
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetection methods for Pseudomonas aeruginosa: history and future perspective
Yongjun Tang
*a,
Zeeshan Ali
 a,
Jun Zou
b,
Gang Jin
a,
Junchen Zhu
a,
Jian Yang
a and
Jianguo Dai
*a
a,
Jun Zou
b,
Gang Jin
a,
Junchen Zhu
a,
Jian Yang
a and
Jianguo Dai
*a
aSchool of Applied Chemistry and Biotechnology, Shenzhen Polytechnic, Liuxian Avenue, No. 7098, Nanshan District, Shenzhen 518055, Guangdong Province, China. E-mail: tangyongjun32@163.com; jgdai@szpt.edu.cn; Fax: +86-755-26019169; Tel: +86-755-26019359
bSchool of Chemistry and Chemical Engineering, Hunan Institute of Engineering, Xiangtan 411104, China
First published on 8th November 2017
Abstract
Pseudomonas aeruginosa is a prevalent, opportunistic, Gram-negative bacterium that infects immunocompromised individuals, frequently causing hospital-acquired and community-acquired infections. Currently, Pseudomonas aeruginosa is one of the most widespread and fatal agents among the various causes of nosocomial infections. P. aeruginosa has been associated with increased mortality relative to Staphylococcus aureus or other Gram-negative in bloodstream infections. As few as 10–100 bacilli are capable of colonizing the intestine of critically ill or immunocompromised patients, therefore, early detection of Pseudomonas aeruginosa is particularly important. Here, we have summarized and analyzed the development of detection techniques for Pseudomonas aeruginosa over the past 50 years. We also discuss the prospects for future research on Pseudomonas aeruginosa detection methods in the hope of providing a reference for relevant studies.
 Yongjun Tang | Dr Yongjun Tang obtained his B.S. Degree and M.S. at Hunan Agriculture University in China in 2000 and 2003, respectively and completed PhD in biomedical engineering at Southeast University in China in May 2015. He is engaged in research and teaching at Shenzhen Polytechnic as an Associate Professor since June 2015. To date, he has published 5 patent applications and more than 20 papers. His current research is focused on the establishment of nanotechnology-based rapid molecular detection techniques to detect food pathogens. |
 Zeeshan Ali | Zeeshan Ali completed his PhD in Biomedical Engineering at School of Biological Sciences & Medical Engineering, Southeast University, China in December 2015. Since then, he is a postdoctoral researcher at Postdoctoral Innovation Practice Base, Shenzhen Polytechnic, China. His research focused on the development of simple, automated biosensors for the detection of pathogens. |
 Jun Zou | MS Jun Zou obtained her B.S. and M.S. Degree from Hunan Agriculture University, China in 2002 and 2005, respectively. After her M.S., She joined Hunan Institute of Engineering and continued her research activities. In addition to research activities, she is also teaching microbiology, biochemistry and cell biology to undergraduate students for the last 11 years. Her current research is focused on the detection methods for microorganisms, strains filtration and microorganism fermentation. |
 Gang Jin | Dr Gang Jin obtained his B.S. Degree at Huazhong Agriculture University, China in 1986, and completed his M.S. and PhD at Institute of Hydrobiology, the Chinese Academy of Sciences, China, in 1989 and 2001, respectively. Since June 2004, He is working as a Professor at Shenzhen Polytechnic, China. He has published over 100 patents and 60 papers in various journals. His research is focused on food safety management and brain cancer cell biology. |
 Junchen Zhu | Dr Junchen Zhu Tang graduated in 1988 from Jiangxi University of Technology (now renamed as Nanchang University) in food engineering. He obtained a masters degree in food engineering from Heilongjiang Business School in 1993 and completed his PhD degree in botany at South China Normal University in 2006. He has published over 30 research papers. His current research is focused on the screening of probiotics, and identification and development of probiotic products. |
 Jian Yang | Dr Jian Yang obtained his B.S. Degree and M.S. in biology at Hubei University in 1988 and 1991, respectively and earned a PhD degree in environmental science and engineering at Xiamen University in 2008. He was a Visiting Scholar at University of Nagoya, Japan (2000–2001). Currently, he is serving as a professor at Shenzhen Polytechnic. His research focused on the purification, nanoencapsulation, absorption mechanism, in vitro ACE inhibition and in vivo antihypertensive activity of bioactive peptides. He has published more than 30 papers and 12 patents. |
 Jianguo Dai | Dr Jianguo Dai obtained his PhD degree in animal nutrition and food science from China Agricultural University in 2002. Currently, he is a professor of food nutrition at Shenzhen Polytechnic. He has served as Pearl River Scholar Distinguished Professor of Guangdong Province, China (2012–2017) and was a Visiting Scholar at the University of British Columbia, BC, Canada (2016–2017). He has delivered lectures on inorganic chemistry, biochemistry, instrument analysis, food nutrition and inspection to college students during the past 15 years. His current research is focused on antimicrobial peptides. He has published more than 30 papers and 10 patents. |
1. Introduction
Pseudomonas aeruginosa is a widespread, opportunistic, Gram-negative bacillus that causes various clinical infections and severe infectious diseases.1–3 Currently, it is one of the most troublesome multidrug-resistant bacterial causes of nosocomial infections; infection with Pseudomonas aeruginosa can cause long-term chronic diseases, particularly affecting immunocompromised (especially neutropenic) patients or those admitted to the intensive care unit (ICU).4–6 In food testing, the WHO has identified Pseudomonas aeruginosa as an indicator of drinking water quality while the European Communities and the Codex Alimentarius Commission also stipulated that Pseudomonas aeruginosa should not be detected in water.7,8 In addition, Pseudomonas aeruginosa can cause infections at very low concentrations. Hence, early detection is critical for treating Pseudomonas aeruginosa infection. Scientists have long been committed to establishing a rapid and sensitive detection method for Pseudomonas aeruginosa. Currently, numerous modern detection approaches have been developed, such as flow cytometry (Rüger et al., 2014),9 immunological detection methods10 and molecular biology-based detection methods.11 However, conventional culture methods are still the most commonly applied methods in clinical practice at present. Therefore, these new detection approaches still need to be further improved to achieve large-scale applications in clinical practice. Here, we review the development, advantages, and disadvantages of Pseudomonas aeruginosa detection methods.2. Conventional bacterial culture methods
