigma-Aldrich (Dorset, UK) unless otherwise indicated. DOTA, NOTA and NOTP were purchased from Macrocyclics (Dallas, USA). TRAP was purchased from CheMatech (Dijon, France). HBED was purchased from Santa Cruz Biotechnology (Dallas, USA). DFO was purchased from Sigma-Aldrich. THP was synthesised in our laboratory according to a previously reported procedure.33,56 The purchased chemicals were used without further purification. NMR spectra were acquired on Bruker Avance 400 MHz spectrometers (either narrow-bore or wide-bore) (Bruker, Germany) equipped with either a 5 mm QNP probe or a 5 mm BBO probe at 298 K. Spectra were referenced to residual solvent signals or TMS. High-performance liquid chromatography (HPLC) analysis was carried out using an Agilent 1200 LC system with in-line UV and gamma detection (Flow-Count, LabLogic). Instant thin layer chromatography plates (iTLC-SG) were obtained from Agilent Technologies (California, USA) and iTLC strips were visualized and quantified using a Cyclone Plus Storage Phosphor System (Perkin Elmer) interfaced with OptiQuant V5.0 software (Perkin Elmer). Analytical reverse phase HPLC were acquired using an Agilent Eclipse XDB-C18 column (9.4 × 250 mm, 5 μm) and UV spectroscopic detection at 220 nm. Aliquots (50 μL) of each radiolabelled sample were injected onto the column, using a flow rate of 1 mL min−1, and the following gradient: 0–5 min: 100% A/0% B; 5–25 min: 100% A/0% B to 60% A/40% B. Mobile phase A comprised water with 0.1% trifluoroacetic acid and mobile phase B comprised acetonitrile with 0.1% trifluoroacetic acid. Analytical LC-MS were recorded in the positive ion mode on an Agilent 6510 Q-TOF LC/MS mass spectrometer coupled to an Agilent 1200 LC system (Agilent, Palo Alto, CA) and a LabLogic scintillation detector Flow-count system (Sheffield, UK). An Agilent Eclipse XDB-C18 column (9.4 × 250 mm, 5 μm) and UV spectroscopic detection at 220 nm was used with a flow rate of 1 mL min−1, and the following gradient: 0–5 min: 100% A/0% B; 5–25 min: 100% A/0% B to 0% A/100% B. Mobile phase A comprised water with 0.1% formic acid and mobile phase B comprised acetonitrile with 0.1% formic acid. Elemental analysis was performed by the Science Centre, London Metropolitan University.68Ga radiolabelling and iTLC quantification
68Ga was eluted from an Eckert & Ziegler 68Ge/68Ga generator system (Berlin, Germany). Aqueous HCl solution (0.1 M, 5 mL) was passed through the generator and the eluate was collected in 5 × 1 mL fractions. Aliquots of the second fraction (1 mL, containing 130–230 MBq 68Ga) were used directly for radiolabelling reactions.Chelators were dissolved in aqueous solutions of sodium acetate (0.2 M) or ammonium acetate (0.2 M) to provide solutions with chelator concentrations ranging from 50 nM to 1 mM (50 nM, 500 nM, 5 μM, 50 μM, 500 μM, and 1 mM). Ligand solutions were freshly prepared from stock solutions for each experiment. 68Ga (10 μL, approx. 2 MBq in 0.1 M aqueous HCl) was added to chelator solutions (100 μL) and the reaction solution was incubated at either 25 or 90 °C. The final pH of the reaction solutions was 3.5 and pH 6.5 for the sodium acetate and ammonium acetate solutions respectively. After 10 min, the reaction solution was analysed by iTLC (glass microfiber chromatography paper impregnated with silica gel, 80 × 10 mm).
Separately, solutions of 68Ga3+ (10 μL, approx. 2 MBq in 0.1 M aqueous HCl) were added to aqueous solutions of sodium acetate (0.2 M) or ammonium acetate (0.2 M), and incubated at either 25 or 90 °C. After 10 min, the solutions were analysed by iTLC.
Three different mobile phases were employed for iTLC:
(1) For THP, DFO and chelator-free reactions under all reaction conditions, aqueous sodium citrate solution (0.1 M, pH 5.5) was used. [68Ga(chelator)] Rf < 0.1; non-chelated, soluble 68Ga3+ Rf > 0.9; 68Ga colloids: <0.1. Based on quantification of 68Ga colloid and soluble 68Ga3+ in chelator-free reactions, RCY values were adjusted to account for coincident Rf values of 68Ga colloid, [68Ga(THP)] and [68Ga(DFO)].
(2) For NOTP under all reaction conditions, and DOTA, NOTA, TRAP and HBED, under all conditions except pH 3.5, 90 °C, aqueous sodium phosphate solution (0.4 M, pH 4) was used. [68Ga(NOTP)] Rf = 0.6–0.7; DOTA, NOTA, TRAP and HBED: [68Ga(chelator)] Rf = 0.8–1; non-chelated 68Ga3+ Rf < 0.1.
(3) For DOTA, NOTA, TRAP and HBED at pH 3.5 and 90 °C, an ammonium acetate solution (1 M in 80% methanol, 20% water) was used. [68Ga(DOTA)] Rf = 0.65–0.75; [68Ga(NOTA)] Rf = 0.8–0.9; [68Ga(HBED)] Rf = 0.9–1; [68Ga(TRAP)] Rf = 0.4–0.6; non-chelated 68Ga3+ Rf = <0.3. These conditions were selected because after heating 68Ga solutions at pH 3.5 and 90 °C, two distinct compounds were observed for non-chelated 68Ga3+: Rf = 0–0.1 and 0.7–0.9 using mobile phase (2). Thus, non-chelated 68Ga3+ could not be distinguished from [68Ga(chelator)] using mobile phase (2) after heating at pH 3.5 and 90 °C (except in the case of [68Ga(NOTP)]).
iTLC conditions and Rf values are also summarised in Tables S3–S6.†
iTLC plates were imaged and quantified by digital autoradiography using instruments and software described above.
ICP-MS analysis of 68Ga generator eluate
Fractionated eluate (as described above) was allowed to decay for several days before it was analysed by ICP-MS. The quantification of metal contaminants was carried out on a PerkinElmer NexION 350D Inductively coupled plasma mass spectrometer (ICP-MS) running Syngistix v1.0 software with a CETAC ASX520 autosampler (King's College London, UK). The acquisition mode included 5 replicates averaged to give reported values (Fig. 2, S12 and S13†) for 27Al, 59Co, 52Cr, 65Cu, 56Fe, 69Ga, 72Ge, 55Mn, 60Ni, 208Pb, 45Sc, 118Sn, 47Ti, 51V, 66Zn and 68Zn. The dwell time was 50 ms per isotope, with 18 L min−1 main argon flow, 1.2 L min−1 auxiliary argon flow, 0.97 L min−1 nebuliser argon flow (optimised), 1600 W RF power, 0.2 mL min−1 sample flow, and KED cell mode with 1.2 mL min−1 helium flow.HPLC analysis of [68Ga(chelator)] complexes and competition studies
68Ga3+ generator eluate (10 μL in 0.1 M aqueous HCl, approx. 2 MBq) was added to chelator solutions (1 mM chelator, 100 μL in either 0.2 M ammonium acetate, or 0.2 M sodium acetate) and the reaction mixtures were incubated at either 25 °C or 90 °C for 10 min, after which they were applied to an analytical reverse phase C18 HPLC column.For competition studies, 68Ga3+ generator eluate (20 μL in 0.1 M aqueous HCl, approx. 4 MBq) was added to a solution containing equimolar concentrations of two chelators (each 500 μM in either 200 μL 0.2 M ammonium acetate, or 200 μL 0.2 M sodium acetate) and the reaction mixtures were incubated at either 25 °C or 90 °C for 40 min, after which they were applied to an analytical reverse phase C18 HPLC column. Data were processed and analysed using Laura Radiochromatography Software (LabLogic).
Preparation of [natGa(HBED)]
A sample of HBED (40 mg, 0.09 mmol) was reacted with Ga(NO3)3·xH2O (40 mg, 0.15 mmol, 1.6 equiv.) in aqueous ammonium acetate solution (0.2 M, 5–10 mL) and heated at 90 °C for 30 min. The solution was then applied to an Agilent Eclipse semi-preparative reverse phase XDB-C18 column (9.4 × 250 mm, 5 μm) with a 3 mL min−1 flow rate, and the reaction solution was purified using a gradient elution, in which mobile phase A consisted of water containing 0.1% TFA and mobile phase B consisted of acetonitrile containing 0.1% TFA. The concentration of B increased at a rate of 1% min−1. [Ga(HBED)] eluted with a retention time of 32 min. Fractions containing pure [Ga(HBED)] were combined, lyophilised, and 1H and 13C{1H} NMR spectra (in both D2O/CD3OD (50%/50%) and D2O/CD3CN (60%/40%)), and LC-MS chromatograms were acquired. Data are reported in Fig. 4, 5 and S4–S8.†Crystals of Ga–HBED of were obtained from a solution of D2O and CD3CN. Anal. calc. for [Ga(HBED)(H2O)]·CH3CN (C22H26GaN3O7): C, 51.39; H, 5.10; N, 8.17. Found: C, 51.23; H, 5.25; N, 8.10. A suitable crystal containing [Ga(HBED)(H2O)] was selected and mounted on a nylon loop on a SuperNova Atlas diffractometer with Cu Kα radiation (λ = 1.54184 Å). The crystal was kept at 150.4(5) K during data collection. Using Olex2,57 the structure was solved with the ShelXS58 structure solution program using Direct Methods, and refined with the ShelXL59 refinement package using Least Squares minimisation.