Conventional Pseudomonas aeruginosa detection methods are based on the biological characteristics of the bacterium under certain culture conditions, such as Gram-negative or Gram-positive status, or the activities of bacterial molecules such as oxidase, acetamidase, arginine dihydrolase, and pyocyanin. In 1955, Lowbury et al. developed a selective medium for Pseudomonas aeruginosa that is supplemented with 0.03% cetrimide. The medium has very high selectivity and subsequently became one of the most common selective media for Pseudomonas aeruginosa.12 In 1965, Brown et al.13 developed an improved selective medium containing cetrimide, based on previous studies, and demonstrated that the Lemco-based selective medium (CTA1) promoted better pyocyanin production. Growth in selective medium with King's medium B as the base (CTA2) resulted in stronger fluorescence at an incubation temperature of 42 °C, while other (non-Pseudomonas aeruginosa) species did not produce fluorescence at this temperature. Therefore, the CTA2 medium can serve as a diagnostic medium specific for Pseudomonas aeruginosa. Pseudomonas aeruginosa can produce ammonia as a nutrient by breaking down acetamide, an ability that is absent in other bacteria. Based on this mechanism, Szita et al.14 (1990) developed a liquid synthetic medium in 1990 that enables higher detection selectivity, sensitivity, and speed than culture medium supplemented with cetrimide. In addition, many other researchers have investigated the feasibility of using cetrimide as an additive to identify Pseudomonas aeruginosa.15–17 Infection with Pseudomonas aeruginosa can cause long-term chronic diseases. Long diagnostic turnaround time can reduce patient outcomes and increase hospital costs. The automated systems promise shorter turnaround times to diagnostic results and are widely used in many clinical laboratories for identification of bacterial species and antimicrobial susceptibility testing (AST). The automated systems have many advantages, such as high degree of automation with a simple operating procedure, improved specimen handling, good reproducibility and accuracy, etc. Vitek 2 (BioMérieux, France),18 Phoenix 100 (BD Biosciences, USA)19 and MicroScan WalkAway (Dade Behring, Inc., USA)20 are the common automated identification systems currently used in China. The Vitek 2 is one of the earliest and most commonly used automated identification systems.21 These automated systems not only identify Pseudomonas aeruginosa but are also capable of performing AST. Many scientists have used these instruments to analyze different sources of Pseudomonas aeruginosa and tested their identification and misidentification by comparing them with conventional methods (Table 1). For example, Bruins and his colleagues compared direct inoculation by Vitek 2 and the standard method; a total of 33 isolates of Pseudomonas aeruginosa were tested, and 78.8% were correctly identified (Bruins et al., 2004).22 Saiman's result showed that only 57% (108 of 189) of nonmucoid strains and 40% (24 of 60) of mucoid strains were definitively identified as Pseudomonas aeruginosa with MicroScan Autoscan.20 In 2003, a high rate of agreement between the Phoenix and the conventional methods was observed for Pseudomonas aeruginosa identification, and the rate of agreement was 90.9%.23 The same system showed different results in different studies, which might be due to different sample sources or experimental conditions.25–28 Although automated identification systems have been clinically used to identify a variety of microbial species, these systems have a low rate of accuracy in the identification of Pseudomonas aeruginosa.18,20,24 Clinical laboratories should be aware of the problem with the automated systems in testing Pseudomonas aeruginosa and seek alternatives, validated methods for routine use.| Vitek-2 | Phoenix 100 | MicroScan WalkAway | References | |||
|---|---|---|---|---|---|---|
| Correctly/tested identified | Correct identification rate (%) | Correctly/tested identified | Correct identification rate (%) | Correctly/tested identified | Correct identification rate (%) | |
| a The correct identification don't contained low discrimination. | ||||||
| 26/33 | 78.8 | 22 | ||||
| 21/21 | 100 | 21 | ||||
| 98/105 | 93.3 | 26 | ||||
| 88/146 | 60.3 | 18 | ||||
| 8/9 | 88.9 | 6/9 | 66.7 | 24 | ||
| 132/249 | 53.0 | 20 | ||||
| 8/10 | 80.0 | 25 | ||||
| 12/12 | 100 | 19 | ||||
| 20/22 | 90.9 | 23 | ||||
| 5/7 | 71.4 | 27 | ||||
| 55/55 | 100 | 28 | ||||
3. Immunological assays
Immunological techniques utilize the highly specific binding between antibodies and antigens and facilitate qualitative or quantitative detection based on specific reactions resulted from antigen–antibody binding. Modern immunoassay techniques have achieved high-sensitivity detection and have been developed into multiple types of immunoassay methods by introducing enzyme-catalyzed reactions, fluorescence, or isotope labeling as a specific measure of antigen–antibody binding.29,30 While reviewing the literature, we found that the main immunoassays currently applied for detecting Pseudomonas aeruginosa include enzyme-linked immunosorbent assays (ELISAs),31 immunoblotting (IBT),32 immunofluorescence,33 immunoelectrophoresis34 and solid-phase radioimmunoassay,35 which are discussed briefly below.3.1 Enzyme-linked immunosorbent assays (ELISAs)
ELISAs are based on an immunological technique that utilizes enzyme-catalyzed reactions to enhance the sensitivity of the specific antigen–antibody reaction.30 The underlying principle is that antigens or antibodies with immunological activity are immobilized on the surface of a solid-phase carrier, and then the antigen or antibody sample to be detected is reacted with the immobilized antigen or antibody molecules, followed by the addition of enzyme-labeled antigen or antibody molecules prior to incubation. Finally, enzyme-catalyzed substrates are added for luminescence detection31 and the enzymatic reaction plays a role in signal amplification. Currently, ELISAs has been widely applied in the detection of pathogens and food-borne bacteria. It is also considered as one of the most successful detection techniques over the past few decades.ELISAs were first used by Ueda et al. to detect immunoglobulin M (IgM) and immunoglobulin G (IgG) in horse serum against common serological Pseudomonas aeruginosa antigens (protease and elastase).36 With their method, horseradish peroxidase (HRP)-labeled rabbit anti-horse IgM and IgG were used as conjugated enzyme-labeled antibodies, while 5-aminosalicylic acid and H2O2 were used as HRP substrates for signal amplification, thereby enabling the detection of IgM and IgG in colt and racehorses. Granstrom et al. developed a specific ELISA-based detection method against antibodies from patients infected with Pseudomonas aeruginosa, using 4 extracellular proteins as antigens: exotoxin A, elastase, alkaline protease, and phospholipase C.37 The method was tested on samples from 39 burn patients, and the results showed that the detection of antibodies against exotoxin A and phospholipase C by ELISA could be used to monitor Pseudomonas aeruginosa infection in patients. Fomsgaard et al.38 reported the use of anti-lipopolysaccharide (LPS) antibodies to develop an ELISA method for the quantitative detection of Pseudomonas aeruginosa in cystic fibrosis (CF) patients with chronic infection. The results of their study showed that expression levels of anti-LPS antibodies (IgG and IgA) significantly increased during the early stage of Pseudomonas aeruginosa lung infection, and they continued to increase to very high levels during later stages of infection. IgM levels increased during early infection but did not continue to increase in the later stages of infection. Fomsgaard et al. also assessed the diagnostic efficiency of this method for detecting early chronic infection with Pseudomonas aeruginosa by comparison with the immunoelectrophoresis method. Their results showed that the method targeting IgG and IgA antibodies had 86% and 89% positive predictive values, respectively, and has 98% and 97% negative predictive values, respectively. The detection rate using IgM as the target was low. Dogru et al. (2013) assessed the utility of ELISA in detecting early Pseudomonas aeruginosa infection in CF patients through comparison with culture methods. The study involved 90 CF patients with long-term follow-up.39 The results showed that the ELISA method had a higher sensitivity in detecting anti-Pseudomonas aeruginosa antibodies, and thus, these antibodies can be used as markers for early diagnosis. Antibodies against alkaline protease showed the highest specificity, while anti-elastase antibodies showed the highest sensitivity. Although specificity is one of the major advantages of ELISA, many secreted Pseudomonas aeruginosa proteins share high homology with those of other bacteria, raising the possibility of false-positive detection. For example, Beutin et al. (1996) reported that toxins from Pseudomonas aeruginosa could induce false-positive results when assaying for Shiga-like cytotoxins, using a commercial ELISA kit.40
ELISA methods have not only been applied for PA detection but also been used for the diagnosis of other lethal infections as well. For example, Cyrille et al. (2017)41 reported the detection of HIV, HBV and HCV with 93.39%, 98.90% and 91.86% sensitivity, respectively. In another study, Bouba et al. (2017)42 successfully detected Zika virus (ZIKV) in 1084 blood donors from Cameroon by using the EuroImmun anti-NS1 IgG ELISA detection kit. Thus, despite the specificity issue, ELISA is an important technique which has been widely used for the detection of pathogens.
3.2 Enzyme-linked immunoelectrotransfer blots (IBTs)
IBTs, also known as enzyme-linked immunoelectrotransfer blots, are immunoassays involving the integration of gel electrophoresis and immunohistochemistry.43 IBTs involve the separation of protein samples by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transfer to a nitrocellulose membrane as the solid-phase carrier, using electric transfer system.44 Subsequently, specific antibodies and enzyme-labeled antibodies are added successively, and finally, an enzyme-catalyzed substrate is added to produce a colored or luminescent product for detection purposes.45 This method integrates the high resolution of SDS-PAGE and high specificity of immunoassays; therefore, it is very suitable for the highly sensitive detection of target proteins among different complexes (Hamid et al., 2009; Pârishamelin et al., 1999; Chin et al., 1991; Ayala-Sulca et al., 2015).43–46 The chronic infection caused by Pseudomonas aeruginosa can be fatal for CF patients. Shand et al. (1988) prepared a standard antigen (StAg) of Pseudomonas aeruginosa to serve as a basis for developing the IBT method with serum antibodies. A comparison with the crossed immunoelectrophoresis (CIE) method showed that the sensitivity of the IBT method was approximately 2 orders of magnitude higher than that of the CIE method, with good reproducibility. The results of their study also showed that IBT could be developed into a highly sensitive assay for detecting early Pseudomonas aeruginosa infection in CF patients. Caballero47 reported an improved IBT method on the basis of Shand's study,32 which was successfully tested with anti-Pseudomonas aeruginosa antibodies in patients with bronchiectasis. Weisner et al.48 utilized the IBT technique to develop a method that could detect the A-band LPS antibody of Pseudomonas aeruginosa in oral fluid samples.Besides many applications of IBTs in Pseudomonas aeruginosa infection, recent past also illustrates the pace at which the IBTs based detection of other diseases obtained valuable implementations and is advancing. For example, Eduardo et al. (2015) detected human cysticercosis which utilized purified native antigen mix from cysticercus fluid of Taenia solium by using ITBs techniques, and results showed that the purified native antigen mix improved the diagnostic efficiency of the IBTs test.46
3.3 Immunofluorescence methods
Although immunoenzyme techniques have good sensitivity, their relatively tedious procedures have limited further development. However, immunofluorescence methods developed on the basis of immunological techniques involve simpler operating procedures with a good sensitivity and specificity, thus, they are considered to be among the most promising assays for pathogens (Pemberton et al., 2014; Miyazaki et al., 2014; Parks et al., 1983).49–51 In addition, the development of nanotechnology has brought new opportunities for fluorescence assays. For instance, nanomaterials developed in recent years, such as fluorescence quantum dots,52 nanoclusters,53 fluorescent rare-earth complexes,54 and internalizable fluorescent materials,55 offer the advantages of high fluorescence efficiency, durable fluorescence, and tunable fluorescence emission wavelengths.56 Shibuya et al. (1987) used the immunofluorescence technique for the first time to perform rapid in situ detection of Pseudomonas aeruginosa in sputum samples.33 Pfaller et al.57 developed and evaluated a 1-step immunofluorescence method for the direct detection of Pseudomonas aeruginosa in a blood culture sample and used this method to perform statistical analysis on the sample. The method enabled effective and rapid detection of Pseudomonas aeruginosa. Tang et al. (1992)58 established a direct immunofluorescent antibody-staining method for detecting Pseudomonas aeruginosa in sputum samples using serum-specific monoclonal antibodies, in order to assess the status of pulmonary infection. The results showed that this method had a detection limit of 10 colony-forming units (CFU) per mL and could be completed within 3 h. However, the greatest drawback of immunofluorescence methods is a low signal-to-noise ratio, which may lower its detection specificity. Immunofluorescence technique is steadily improving with the passage of time. Recently, David et al. (2014) demonstrated that automated-multiplex immunofluorescence immuno-assay (InoDiag) test owned similar sensitives (65.5%) between immunofluorescence, but better specificities (95.1–98%) than enzyme-linked immunosorbent assays (ELISAs), through detecting specific anti-C. trachomatis immunoglobulin G.59 Furthermore, Helle et al. (2016) verified that immunofluorescence assay was significantly more sensitive than microscopy of iodome-stained concentrates using either formol-ethylacetate assay or salt-sugar flotation.60The developed immunoassays for Pseudomonas aeruginosa also include convective immunoelectrophoresis, which was developed by Wagner et al.34 and solid-phase radioimmunoassay, established by Kohler et al.35 These immunoassays have provided abundant approaches for the highly sensitive detection of Pseudomonas aeruginosa.