Crystal structure determination
Crystal data for [Ga(HBED)(H2O)]·CH3CN (C22H26GaN3O7) (M = 514.18 g mol−1): monoclinic, space group P21/c (no. 14), a = 12.96001(14) Å, b = 7.01939(10) Å, c = 25.0910(3) Å, β = 97.7700(10)°, V = 2261.60(5) Å3, Z = 4, T = 150.4(5) K, μ(CuKα) = 2.093 mm−1, Dcalc = 1.510 g cm−3, 34357 reflections measured (6.884° ≤ 2Θ ≤ 147.354°), 4541 unique (Rint = 0.0461, Rsigma = 0.0263) which were used in all calculations. The final R1 was 0.0292 (I > 2σ(I)) and wR2 was 0.0747 (all data). The identification code is xstr0762. Deposit number for [Ga(HBED)(H2O)]·CH3CN: CCDC 1564603.†
Results
A comprehensive selection of chelators was reacted with generator-produced solutions of 68Ga3+ at a range of chelator concentrations, at high (90 °C) and room (25 °C) temperatures, and in acidic (pH 3.5) and near neutral (pH 6.5) aqueous acetate solutions. In all cases, the reaction time was 10 min. The chelators are: macrocyclic DOTA, NOTA, NOTP and TRAP, and acyclic HBED, DFO and THP (Chart 1). [Note: for ease of nomenclature, charge and protonation states are not included in these abbreviations of the ligands or complexes.]Quantifying the efficiency of 68Ga3+ chelation
Whilst many ligands will chelate a metal quantitatively if the concentration is high enough, only the most efficient will continue to do so as the concentration is reduced.12,21,36,46,60,61 A series of reactions was undertaken in which a solution of generator-produced 68Ga3+ (approx. 2 MBq in 0.1 M aqueous HCl, 10 μL) was added to a solution of chelator at a concentration in the range 500 μM to 50 nM (100 μL). The final pH of the reaction solution was 3.5 (using 0.2 M sodium acetate) or 6.5 (using 0.2 M ammonium acetate). After 10 min reaction time, radiochemical yields (RCY) were measured using instant thin layer chromatography (iTLC). Experimental data are supplied for each chelator in Fig. 1, S1, S2† and Table 2. A summary of iTLC conditions is provided in Tables S3–S6.†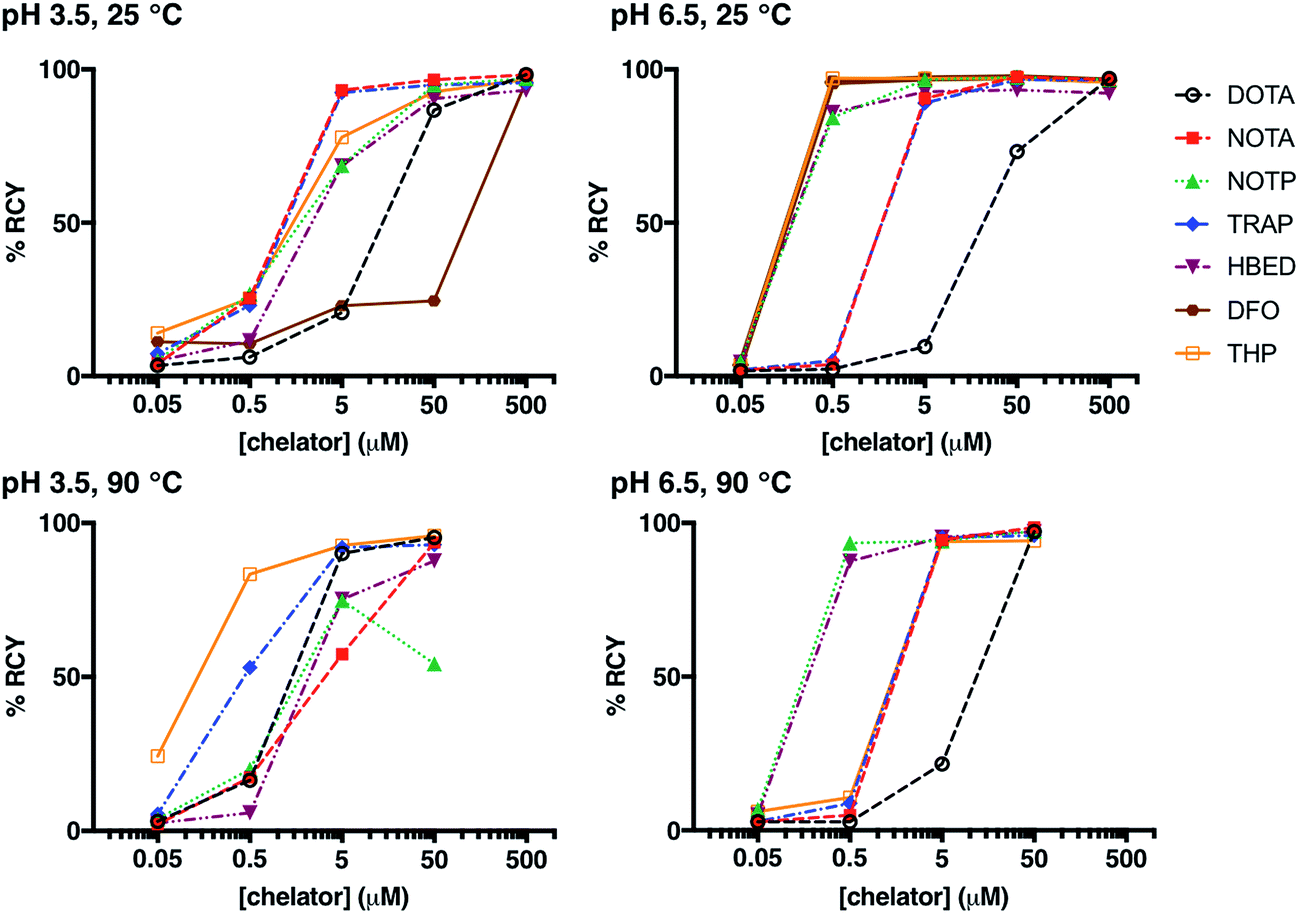 | ||
| Fig. 1 Radiochemical yields for the reaction of 68Ga3+ with DOTA, NOTA, NOTP, TRAP, HBED, DFO and THP under different concentrations of chelator (500 μM to 50 nM); different pH conditions (pH 3.5 or pH 6.5); and different temperatures (25 °C or 90 °C), after 10 min reaction. | ||
| Chelator | Concentration (μM) | pH 3.5, 25 °C | pH 3.5, 90 °C | pH 6.5, 25 °C | pH 6.5, 90 °C |
|---|---|---|---|---|---|
| DOTA | 500 | 98.3 ± 0.4 | 97.0 ± 0.7 | ||
| 50 | 86.7 ± 5.0 | 95.3 ± 0.9 | 73.2 ± 6.4 | 97.2 ± 0.3 | |
| 5 | 20.7 ± 5.6 | 90.1 ± 1.5 | 9.6 ± 5.9 | 21.6 ± 2.5 | |
| 0.5 | 6.2 ± 2.8 | 16.5 ± 2.2 | 2.3 ± 0.7 | 2.8 ± 0.2 | |
| 0.05 | 3.5 ± 1.1 | 3.1 ± 0.7 | 1.7 ± 0.5 | 2.8 ± 0.5 | |
| NOTA | 500 | 98.2 ± 0.6 | 96.6 ± 1.2 | ||
| 50 | 96.2 ± 1.7 | 93.7 ± 0.3 | 97.5 ± 0.1 | 98.5 ± 0.1 | |
| 5 | 93.2 ± 2.0 | 57.3 ± 2.6 | 90.6 ± 4.5 | 94.4 ± 0.4 | |
| 0.5 | 25.4 ± 35.6 | 17.4 ± 2.1 | 3.8 ± 0.3 | 4.9 ± 0.4 | |
| 0.05 | 4.0 ± 2.2 | 2.2 ± 0.5 | 1.9 ± 0.7 | 2.8 ± 0.2 | |
| NOTP | 500 | 97.0 ± 0.8 | 96.7 ± 1.8 | ||
| 50 | 95.0 ± 1.8 | 54.3 ± 2.4 | 97.5 ± 0.1 | 97.3 ± 0.4 | |
| 5 | 68.7 ± 25.0 | 74.9 ± 3.4 | 96.6 ± 0.4 | 94.2 ± 0.9 | |
| 0.5 | 26.8 ± 33.0 | 20.0 ± 1.9 | 84.3 ± 0.9 | 93.5 ± 0.8 | |
| 0.05 | 5.2 ± 3.7 | 3.9 ± 0.7 | 4.8 ± 0.4 | 6.8 ± 1.2 | |
| TRAP | 500 | 95.6 ± 0.7 | 96.5 ± 0.4 | ||
| 50 | 95.0 ± 1.3 | 93.0 ± 0.6 | 96.6 ± 0.1 | 96.0 ± 1.4 | |
| 5 | 92.5 ± 2.4 | 92.0 ± 1.5 | 89.0 ± 1.4 | 95.2 ± 0.4 | |
| 0.5 | 23.0 ± 9.5 | 53.1 ± 4.8 | 5.1 ± 0.8 | 8.8 ± 2.0 | |
| 0.05 | 7.4 ± 1.5 | 5.5 ± 0.6 | 2.0 ± 0.5 | 2.8 ± 0.5 | |
| HBED | 500 | 93.2 ± 3.8 | 92.2 ± 0.6 | ||
| 50 | 90.4 ± 8.1 | 87.7 ± 0.5 | 93.3 ± 3.0 | 97.2 ± 0.4 | |
| 5 | 68.6 ± 23.5 | 75.2 ± 0.8 | 92.7 ± 2.2 | 95.4 ± 0.8 | |
| 0.5 | 11.6 ± 11.9 | 5.8 ± 0.4 | 86.0 ± 3.9 | 87.5 ± 4.4 | |
| 0.05 | 4.9 ± 2.2 | 2.5 ± 0.4 | 4.6 ± 0.7 | 5.1 ± 0.8 | |
| DFO | 500 | 96.0 ± 0.8 | 96.4 ± 1.4 | ||
| 50 | 24.5 ± 1.9 | 97.5 ± 0.7 | |||
| 5 | 23.0 ± 12.9 | 97.0 ± 1.0 | |||
| 0.5 | 10.5 ± 5.4 | 95.8 ± 1.5 | |||
| 0.05 | 11.2 ± 5.0 | 3.6 ± 1.3 | |||
| THP | 500 | 96.7 ± 1.4 | 96.2 ± 0.8 | 95.8 ± 1.6 | 94.3 ± 0.7 |
| 50 | 92.7 ± 4.2 | 95.9 ± 0.5 | 97.1 ± 1.1 | 94.2 ± 0.6 | |
| 5 | 77.8 ± 13.5 | 92.7 ± 1.1 | 97.1 ± 0.1 | 94.0 ± 0.3 | |
| 0.5 | 25.4 ± 6.8 | 83.4 ± 1.1 | 97.1 ± 0.6 | 10.7 ± 2.6 | |
| 0.05 | 14.0 ± 6.8 | 24.3 ± 3.4 | 3.8 ± 3.6 | 6.1 ± 1.1 |
At pH 6.5, 25 °C, and a chelator concentration of 5 μM, RCYs were greater than 85% for all chelators except for DOTA. At pH 6.5 and 25 °C, at very low chelator concentrations of 500 nM and 5 μM, the best performing chelators were DFO and THP. At concentrations of 5 μM, RCY of [68Ga(DFO)] was 97 ± 1.0%, and RCY of [68Ga(THP)] was 97 ± 0.1%. At 500 nM, RCY of [68Ga(DFO)] was 96 ± 1.5% and the RCY of [68Ga(THP)] was 97 ± 0.6%.