4. Molecular biology assays
Alongside the development of polymerase chain reaction (PCR) in the 1980s and the continuous progression of molecular biology methods,61 scientists have established numerous effective molecular biology assays that have been widely applied for detecting pathogens and have provided a technical basis for epidemiological investigations,62 pathogen-specific detection,63 and pathogen genotyping.64 With the development of molecular biology techniques, molecular biology-based methods for rapidly detecting Pseudomonas aeruginosa have also developed rapidly.65,66 Here, we summarize the development of molecular biology-based Pseudomonas aeruginosa assays.4.1 Conventional PCR methods
The PCR technique is one of the most important inventions over the past 3 decades. In recent years, PCR-based assays for Pseudomonas aeruginosa also have developed progressively. Kingsford et al.65 established a PCR detection method for Pseudomonas aeruginosa that specifically targets the 16S rRNA gene. The method allows the detection of 1 pg chromosomal DNA or 1 × 105 CFU mL−1 of Pseudomonas aeruginosa. Detection sensitivity can be increased up to 1 fg chromosomal DNA or 10 CFU mL−1 if detection is based on the use of a fluorescent probe. Hummel and Unger66 established the first PCR method that specifically detected Pseudomonas aeruginosa based on the exotoxin A gene and evaluated the effectiveness of this method for rapid Pseudomonas aeruginosa detection in mechanically ventilated patients. The results showed that the PCR method targeting exotoxin A gene detected 57 positive samples out of 364 total samples, whereas the conventional culture method only detected 36 positive samples, indicating that the exotoxin A gene-based PCR method had higher sensitivity. Alongside the continuous development of PCR assays for Pseudomonas aeruginosa, an increasing number of specific genes have been discovered, such as ecfX, gyrB, algD GDP mannose, oprL, and fliC.67 Specificity is critical for the success of conventional PCR, but is also the most important cause of failure in PCR detection. Therefore, many researchers have investigated the specificity of different Pseudomonas aeruginosa genes. For example, Dev Vos et al. (1997) examined the specificity of the oprL gene in Pseudomonas aeruginosa detection.68 Further information on the specificity of tests for Pseudomonas aeruginosa genes is presented in Table 2. It can be seen from Table 2 that except for fliC gene, which shows low specificity, all of the other genes have very high specificity, especially the ecfX and gyrB genes, whose detection was not associated with false positive or false negative results.70,73| PCR target gene | Accuracy of P. aeruginosa-detection assay | Year | Ref. | |
|---|---|---|---|---|
| Positive samples | Negative samples | |||
| a Statistical analysis was performed according to Deschaght et al. (2011). | ||||
| 16s rDNA | 59/59 | 1/15 | 2007 | 69 |
| 63/63 | 2/28 | 2009 | 73 | |
| algD GDP mannose | 101/103 | 0/87 | 2003 | 71 |
| 176/176 | 0/26 | 1999 | 72 | |
| ETA | 62/63 | 0/28 | 2009 | 73 |
| 108/113 | 0/87 | 2003 | 71 | |
| ecfX | 63/63 | 0/28 | 2009 | 73 |
| 59/59 | 0/15 | 2007 | 69 | |
| fliC | 59/59 | 13/15 | 2007 | 69 |
| gyrB | 59/59 | 0/15 | 2007 | 69 |
| 63/63 | 0/28 | 2009 | 73 | |
| 113/113 | 0/87 | 2003 | 71 | |
| 102/104 | 0/120 | 2007 | 74 | |
| oprL | 40/40 | 0/18 | 2001 | 75 |
| 63/63 | 4/28 | 2009 | 73 | |
| 59/59 | 2/15 | 2007 | 69 | |
| 128/138 | 2016 | 76 | ||
| toxA | 55/59 | 0/15 | 2007 | 69 |
| oprI | 128/138 | 2016 | 76 | |
Conventional PCR was utilized widely for its developed procedure to obtain consequence of reliability and stability. Recently, PCR-based assays for clinical diagnosis and ensuring food safety have been developed progressively. For instance, Bobbi et al. (2016) detected routine clinical diagnostic specimens from patients in the USA with PCR through targeting the oppA1 gene of B burgdorferi sensu lato and identified positive specimens.77 The combined use of conventional PCR, magnetic particles and chemiluminescence technology for the development of efficient biosensors for clinical purpose is an emerging trend. Highly sensitive and rapid detection of Pseudomonas aeruginosa was achieved using conventional PCR, magnetic particles (MNPs), and chemiluminescence technology.78,79 The results showed that Pseudomonas aeruginosa was successfully detected with detection limit as low as 7.5 fM in term of gyrB fragments. Furthermore, Tang et al. (2014) devised a nested PCR method based on MNPs to detect Pseudomonas aeruginosa.80
4.2 Real-time, fluorescence-based quantitative PCR
In 1996, Applied Biosystems, Inc. (USA) invented the real-time, fluorescence-based quantitative PCR (real-time qPCR) method that enables quantitative detection via real-time monitoring of PCR reactions by introducing fluorescent molecules into the reaction mixture. Currently, it has become one of the most widely used nucleic acid-based molecular detection techniques for pathogens.81 Qin et al.71 established a multiplex real-time PCR assay that simultaneously and specifically detected the 16S rRNA and gyrB genes of Pseudomonas aeruginosa in CF patients, and validated their reliability and sensitivity as compared with other specific genes, such as oprI, exoA, and algD. The method enabled detection of Pseudomonas aeruginosa in CF patients within a shorter period. Motoshima et al.74 developed a real-time PCR method using melting curve analysis (MCA), which specifically targeted the gyrB gene. As shown in Fig. 1, this method could readily distinguish Pseudomonas aeruginosa from other bacteria. Comparison between the performance of the real-time PCR method and the Vitek detection system, using 224 Gram-negative bacterial samples, confirmed that this method could accurately distinguish Pseudomonas aeruginosa from other Gram-negative bacteria within 3 h. A duplex real-time PCR assay was established against 2 specific genes, ecfX and gyrB, to effectively address the false-positive and false-negative issues in detecting Pseudomonas aeruginosa.60,73 Le Gall et al. (2013)82 demonstrated that multiplex real-time PCR could effectively solve the contradiction between sensitivity and specificity by analyzing the specificity and sensitivity of different genes, in addition to enabling detection of Pseudomonas aeruginosa from a mixed bacterial sample. Real-Time fluorescence-based PCR was also a highly sensitive, powerfully rapid, widely useful and prospectively detected tool in respiratory tract infections and pneumonia leading pathogen. Naoyuki et al. (2015) tested diagnostic sensitivity of immunochromatographic assay which was a rapid antigen kit for detection of Mycoplasma pneumonia, and compared with real-time PCR as gold standard.83 Idrissa et al. (2016) described a duplex real-time PCR assay based on SYBR dye for detection of Neisseria meningitides and Streptococcus pneumonia in the cerebrospinal fluid.84 Although most Pseudomonas aeruginosa tests have been performed with samples from CF patients, numerous researchers use real-time PCR to detect Pseudomonas aeruginosa in other samples. For example, Schwartz et al. have detected Pseudomonas aeruginosa in environmental wastewater,85 Feizabadi et al. tested saliva and bronchial secretions,86 and Lee et al. established a Pseudomonas aeruginosa-detection method for chlorinated water and aerosols.87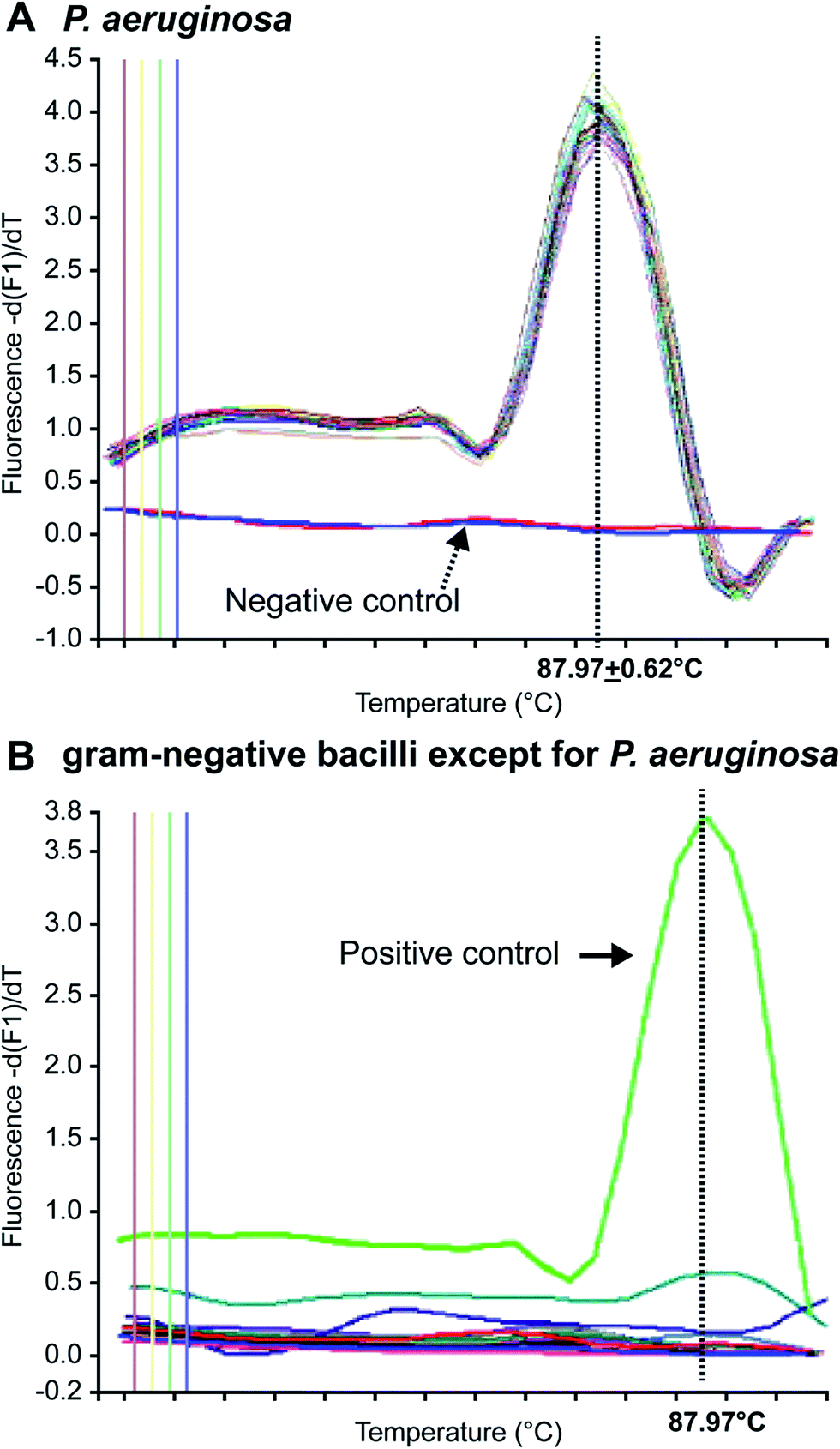 | ||
| Fig. 1 MCA of Gram-negative bacilli. (A) Pseudomonas aeruginosa-positive samples provided sharp peaks with a constant Tm of 87.9 °C, and (B) non-Pseudomonas aeruginosa samples showed no peak at 87.9 °C. (adapted from Motoshima et al., 2007). Order number from the Copyright Clearance Center: 4066350162315. | ||