The pKa values for deprotonation of coordinating O donor atoms of NOTA and TRAP are substantially lower than those of DFO and THP (Table 1). At pH 3.5, both NOTA and TRAP complexed 68Ga3+ with efficiency comparable to that achieved at pH 6.5. At pH 3.5, at concentrations below 500 μM, RCYs of THP and DFO with 68Ga3+ were relatively poor, however with an increase in pH, Ga3+ competed more effectively with protons for coordination to THP and DFO at lower concentrations of chelator. Thus, at pH 6.5, the lowest concentration at which a RCY greater than 95% was reached was 500 nM, achieved using DFO and THP.
At pH 3.5, 90 °C, and a chelator concentration of 50 μM, RCYs were greater than 85% for all chelators except NOTP, and the most efficient chelators were THP, DOTA and TRAP. RCY of [68Ga(THP)] was 96 ± 0.5%, RCY of [68Ga(DOTA)] was 95 ± 0.9% and RCY of [68Ga(TRAP)] was 93 ± 0.6%. At 5 μM, RCY of [68Ga(THP)] was 93 ± 1.1%, RCY of [68Ga(DOTA)] was 90 ± 1.5% and RCY of [68Ga(TRAP)] was 92 ± 1.5%. Thus, at pH 3.5, heating substantially improves RCY at chelator concentrations of 500 nM to 5 μM for DOTA and THP. The increased RCY observed for [68Ga(DOTA)] at 5 μM at 90 °C (90 ± 1.5%) compared to 25 °C (21 ± 5.6%) is consistent with previous reports.8,9,19 This suggests that the labelling efficiencies of these ligands at pH 3.5 at room temperature are limited by kinetic barriers.
Interestingly, the RCY (57 ± 2.6%) of [68Ga(NOTA)] at 5 μM, 90 °C, pH 3.5 was substantially decreased compared to that observed at 25 °C (93 ± 2.0%). It is possible that contaminating metal ions present in generator eluate (see below) effectively compete with Ga3+ for NOTA binding at high temperature, but that at lower temperature, the kinetic barriers to complexation of these other metal ions prevent them from competing with Ga3+.
At pH 6.5, 90 °C, and a chelator concentration of 5 μM, RCYs were greater than 94% for all chelators except DOTA. In contrast to results observed at room temperature, the best performing chelators were NOTP and HBED. At a chelator concentration of 500 nM, RCY of [68Ga(NOTP)] was 94 ± 0.8% and RCY of [68Ga(HBED)] was 88 ± 4.4%.
The RCY of [68Ga(THP)] at 500 nM and pH 6.5 was substantially reduced at 90 °C (11 ± 2.6%) compared to RCY at 25 °C (97 ± 0.6%). Again, it is possible that THP complexes of metal ion contaminants are formed at high temperature, but not low temperature.
In most eluate fractions, including the second fraction used for radiolabelling, Al, Ti, Fe, Zn, Ga and Pb were present at concentrations of 0.1 μM to 5 μM (Fig. 2, S12 and S13†). Concentrations of 68Zn (arising from decay of 68Ga) were significantly higher in eluate B than eluate A. This is expected: eluate B contained decay products of 150 h of 68Ge/68Ga decay, whereas eluate A contained decay products of only 5 h of 68Ge/68Ga decay. Concentrations of natGa were also higher in eluate B compared to eluate A. Although the sampling size here is very low, the measured metal contaminant levels fall within a similar range to that of prior reports.9 The source of these metal ion contaminants includes the HCl solution used as eluate (contributing a proportion of measured Al, Fe, Zn and Pb), and components of the 68Ga generator including titanium dioxide on a borosilicate glass column, lead shielding and tubing.
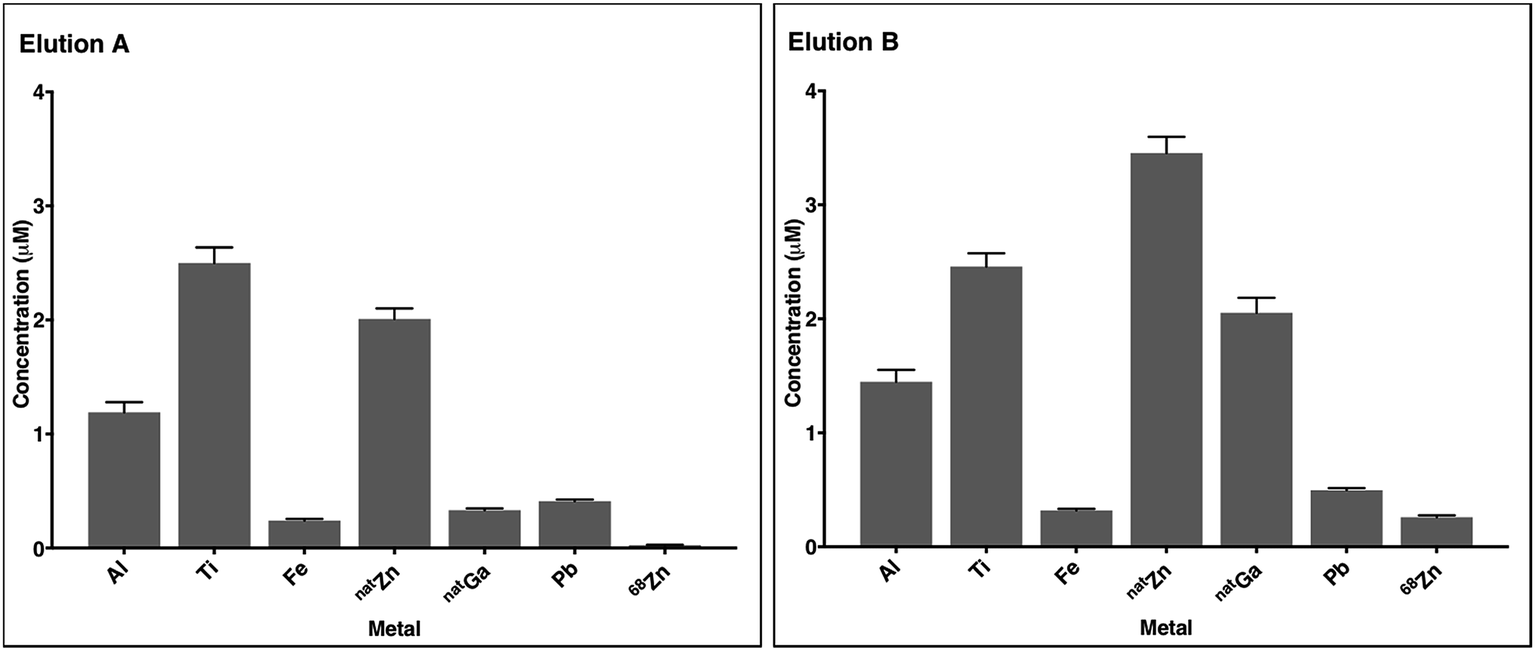 | ||
| Fig. 2 The concentrations of selected metals in 68Ga generator eluate from the second 1 mL fraction (measured by ICP-MS). Elution A was obtained 5 hours after the previous elution, and elution B, 150 hours (five days) after the previous elution. Error bars represent standard deviation of the measurement (n = 5). 68Zn concentrations correspond to 68Zn arising from 68Ga decay. | ||
Complexes of Al3+, Ti4+, Fe3+, Zn2+, Ga3+ and Pb2+ with many of these chelators or their derivatives have been described and characterised. Some of these chelators (for example, HBED,52 hydroxypyridinones,63 NOTA18 and TRAP18,19) have demonstrated selectivity for Ga3+ over divalent ions. However, it is likely that the presence of Al3+, Fe3+ and natGa3+, and the high concentrations of Ti4+, Zn2+ and Pb2+, will decrease RCY of the desired 68Ga–chelator complex for all chelators used in these experiments. It is also likely that differences in metal concentrations in different eluates leads to variability in RCYs.