4.3 Multiplex PCR
The main advantage of multiplex PCR is its ability to simultaneously amplify multiple PCR products in a single reaction, thereby enabling multiplex detection and significantly reducing the detection cost and time requirements.88 Microorganism-induced diseases are usually complicated with infections by multiple pathogens, for instance, bacteria can easily enrich and grow on the walls of catheters in patients with mechanical ventilation and renal intubation, leading to mixed infections with multiple bacteria, but a singleplex PCR assay cannot be used to simultaneously detect multiple bacterial species.89,90 Several studies have been conducted to establish multiplex PCR assays that include Pseudomonas aeruginosa. De Vos et al.68 simultaneously detected Pseudomonas fluorescens and Pseudomonas aeruginosa in clinical samples using a duplex PCR assay. This method had 100% sensitivity and 74% specificity, with a detection limit of 102 cells per mL in skin biopsy specimens from patients with burns and sputum samples from CF patients. Da Silva Filho et al. developed a multiplex PCR assay that simultaneously detected Pseudomonas aeruginosa, Burkholderia cepacia, and Stenotrophomonas maltophilia in the saliva and respiratory samples from CF patients (Da et al., 2004).91 British scholars, Fothergill et al. (2008)92 reported development of a multiplex PCR method that differentiated Liverpool epidemic strain (LES), Midlands1 (Mid1) strain, and Manchester epidemic strain (MES) with 100% specificity and sensitivity. Thong et al. developed a multiplex PCR assay that simultaneously detected methicillin-resistant Staphylococcus aureus (MRSA), Acinetobacter baumanni, Escherichia coli, Klebsiella pneumonia, and Pseudomonas aeruginosa.93 They also optimized the PCR conditions to ensure the specificity of amplification and confirmed that this method had 100% accuracy for both positive and negative samples by testing it with 50 culture samples. To better guarantee the detection specificity, Salman et al. (2013) developed a multiplex PCR assay for simultaneous amplification of 4 specific genes: gyrB, 16s rRNA, oprL, and ETA.94 Aghamollaei et al. (2015) developed a highly specific and sensitive method to detect Pseudomonas aeruginosa based on triplex PCR that amplifies the lasI, lasR, and gyrB genes.95 These methods could improve the specificity, but are inconvenient and have a relatively high cost in clinical applications. In 2017, Jiang et al. developed a combination of multiplex polymerase chain reaction (PCR) and capillary electrophoresis (MPCE) assay to detect thirteen bacterial pathogens responsible for lower respiratory tract infections. The result showed that the specificity and sensitivity for Pseudomonas aeruginosa were 100%.96 Multiplex PCR was a widely useful tool for detection of respiratory pathogens and transmitted infection in clinical specimens as well. Tayoun et al. (2015) described a highly sensitive, rapid and affordable sample-to-answer multiplex PCR assay for simultaneous detection of Trichomonas vaginalis, Nesserria gonorrhoeae, and Chamydia trachomatis resulting in sexually transmitted infections (STIs).974.4 Isothermal amplification techniques
Isothermal amplification techniques have overcome the shortcomings of conventional PCR methods, which require thermocycling for amplification. Isothermal amplification enables the rapid amplification of nucleic acid molecules at a constant temperature, which has not only reduced the equipment demand, but also has a higher sensitivity.98 Some recent studies have reported use of the loop-mediated isothermal amplification (LAMP) technique to detect Pseudomonas aeruginosa. Goto et al. developed a LAMP assay based on a hydroxynaphthol blue (HNB) colorimetric assay (Fig. 2), which targeted the oprL gene of Pseudomonas aeruginosa specifically. The results showed that the LAMP assay had 100% specificity for the serogroup with a >10-fold greater sensitivity than conventional PCR. It could detect 130 CFU per 0.1 g of mouse feces or 3.25 CFU per reaction. In addition, performing the assay only required 2 h from DNA extraction to detection.99 Moreover, this technique was also used to detect other organisms. For example, Liu et al. (2017) described a sensitive, reliable LAMP method to detect Neisseria gonorrhoeae porA pseudogene, and their results showed that the sensitivity and specificity of the LAMP assay were 94.7 and 85.7%, related to traditional culture, respectively.100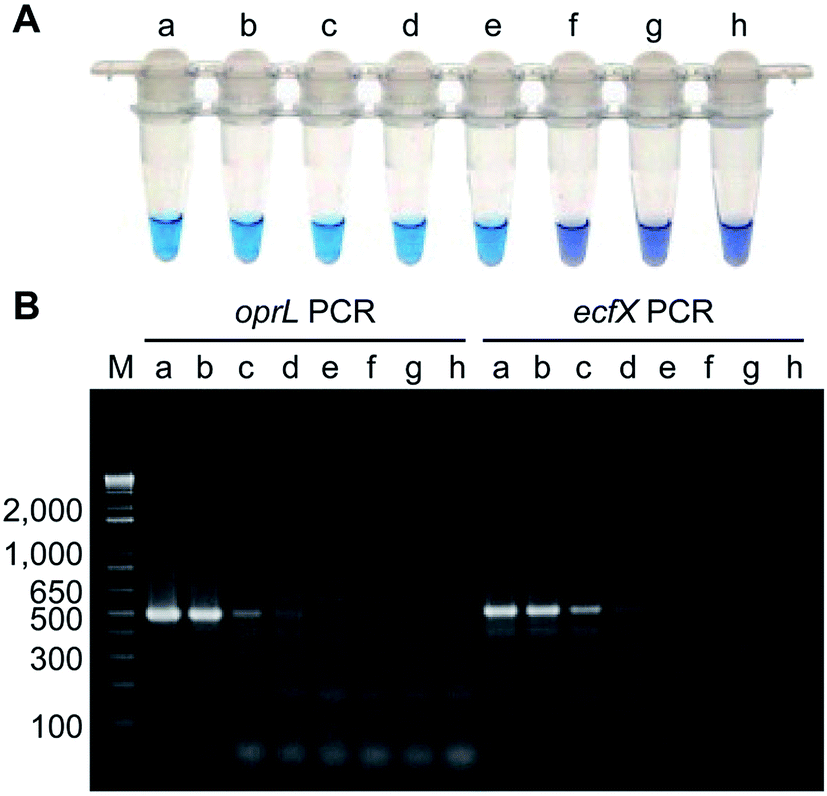 | ||
| Fig. 2 Comparison of the sensitivities of LAMP and PCR assays using Pseudomonas aeruginosa-inoculated fecal samples. (A) Sensitivity of LAMP for Pseudomonas aeruginosa as determined by measuring HNB color changes. A positive reaction was indicated by the change in color from violet to sky blue. (B) Agarose gel electrophoretic analysis of PCR products obtained after 35 cycles. The expected sizes of the PCR products obtained by PCR-amplification of the oprL and ecfX genes were 504 bp and 528 bp, respectively. Lanes (tubes) (a) 1.3 × 106; (b) 1.3 × 105; (c) 1.3 × 104; (d) 1.3 × 103; (e) 130; (f) 13; (g) 1.3 CFU; h, no Pseudomonas aeruginosa; M, DNA size marker. (adapted from Goto et al., 2010). Order number from the Copyright Clearance Center: 4066350607817. | ||
4.5 Fluorescence in situ hybridization (FISH) technique using peptide nucleic acid (PNA) probes
FISH based on PNA probes is a rapid diagnostic technique for pathogens. PNA probes are more conducive to hybridization than oligonucleotides owing to their neutral charges. The targets of PNA probes are the rRNA molecules, which are abundant in nature, and thus do not need to be amplified during the detection of single microorganisms (Parcell et al., 2013).101 In addition, PNA probes are barely affected by impurities in clinical samples; therefore, they have higher efficiency in directly detecting pathogens in clinical samples, such as blood, saliva, and wound cultures (Almeida et al., 2013).102 Søgaard and his colleagues evaluated the feasibility of using FISH with PNA probes to detect E. coli and Pseudomonas aeruginosa in clinical blood cultures.103 The results showed that the method had 94.1% sensitivity and 99% specificity in detecting Pseudomonas aeruginosa. Peleg et al. developed a multiplex PNA FISH technique for the simultaneous detection of Acinetobacter and Pseudomonas aeruginosa, using dual-color fluorescence, which could detect the target bacteria in a mixture consisting of 10 different bacterial species.104 The technique was tested with 60 clinical samples, and the results showed that its sensitivity and specificity for detecting Pseudomonas aeruginosa were 100% and 95%, respectively. Although this method is very simple, it still involves a culture period for enrichment to aid the detection of Pseudomonas aeruginosa during early infection.4.6 Aptamer-based in situ detection methods
Aptamers, also known as “artificial antibodies,” are short nucleic acid or peptide sequences capable of binding to a target molecule with high specificity and affinity (Ruff et al., 2012).105 Aptamers have several advantages over antibodies, such as easy production, easy storage, easy modification, and low cost. In addition, aptamers exhibit greater affinity and specificity for targets. Therefore, aptamers have been widely applied for detecting pathogens in recent years.106,107 Wang et al. (2011) selected a bacteria-targeting ssDNA aptamer that specifically bound Pseudomonas aeruginosa and utilized its specific binding capacity to develop a rapid FISH method for detecting Pseudomonas aeruginosa (Fig. 3). The results showed that the method had excellent specificity, and the entire detection process took 1.5–2 h. Hence, the method can potentially facilitate clinical Pseudomonas aeruginosa detection.108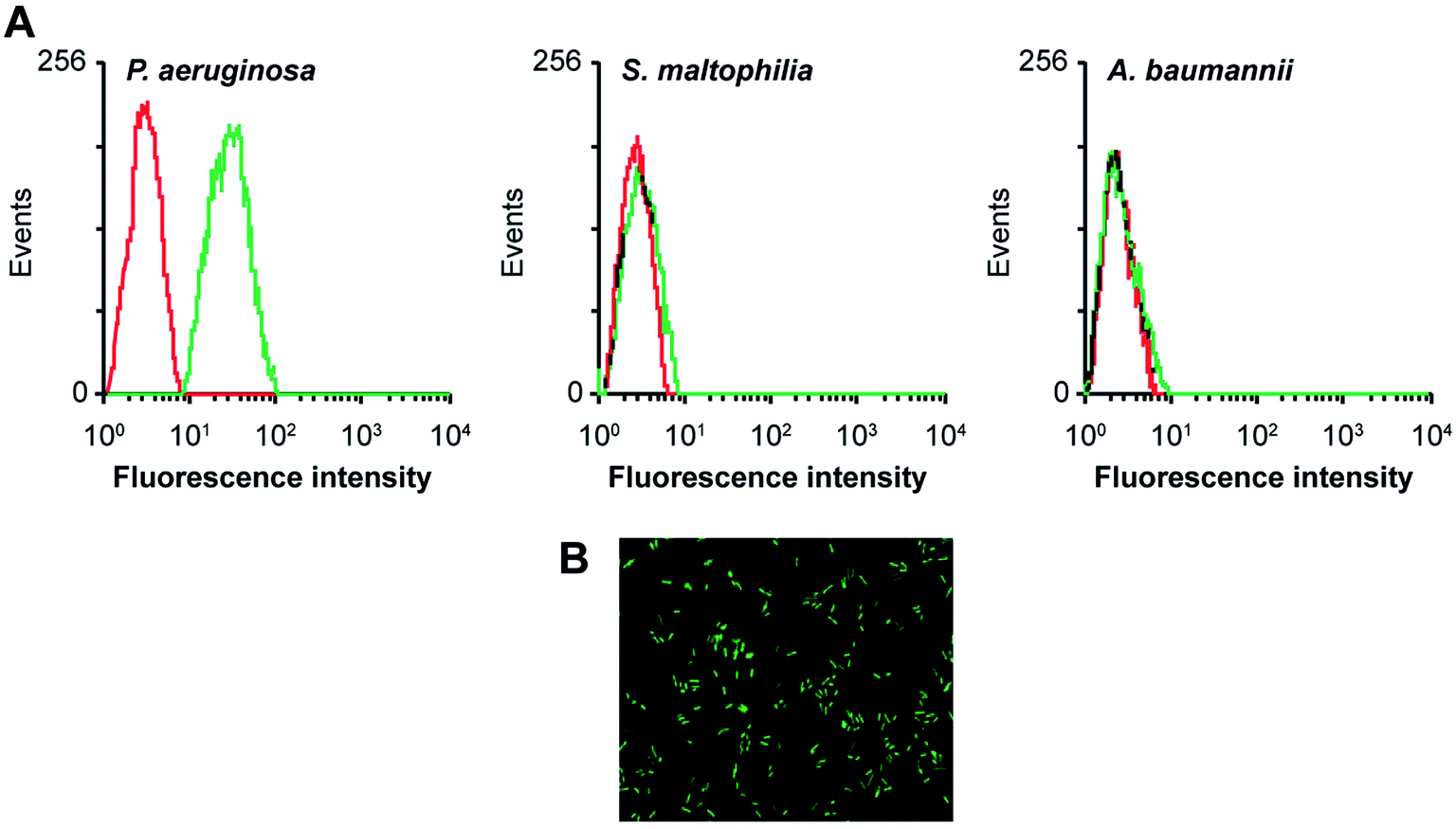 | ||