HPLC radiochromatograms of 68Ga complexes
Before performing 68Ga competition experiments with reverse-phase HPLC, each chelator was reacted with 68Ga3+ solution, and HPLC radiochromatograms (Fig. S3†) were acquired to determine the retention times and chromatographic behaviour of each complex. For [68Ga(DFO)] at 90 °C, multiple signals were observed in the radiochromatograms at both pH 3.5 and 6.5, with wide-ranging retention times. We did not pursue further experiments to elucidate the nature of these species, nor did we study any further reactions of DFO at 90 °C. Others have previously described the structure of [Ga(DFO)].64 We postulate that the ligand or complex are not stable at 90 °C, and that the signals correspond to decomposition products.Ga–HBED complexes
Prior studies have reported that derivatives of the HBED chelator form isomers when complexed to Ga3+ in solution,10,65 and we have previously suggested that these correspond to geometric isomers (Fig. 3),66 however little empirical evidence is available to support this. Consistent with all previous reports of Ga3+-bound HBED (Ga–HBED) derivatives, multiple species with distinct HPLC retention times were formed (Fig. 4). At 25 °C, at least three distinct signals in the radiochromatogram of 68Ga3+–HBED could be distinguished but at 90 °C, only two of these signals were observed (Fig. S4†). The distribution of these species at each temperature was the same whether they were synthesised at pH 3.5 or 6.5.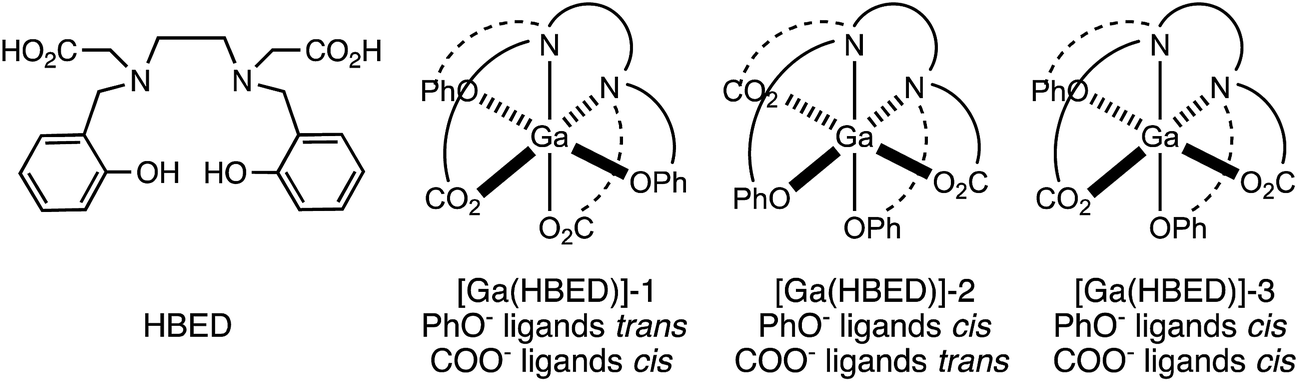 | ||
| Fig. 3 Possible geometric isomers for hexadentate [Ga(HBED)]. Note that each isomer depicted here is one of a pair of enantiomers. | ||
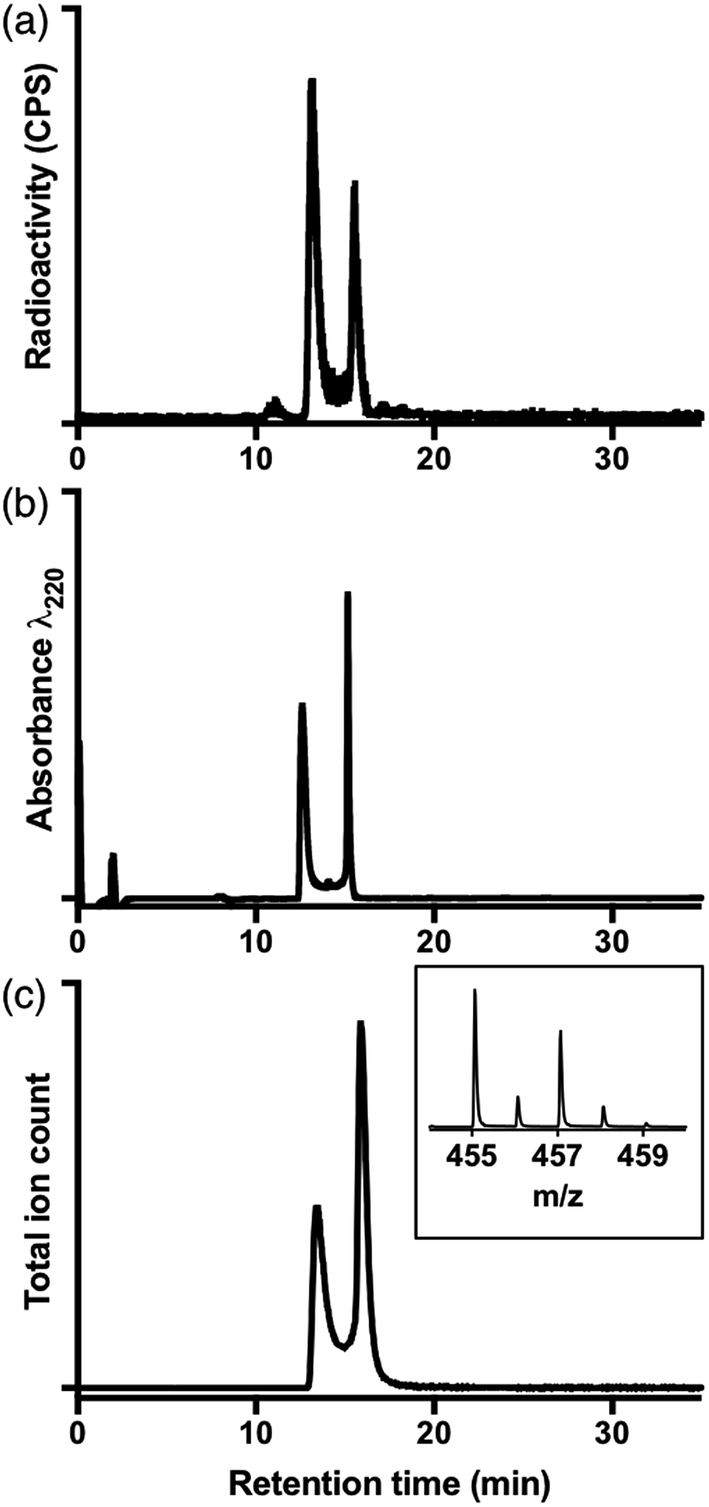 | ||
| Fig. 4 (a) Radiochromatogram of [68Ga(HBED)] (prepared at 90 °C); (b) UV chromatogram of [Ga(HBED)]; (c) extracted ion chromatogram of isolated [Ga(HBED)] (m/z of monoisotopic signal = 455.07); inset positive ion MS spectrum of {[Ga(HBED)] + 2H+}: [Ga(C20H22N2O6)]+. | ||
To further characterise these reaction products, a solution of HBED was reacted with 1.6 equivalents Ga(NO3)3 at 90 °C, and the resulting complex was isolated (using semi-preparative reverse phase C18 HPLC as a mixture of species, including stereo- and possible geometric-isomers). The isolated material was characterised by NMR and LC-MS.
The LC-MS retention times (absorbance at λ = 220 nm and total ion count) were coincident with HPLC signals observed from reaction solutions of 68Ga3+ with HBED at 90 °C (Fig. 4). The product was resolved into two distinct molecular ions by LC-MS, with both corresponding to the expected isotopic pattern for {[Ga(HBED)]− + 2H+} (Fig. 4c inset). In the 13C{1H} spectrum of Ga–HBED, there are four signals corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups, four signals corresponding to C–O phenolic groups and ten signals (rather than the expected twelve) corresponding to methylene groups. Presumably there are two pairs of coincident signals in the case of the methylene groups. In the 1H NMR spectra (including COSY and heteronuclear HSQC 1H–13C spectra, Fig. S5–S8†), the chemically distinct methylene protons display geminal coupling, and the spectra are also consistent with formation of multiple species. 13C{1H} and 1H spectra were acquired in a mixture of D2O/CD3CN (60%/40%), and separately, in a mixture of D2O/CD3OD (50%/50%). Similar spectra were observed for both samples, with no notable differences in the number of signals, nor their chemical shifts and relative intensities. In the 71Ga spectrum (acquired in D2O/CD3CN), a very broad signal is observed, likely arising from overlaid broad resonances corresponding to different Ga–HBED complexes (Fig. 4). A small amount of unchelated Ga3+, presumably [Ga(H2O)6]3+, is also present. The broad signal from 71Ga reflects the asymmetric environment in all Ga3+–HBED species, causing significant quadrupolar relaxation, compared to the symmetric aqua ion.
O groups, four signals corresponding to C–O phenolic groups and ten signals (rather than the expected twelve) corresponding to methylene groups. Presumably there are two pairs of coincident signals in the case of the methylene groups. In the 1H NMR spectra (including COSY and heteronuclear HSQC 1H–13C spectra, Fig. S5–S8†), the chemically distinct methylene protons display geminal coupling, and the spectra are also consistent with formation of multiple species. 13C{1H} and 1H spectra were acquired in a mixture of D2O/CD3CN (60%/40%), and separately, in a mixture of D2O/CD3OD (50%/50%). Similar spectra were observed for both samples, with no notable differences in the number of signals, nor their chemical shifts and relative intensities. In the 71Ga spectrum (acquired in D2O/CD3CN), a very broad signal is observed, likely arising from overlaid broad resonances corresponding to different Ga–HBED complexes (Fig. 4). A small amount of unchelated Ga3+, presumably [Ga(H2O)6]3+, is also present. The broad signal from 71Ga reflects the asymmetric environment in all Ga3+–HBED species, causing significant quadrupolar relaxation, compared to the symmetric aqua ion.
1H, 13C{1H}, COSY, HSQC and 71Ga NMR spectra (Fig. 5 and S5–S8†) are consistent with formation of at least two different species. We postulate that these species could include the three possible geometric isomers of a hexadentate N2O4 species (Fig. 3), as well as complexes in which the HBED ligand coordinates to Ga3+ with lower density, (with each species consisting of NMR-indistinguishable Δ and Λ enantiomers). For each of the geometric isomers designated [Ga(HBED)]-1 and [Ga(HBED)]-2, there are three chemically distinct methylene environments, one phenolic environment and one carboxylate environment. For the geometric isomer designated [Ga(HBED)]-3, there are six chemically distinct methylene environments, two phenolic environments and two carboxylate environments. For a Ga–HBED pentadentate species (with a monodentate ligand occupying the sixth coordination site), there are six chemically distinct methylene environments, two phenolic environments and two carboxylate environments.
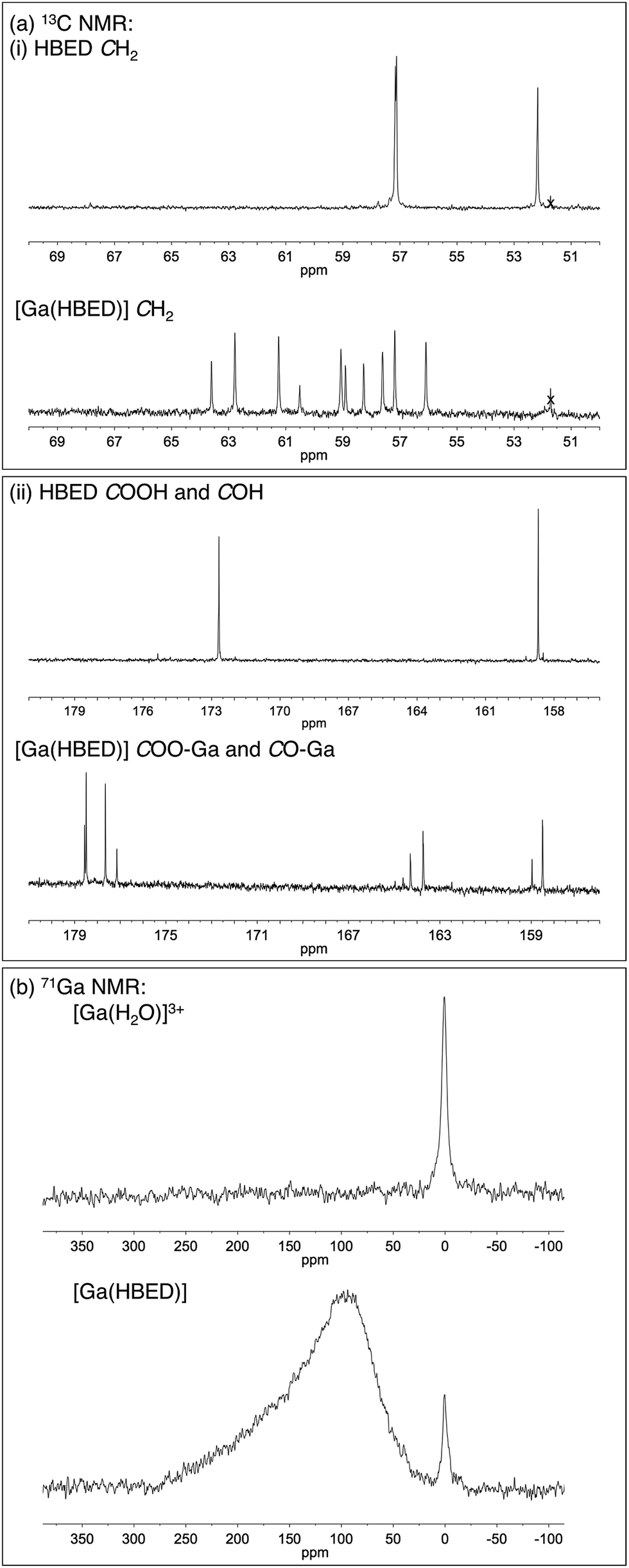 | ||
| Fig. 5 Regions from (a) 13C{1H} NMR spectra of HBED and [Ga(HBED)], and (b) 71Ga NMR spectrum of [Ga(HBED)] (60% D2O/40% CD3CN). In the 13C{1H} NMR spectrum of HBED, three CH2 resonances, one COH resonance and one COOH resonance are detected, but upon coordination to Ga3+, an increase in the number of signals is observed. A residual methanol signal is marked (x). In the 71Ga NMR spectrum of [Ga(HBED)], a broad, asymmetric peak is observed, distinct from that of unchelated [Ga(H2O)6]3+. | ||
Slow evaporation of a solution of isolated Ga–HBED material in water and acetonitrile provided crystals suitable for X-ray diffraction (Table S1†). The selected crystal contained the neutral [Ga(HBED)(H2O)] complex crystallised with a molecule of acetonitrile, and the unit cell contained four symmetry-related equivalents of [Ga(HBED)(H2O)]·CH3CN, including both Δ and Λ enantiomers of the Ga3+ complex. In [Ga(HBED)(H2O)], HBED is bound to Ga3+ in a pentadentate N2O3 environment, with only one phenolic group coordinated to Ga3+ (Fig. 6). The non-coordinating phenolic group is protonated and uncharged. A water molecule occupies the remaining Ga3+ coordination site to give an octahedral Ga3+ complex in which the two carboxylate groups are coordinated trans to each other, and the single coordinating phenolic group and H2O ligand are cis to each other. There is some distortion of the octahedral environment. N–Ga–O bond angles of the five-membered chelate rings, formed by the coordinating carboxylate and amine ligands, are significantly smaller (81.73° and 82.30°) than other bond angles about the metal centre (Table S2†), likely owing to the steric strain in the ligand.
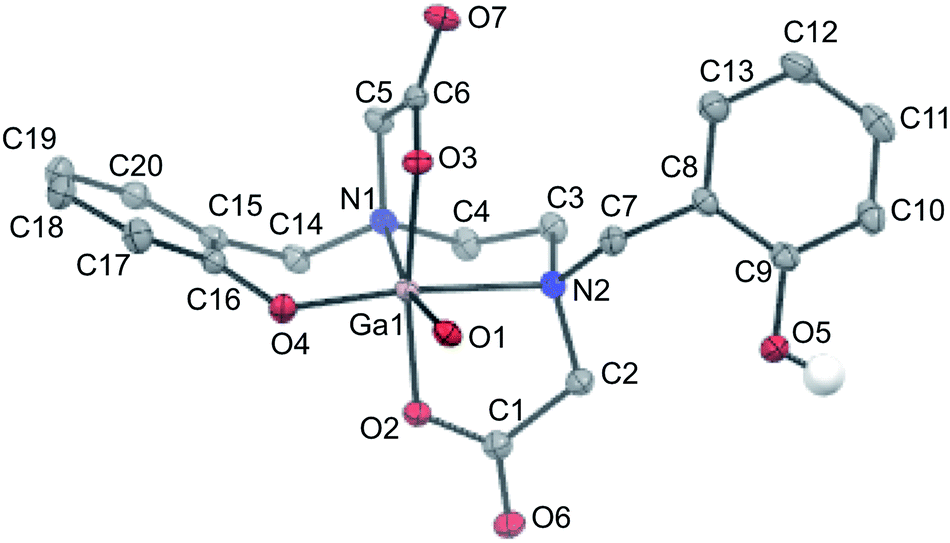 | ||
| Fig. 6 ORTEP representation of [Ga(HBED)(H2O)]·CH3CN. Ellipsoids are at the 50% probability level. Hydrogen atoms (with the exception of the proton of the non-coordinating phenolic group) and solvent molecules are omitted for clarity. | ||
Elemental analysis of the isolated crystalline material indicated that the bulk crystalline material has the same elemental (C, H, N) composition as the single crystal used to acquire X-ray diffraction data (see Experimental section).
Similar geometric arrangements of the HBED ligand have been observed for HBED complexes of Ti4+ and Fe3+, although in these structures, both phenolic groups are coordinated to the metal centre.67,68 In hexadentate [Ti(HBED)] and [Fe(HBED)]−, the two carboxylate groups are trans to each other, and the two phenolic groups are cis to each other. The metal–HBED ligand bond lengths in these octahedral complexes are similar to corresponding metal–HBED ligand bond lengths of [Ga(HBED)(H2O)].
HPLC chromatograms of solutions of isolated [Ga(HBED)(H2O)]·CH3CN gave the same peak shapes as previously observed (Fig. S4†).
Competition studies
Competition studies36 were undertaken, in which two chelators were allowed to compete for 68Ga3+ binding, at different temperatures (90 °C and 25 °C) and different pH conditions (pH 3.5 and 6.5). Generator eluate containing 68Ga3+ was added to solutions containing equimolar concentrations of two chelators. The concentration of each chelator was 500 μM. At this concentration, the chelators were in large excess over 68Ga3+ and the above iTLC studies demonstrated that RCYs for every chelator were near-quantitative. After 40 min reaction (to allow for equilibration), solutions were analysed by reverse phase HPLC (for example, Fig. 7). Radio-chromatographic signals for each 68Ga species were integrated, and results summarised as a percentage of total radioactivity (Tables 3 and 4).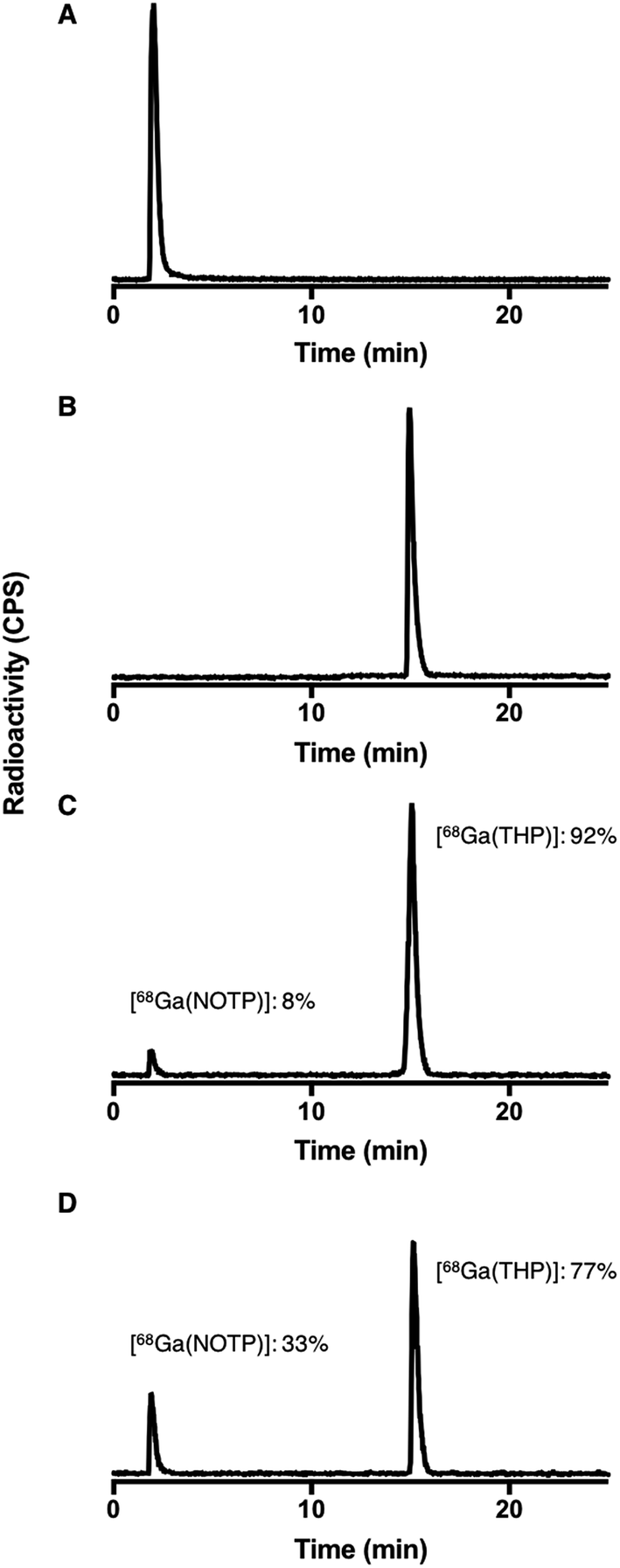 | ||
| Fig. 7 Exemplar HPLC radiochromatograms from competition studies: (A) [68Ga(NOTP)] standard; (B) [68Ga(THP)] standard; reaction solutions (pH 6.5) containing equimolar concentrations (500 μM) of THP and NOTP with 68Ga3+ eluate at (C) 25 °C and (D) 90 °C. Radiochromatograms from competition studies are included in ESI, Fig. S9–S11.† | ||
| 25 °C | 90 °C | ||||
|---|---|---|---|---|---|
| HBED | THP | DFO | HBED | THP | |
| DOTA | 99% HBED | 100% THP | 100% DFO | 96% HBED | 100% THP |
| NOTA | 43% HBED | 97% THP | 38% DFO | 32% HBED | 79% THP |
| NOTP | 0% HBED | 94% THP | 24% DFO | 0% HBED | 64% THP |
| TRAP | 0% HBED | 100% THP | 30% DFO | 0% HBED | 82% THP |
| HBED | — | 100% THP | 17% DFO | — | 100% THP |
| THP | 0% HBED | — | 0% DFO | 0% HBED | — |
| 25 °C | 90 °C | ||||
|---|---|---|---|---|---|
| HBED | THP | DFO | HBED | THP | |
| DOTA | 99% HBED | 100% THP | 100% DFO | 96% HBED | 100% THP |
| NOTA | 41% HBED | 99% THP | 33% DFO | 38% HBED | 87% THP |
| NOTP | 0% HBED | 92% THP | 26% DFO | 0% HBED | 77% THP |
| TRAP | 38% HBED | 100% THP | 25% DFO | 20% HBED | 88% THP |
| HBED | — | 100% THP | 18% DFO | — | 100% THP |
| THP | 0% HBED | — | 0% DFO | 0% HBED | — |
Relative labelling efficiency among DOTA, NOTA, NOTP and TRAP was not compared, as their complexes could not be adequately separated from each other by HPLC or iTLC. 68Ga radiolabelled complexes of HBED, DFO and THP showed distinct retention times under the HPLC conditions employed, and labelling efficiencies of these chelators could be compared with each other and with each of DOTA, NOTA, NOTP and TRAP. These competitive comparisons were repeated at pH 3.5 and 6.5, and 25 and 90 °C, except for those involving DFO at 90 °C.
Under all conditions, THP “competed” most effectively for 68Ga3+ in comparison with all other chelators (Fig. S9†). In each competition study involving THP at 25 °C, RCY of [68Ga(THP)] was in the range 92–100%. At 90 °C, the proportion of 68Ga3+ complexed by the three triazacyclononane (tacn) derivatives (NOTA, NOTP and TRAP) in competition with THP was significantly higher than at 25 °C, although RCYs of [68Ga(THP)] still exceeded 60% in these reactions. This suggests that, to some extent, these 68Ga reaction products at 25 °C are a result of kinetic preferences.
For competition reactions between THP and DOTA, and between THP and HBED, RCYs of [68Ga(THP)] were 100% under all conditions.
The three tacn derivatives competed favourably for 68Ga3+ in reactions with either HBED or DFO (Fig. S10 and S11†). Indeed in all such competitive reactions, RCYs of 68Ga with either NOTA, NOTP or TRAP exceeded 55%. Generally, of the three tacn derivatives, radiochemical yields of [68Ga(NOTP)] were highest across all competition studies.
Under the tested conditions, HBED “outcompeted” only DOTA and DFO (Fig. S9 and S10†). At 25 °C under both pH conditions, DFO “outcompeted” only DOTA (Fig. S9–S11†). Compared to all other chelators and under all tested conditions, DOTA was least able to compete for 68Ga3+ complexation.
There were no remarkable differences between competition studies undertaken at pH 3.5 and pH 6.5, except in the case of reactions of 68Ga3+ with HBED and TRAP (Fig. S10†). At pH 3.5, all 68Ga3+ was bound to the TRAP chelator, whereas at pH 6.5, only 60–80% of added 68Ga3+ was bound to TRAP (depending on the temperature).
Discussion
There is a prevailing notion in the radiochemical literature, based largely on knowledge of the pH-dependence of the hydrolytic behaviour of the Ga3+ aqua ion in forming relatively insoluble hydroxides, that 68/67Ga3+ chelation is most effective at pH 5 or lower. Most reactions that assess 68/67Ga complexation are undertaken at low pH values. Our results demonstrate that for 68Ga-radiolabelling of NOTA, NOTP, TRAP, HBED, DFO and THP at 25 °C under these specific reaction conditions, RCYs at pH 6.5 are equal to or greater than RCYs achieved at pH 3.5.Many reported 68Ga radiolabelling experiments have been conducted at temperatures greater than 50 °C. Our results demonstrate that for the majority of chelators, heating the reaction does not significantly increase RCY. The exceptions to this are reactions of 68Ga3+ with DOTA at both pH 3.5 and pH 6.5, and THP at pH 3.5. In our hands, heating and low pH conditions are only favourable in the case of DOTA – the chelator that is currently used the most for clinical 68Ga biomolecule labelling. Even under low pH and high temperature conditions, near-quantitative RCYs for DOTA labelling (greater than 95%) were only achieved at concentrations of 50 μM and above. In these studies, in which low amounts of 68Ga were used, this corresponded to maximum specific activities of 20–40 MBq μmol−1.
In contrast, near-quantitative RCYs were achieved for THP and DFO at pH 6.5 and 25 °C, at chelator concentrations as low as 500 nM, and in aqueous solutions (ammonium acetate solution) that are physiologically compatible. This corresponded to specific activities of approximately 2–4 GBq μmol−1. For reactions that achieved near quantitative RCYs, the maximum specific activity for [68Ga(THP)] and [68Ga(DFO)] (under mild conditions) was two orders of magnitude higher than that achieved for [68Ga(DOTA)] (under low pH and high temperature conditions). High radiochemical yields (>80%) were achieved for NOTP and HBED under the same mild 68Ga-labelling conditions. Identifying chelators such as these, that enable reproducible and near-quantitative 68Ga biomolecular labelling under low chelator concentration, mild conditions and in physiologically compatible solutions will facilitate (i) one-step, kit-based radiosynthesis of 68Ga radiopharmaceuticals; and (ii) 68Ga radiolabelling of small proteins (<50 kDa). Proteins that accumulate at target tissue and clear circulation in less than four hours (including engineered antibody derivatives and recombinant proteins) have utility in imaging in vivo receptor expression,47,69–71 but many are likely to be sensitive to extremes of pH and temperature.
67Ga-labelled DFO–protein conjugates have previously demonstrated in vivo and in vitro instability, with DFO releasing 67Ga3+ (between 20–60% dissociation of 67Ga3+ to serum proteins over three days in solutions containing serum proteins).28 This is likely a result of kinetic instability of the [Ga(DFO)] complex, leading to transmetallation of Ga3+ to endogenous ligands (proteins, peptides and bone mineral) in vivo and in vitro. However, the shorter half-life of 68Ga renders prolonged in vivo stability unnecessary. As DFO complexes 68Ga3+ in near quantitative yields under mild conditions at low concentrations, DFO is possibly very useful for molecular imaging with 68Ga.72
At pH 6.5 and 25 °C, NOTP is more efficient at chelating 68Ga3+ than either of the other tacn derivatives, NOTA and TRAP, and the radiochemical yield for [68Ga(NOTP)] is greater than 95% at chelator concentrations of 5 μM. This corresponded to specific activities of 200–400 MBq μmol−1. NOTP is also potentially very useful for 68Ga biomolecule labelling under mild conditions. Additionally, out of all chelators except THP, NOTP competes most effectively for 68Ga3+ in competition reactions. Whilst other tacn derivatives, TRAP and NOTA, have been extensively studied for 68Ga biomolecule labelling, 68Ga bioconjugates of NOTP have not been reported. Reassessment of its utility would be timely.
At pH 6.5 at both high and low temperatures, HBED efficiently chelates 68Ga3+, achieving 85% RCY at concentrations as low as 500 nM. LC-MS and NMR data for HBED complexes of 68/natGa3+ indicate that under the analytical conditions described here, more than one chemically distinct species exists in solution. It is possible that neutral pentadentate complexes, such as [Ga(HBED)(H2O)] observed by single crystal X-ray diffraction (Fig. 6), exist within this population of Ga–HBED species. In light of the geometric arrangement of donor atoms in crystals of [Ga(HBED)(H2O)]·CH3CN, and existing data on hexadentate HBED complexes of Ti4+ and Fe3+,67,68 we postulate that hexadentate [Ga(HBED)]-2 (Fig. 3) is also present in solution.
The analytical chromatographic and spectroscopic conditions used in this study (and in prior studies10,65) do not mimic physiological conditions. Nonetheless, the data demonstrate the intricate speciation of HBED complexes of Ga3+. Detailed speciation studies under physiologically-relevant conditions are required. The logKa values for phenolic protons of HBED are 12.6 and 11.0,52 and it is possible that the different species of Ga–HBED (and indeed Ga–HBED–PSMA) arise from fluxionality in the coordination/dissociation of the phenolic oxygen ligands. It is likely that in solutions at higher pH values than those studied here, Ga–HBED exists exclusively as a hexadentate complex. It is also possible that a hexadentate complex would be observed in the solid state, if proton counter ions were substituted for other cations.
Competition studies, in which an equimolar solution of two chelators was reacted with 68Ga3+, indicated that out of all tested chelators and under all tested conditions (which were not concentration-limited), THP competed most effectively for Ga3+. These data, alongside THP radiolabelling studies that demonstrate very high 68Ga radiolabelling efficiency, point to THP possessing ideal properties for rapid radiolabelling under mild conditions.33–35,37,39 It is likely that other chelators that have demonstrated suitable properties for kit-based radiolabelling, such as DEDPA,30–32 FSC,27 and DATA,24,25 which we have not tested in this study, possess similar properties. Such properties will enable rapid, one-step kit-based syntheses of 68Ga-biomolecules for molecular PET imaging without the requirement for complex automated equipment or specialist radiochemistry expertise. This will be key to providing many more hospitals and patients with access to 68Ga radiopharmaceuticals.
Whilst thermodynamic and kinetic studies can predict the utility of a chelator for binding very low concentrations of metal ions, it is difficult to compile all necessary data to reliably model the complex reaction matrix of radiolabelling solutions. These solutions contain adventitious metal ions present in concentrations exceeding that of 68Ga, and buffer/salt components, which can coordinate both Ga3+ and other metal ions, and determine metal ion speciation and reactivity. The simple radiolabelling experiments that we have described here enable identification of suitable and efficient chelators for kit-based 68Ga3+ radiolabelling, in a reaction matrix typical of radiopharmaceutical formulations.
Author contributions
M. I. T. and M. T. M. performed radiolabelling experiments, C. E. K. performed crystallography, C. A. F. and M. T. M. synthesised HBED complexes of Ga(III), C. R. M. and A. C. performed ICP-MS analyses, C. I., J. D. Y. and B. M. P. helped to devise experiments and co-authored the manuscript, T. R. E. performed NMR analysis, P. J. B. and M. T. M. conceived of this study and authored the manuscript.Conflicts of interest
P. J. B. is a named inventor on related patents. All other authors have no conflicts to declare.Acknowledgements
This research was supported by the Wellcome Trust through the London Metallomics Facility funded by the Wellcome Trust Multi-user Equipment Grant (202902/Z/16/Z), the Centre of Excellence in Medical Engineering Centre funded by the Wellcome Trust and EPSRC (203148/Z/16/Z), the KCL and UCL Comprehensive Cancer Imaging Centre funded by CRUK and EPSRC in association with the MRC and DoH (England), and by the NIHR Biomedical Research Centre at Guy's and St Thomas' NHS Foundation Trust and King's College London. B. M. P. was supported by a Victorian Postdoctoral Research Fellowship (Victorian Government). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the DoH. We thank Stephen Boyer, Science Centre, London Metropolitan University, for acquiring elemental microanalysis data.References
- I. Velikyan, Molecules, 2015, 20, 12913–12943 CrossRef CAS PubMed.
- W. P. Fendler, D. F. Schmidt, V. Wenter, C. Zach, P. Bartenstein, F. J. Gildehaus, K. M. Thierfelder, C. Stief, C. Gratzke, T. Kirchner and C. Faber, J. Nucl. Med., 2016, 57, 1720–1725 CrossRef PubMed.
- A. Afshar-Oromieh, C. M. Zechmann, A. Malcher, M. Eder, M. Eisenhut, H. G. Linhart, T. Holland-Letz, B. A. Hadaschik, F. L. Giesel, J. Debus and U. Haberkorn, Eur. J. Nucl. Med. Mol. Imaging, 2014, 41, 11–20 CrossRef CAS PubMed.
- A. R. Haug, C. J. Auernhammer, B. Waengler, G. P. Schmidt, C. Uebleis, B. Goeke, P. Cumming, P. Bartenstein, R. Tiling and M. Hacker, J. Nucl. Med., 2010, 51, 1349–1356 CrossRef CAS PubMed.
- M. S. Hofman, G. Kong, O. C. Neels, P. Eu, E. Hong and R. J. Hicks, J. Med. Imaging Radiat. Oncol., 2012, 56, 40–47 CrossRef PubMed.
- R. Srirajaskanthan, I. Kayani, A. M. Quigley, J. Soh, M. E. Caplin and J. Bomanji, J. Nucl. Med., 2010, 51, 875–882 CrossRef CAS PubMed.
- K. P. Zhernosekov, D. V. Filosofov, R. P. Baum, P. Aschoff, H. Bihl, A. A. Razbash, M. Jahn, M. Jennewein and F. Roesch, J. Nucl. Med., 2007, 48, 1741–1748 CrossRef CAS PubMed.
- E. Eppard, M. Wuttke, P. L. Nicodemus and F. Roesch, J. Nucl. Med., 2014, 55, 1023–1028 CrossRef CAS PubMed.
- I. Velikyan, G. J. Beyer and B. Langstroem, Bioconjugate Chem., 2004, 15, 554–560 CrossRef CAS PubMed.
- M. Eder, O. Neels, M. Mueller, U. Bauder-Wuest, Y. Remde, M. Schaefer, U. Hennrich, M. Eisenhut, A. Afshar-Oromieh, U. Haberkorn and K. Kopka, Pharmaceuticals, 2014, 7, 779–796 CrossRef CAS PubMed.
- J.-F. Morfin and É. Tóth, Inorg. Chem., 2011, 50, 10371–10378 CrossRef CAS PubMed.
- I. Velikyan, H. Maecke and B. Langstrom, Bioconjugate Chem., 2008, 19, 569–573 CrossRef CAS PubMed.
- K.-P. Eisenwiener, M. I. M. Prata, I. Buschmann, H.-W. Zhang, A. C. Santos, S. Wenger, J. C. Reubi and H. R. Maecke, Bioconjugate Chem., 2002, 13, 530–541 CrossRef CAS PubMed.
- R. A. Dumont, F. Deininger, R. Haubner, H. R. Maecke, W. A. Weber and M. Fani, J. Nucl. Med., 2011, 52, 1276–1284 CrossRef CAS PubMed.
- C. L. Ferreira, D. T. T. Yapp, D. Mandel, R. K. Gill, E. Boros, M. Q. Wong, P. Jurek and G. E. Kiefer, Bioconjugate Chem., 2012, 23, 2239–2246 CrossRef CAS PubMed.
- M. Fellner, P. Riss, N. S. Loktionova, K. P. Zhernosekov, O. Thews, F. G. C. Geraldes Carlos, Z. Kovacs, I. Lukes and F. Rösch, Radiochim. Acta, 2011, 99, 43–51 CrossRef CAS.
- M. I. M. Prata, A. C. Santos, C. F. G. C. Geraldes and J. J. P. De Lima, J. Inorg. Biochem., 2000, 79, 359–363 CrossRef CAS PubMed.
- J. Simecek, M. Schulz, J. Notni, J. Plutnar, V. Kubicek, J. Havlickova and P. Hermann, Inorg. Chem., 2012, 51, 577–590 CrossRef CAS PubMed.
- J. Notni, P. Hermann, J. Havlickova, J. Kotek, V. Kubicek, J. Plutnar, N. Loktionova, J. Riss Patrick, F. Rosch and I. Lukes, Chem.–Eur. J., 2010, 16, 7174–7185 CrossRef CAS PubMed.
- M. T. Ma, O. C. Neels, D. Denoyer, P. Roselt, J. A. Karas, D. B. Scanlon, J. M. White, R. J. Hicks and P. S. Donnelly, Bioconjugate Chem., 2011, 22, 2093–2103 CrossRef CAS PubMed.
- C. L. Ferreira, E. Lamsa, M. Woods, Y. Duan, P. Fernando, C. Bensimon, M. Kordos, K. Guenther, P. Jurek and G. E. Kiefer, Bioconjugate Chem., 2010, 21, 531–536 CrossRef CAS PubMed.
- M. Eder, M. Schaefer, U. Bauder-Wuest, W.-E. Hull, C. Waengler, W. Mier, U. Haberkorn and M. Eisenhut, Bioconjugate Chem., 2012, 23, 688–697 CrossRef CAS PubMed.
- B. P. Waldron, D. Parker, C. Burchardt, D. S. Yufit, M. Zimny and F. Roesch, Chem. Commun., 2013, 49, 579–581 RSC.
- J. Seemann, B. P. Waldron, F. Roesch and D. Parker, ChemMedChem, 2015, 10, 1019–1026 CrossRef CAS PubMed.
- J. Seemann, B. Waldron, D. Parker and F. Roesch, EJNMMI Radiopharmacy and Chemistry, 2016, 1, 4 CrossRef.
- D. Parker, B. P. Waldron and D. S. Yufit, Dalton Trans., 2013, 42, 8001–8008 RSC.
- P. A. Knetsch, C. Zhai, C. Rangger, M. Blatzer, H. Haas, P. Kaeopookum, R. Haubner and C. Decristoforo, Nucl. Med. Biol., 2015, 42, 115–122 CrossRef CAS PubMed.
- S. V. Govindan, R. B. Michel, G. L. Griffiths, D. M. Goldenberg and M. J. Mattes, Nucl. Med. Biol., 2005, 32, 513–519 CrossRef CAS PubMed.
- J. E. Ryser, K. Rose, R. Jones, A. Pelegrin, A. Donath, R. Egeli, A. Smith and R. E. Offord, Nucl. Med. Biol., 1998, 25, 261–265 CrossRef CAS PubMed.
- E. Boros, C. L. Ferreira, J. F. Cawthray, E. W. Price, B. O. Patrick, D. W. Wester, M. J. Adam and C. Orvig, J. Am. Chem. Soc., 2010, 132, 15726–15733 CrossRef CAS PubMed.
- E. Boros, C. L. Ferreira, D. T. T. Yapp, R. K. Gill, E. W. Price, M. J. Adam and C. Orvig, Nucl. Med. Biol., 2012, 39, 785–794 CrossRef CAS PubMed.
- C. F. Ramogida, J. F. Cawthray, E. Boros, C. L. Ferreira, B. O. Patrick, M. J. Adam and C. Orvig, Inorg. Chem., 2015, 54, 2017–2031 CrossRef CAS PubMed.
- D. J. Berry, Y. Ma, J. R. Ballinger, R. Tavare, A. Koers, K. Sunassee, T. Zhou, S. Nawaz, G. E. D. Mullen, R. C. Hider and P. J. Blower, Chem. Commun., 2011, 47, 7068–7070 RSC.
- M. T. Ma, C. Cullinane, C. Imberti, J. Baguna Torres, S. Y. A. Terry, P. Roselt, R. J. Hicks and P. J. Blower, Bioconjugate Chem., 2016, 27, 309–318 CrossRef CAS PubMed.
- M. T. Ma, C. Cullinane, K. Waldeck, P. Roselt, R. J. Hicks and P. J. Blower, EJNMMI Res., 2015, 5, 52 CrossRef PubMed.
- M. T. Ma, L. K. Meszaros, B. M. Paterson, D. J. Berry, M. S. Cooper, Y. Ma, R. C. Hider and P. J. Blower, Dalton Trans., 2015, 44, 4884–4900 RSC.
- C. Imberti, S. Y. A. Terry, C. Cullinane, F. Clarke, G. H. Cornish, N. K. Ramakrishnan, P. Roselt, A. P. Cope, R. J. Hicks, P. J. Blower and M. T. Ma, Bioconjugate Chem., 2017, 28, 481–495 CrossRef CAS PubMed.
- R. Cusnir, C. Imberti, P. J. Blower, M. T. Ma and R. C. Hider, Int. J. Mol. Sci., 2017, 18, 116 CrossRef PubMed.
- J. D. Young, V. Abbate, C. Imberti, L. K. Meszaros, M. T. Ma, S. Y. A. Terry, R. C. Hider, G. E. Mullen and P. J. Blower, J. Nucl. Med., 2017, 58, 1270–1277 CrossRef PubMed.
- B. Hacht, Bull. Korean Chem. Soc., 2008, 29, 372–376 CrossRef CAS.
- R. E. Weiner and M. L. Thakur, in Handbook of Radiopharmaceuticals: Radiochemistry and Applications, ed. M. J. Welch and C. S. Redvanly, John Wiley & Sons Ltd., 2003, pp. 363–399 Search PubMed.
- G. E. Jackson and M. J. Byrne, J. Nucl. Med., 1996, 37, 379–386 CAS.
- S. M. Moerlein and M. J. Welch, Int. J. Nucl. Med. Biol., 1981, 8, 277–287 CrossRef CAS PubMed.
- S. A. Wood and I. M. Samson, Ore Geol. Rev., 2006, 28, 57–102 CrossRef.
- L. E. McInnes, S. E. Rudd and P. S. Donnelly, Coord. Chem. Rev., 2017 DOI:10.1016/j.ccr.2017.05.011 , advance web publication.
- J. Notni, K. Pohle and H.-J. Wester, EJNMMI Res., 2012, 2(28), 25 Search PubMed.
- M. Eder, A. V. Krivoshein, M. Backer, J. M. Backer, U. Haberkorn and M. Eisenhut, Nucl. Med. Biol., 2010, 37, 405–412 CrossRef CAS PubMed.
- A. Bianchi, L. Calabi, C. Giorgi, P. Losi, P. Mariani, P. Paoli, P. Rossi, B. Valtancoli and M. Virtuani, J. Chem. Soc., Dalton Trans., 2000, 697–705 RSC.
- V. Kubíček, J. Havlíčková, J. Kotek, G. Tircsó, P. Hermann, É. Tóth and I. Lukeš, Inorg. Chem., 2010, 49, 10960–10969 CrossRef PubMed.
- B. Drahos, V. Kubicek, C. S. Bonnet, P. Hermann, I. Lukes and E. Toth, Dalton Trans., 2011, 40, 1945–1951 RSC.
- C. F. G. C. Geraldes, A. D. Sherry and W. P. Cacheris, Inorg. Chem., 1989, 28, 3336–3341 CrossRef CAS.
- C. H. Taliaferro and A. Martell, Inorg. Chim. Acta, 1984, 85, 9–15 CrossRef CAS.
- A. Evers, R. D. Hancock, A. E. Martell and R. J. Motekaitis, Inorg. Chem., 1989, 28, 2189–2195 CrossRef CAS.
- D. J. Clevette, D. M. Lyster, W. O. Nelson, T. Rihela, G. A. Webb and C. Orvig, Inorg. Chem., 1990, 29, 667–672 CrossRef CAS.
- Y.-Y. Xie, Z. Lu, X.-L. Kong, T. Zhou, S. Bansal and R. Hider, Eur. J. Med. Chem., 2016, 115, 132–140 CrossRef CAS PubMed.
- T. Zhou, H. Neubert, D. Y. Liu, Z. D. Liu, Y. M. Ma, X. L. Kong, W. Luo, S. Mark and R. C. Hider, J. Med. Chem., 2006, 49, 4171–4182 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- M. S. Cooper, M. T. Ma, K. Sunassee, K. P. Shaw, J. D. Williams, R. L. Paul, P. S. Donnelly and P. J. Blower, Bioconjugate Chem., 2012, 23, 1029–1039 CrossRef CAS PubMed.
- M. T. Ma, M. S. Cooper, R. L. Paul, K. P. Shaw, J. A. Karas, D. Scanlon, J. M. White, P. J. Blower and P. S. Donnelly, Inorg. Chem., 2011, 50, 6701–6710 CrossRef CAS PubMed.
- E. Oehlke, V. S. Le, N. Lengkeek, P. Pellegrini, T. Jackson, I. Greguric and R. Weiner, Appl. Radiat. Isot., 2013, 82, 232–238 CrossRef CAS PubMed.
- E. T. Clarke and A. E. Martell, Inorg. Chim. Acta, 1992, 191, 56–63 CrossRef CAS.
- B. Borgias, A. D. Hugi and K. N. Raymond, Inorg. Chem., 1989, 28, 3538–3545 CrossRef CAS.
- J. Schuhmacher, G. Klivenyi, W. E. Hull, R. Matys, H. Hauser, H. Kalthoff, W. H. Schmiegel, W. Maier-Borst and S. Matzku, Nucl. Med. Biol., 1992, 19, 809–824 CAS.
- M. T. Ma and P. J. Blower, in Metal Chelation in Medicine, ed. R. R. Crichton, R. J. Ward and R. C. Hider, The Royal Society of Chemistry, 2017, pp. 260–312 Search PubMed.
- S. K. Larsen, B. G. Jenkins, N. G. Memon and R. B. Lauffer, Inorg. Chem., 1990, 29, 1147–1152 CrossRef CAS.
- A. D. Tinoco, C. D. Incarvito and A. M. Valentine, J. Am. Chem. Soc., 2007, 129, 3444–3454 CrossRef CAS PubMed.
- A. C. Freise and A. M. Wu, Mol. Immunol., 2015, 67, 142–152 CrossRef CAS PubMed.
- M. Rosestedt, K. G. Andersson, B. Mitran, V. Tolmachev, J. Loefblom, A. Orlova and S. Staahl, Sci. Rep., 2015, 5, 15226 CrossRef CAS PubMed.
- P. M. Smith-Jones, D. B. Solit, T. Akhurst, F. Afroze, N. Rosen and S. M. Larson, Nat. Biotechnol., 2004, 22, 701–706 CrossRef CAS PubMed.
- P. M. Smith-Jones, B. Stolz, C. Bruns, R. Albert, H. W. Reist, R. Fridrich and H. R. Maecke, J. Nucl. Med., 1994, 35, 317–325 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1564603. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra09076e |
| This journal is © The Royal Society of Chemistry 2017 |