| Fig. 3 Evaluation of the specificity of an aptamer probe. (A) Flow cytometry assay for the binding of the fluorescein isothiocyanate (FITC)-labeled F23 aptamer with Pseudomonas aeruginosa (target bacteria), S. maltophilia (negative bacteria) and A. baumannii (negative bacteria). The red curves represent the background binding of an unselected DNA library. The aptamer concentration in the binding buffer was 400 nM. (B) Evaluation of the specificity of the aptamer probe by FISH. Bacteria were hybridized with the FITC-labeled F23 aptamer probe. A representative image of cultured Pseudomonas aeruginosa stained with the aptamer probe is shown. Green fluorescence signifies positive hybridization. (adapted from Wang et al., 2011). Order number from the Copyright Clearance Center: 4066351239887. | ||
4.7 PCR-ELISA methods
Kurupati et al. used the PCR-ELISA DIG Labellingplus kit (Roche Diagnostics Ltd, Lewes, UK) to establish a method for detecting Pseudomonas aeruginosa in blood cultures by targeting the oprI gene.109 The principle of the method is to first amplify the digoxin-labeled PCR product, which is then hybridized with a biotinylated probe. The resulting hybridization product is immobilized in a microwell plate via a biotin-streptavidin coupling system, followed by incubation with an anti-digoxigenin peroxidase conjugate. Finally, the substrate molecule, 2,2-azino-di-3-ethylbenzithiazoline sulfonate, is added to enable detection. Using this method, the authors correctly identified 73 positive samples and 42 negative samples with 100% specificity and sensitivity. The method could shorten the blood culture time from 2–3 days to 6–8 h. Furthermore, other pathogens were also detected using this technique. For example, Medeiros et al. (2017) reported that kDNA PCR-ELISA exhibited satisfactory precision for detection of Leishmania infantum in 14 peripheral blood samples from immunocompetent patients.1105. Electrochemical assays
Electrochemical analysis is a rapid and sensitive analytical method involving a compact device that enables qualitative and quantitative detection, using the electrochemical properties of materials.111 Liu et al. (2011) designed an electrochemical DNA biosensor based on stem-loop-structured probes to detect Pseudomonas aeruginosa.112 The principle of this method entails opening of the stem-loop after hybridization, so that the biotin molecule on the probe can bind streptavidin-labeled horseradish peroxidase (SA-HRP) in solution, after which HRP catalyzes the production of an electrochemical signal. Sensitivity test results showed that the method could be used to detect 16S rRNA at a concentration as low as 0.012 pg μL−1, with a linear relationship between 0.3–600 pg μL−1. Webster et al. (2014) reported a disposable carbon electrode sensor that could directly detect pyocyanin production by Pseudomonas aeruginosa in samples.113 The electrochemical detection of Pseudomonas aeruginosa pyocyanin in samples could be completed within 5 min, and the entire procedure does not require sample-preparation and -isolation steps. Furthermore, the method has exhibited excellent sensitivity and specificity (Fig. 4). In the same year, Sismaet and colleagues improved the sensitivity of square wave voltammetry (SWV) in analyzing pyocyanin production by supplementing the culture with tyrosine and valine as positive regulators of pyocyanin.114 Sensitive detection of quorum sensing can potentially enable early detection of Pseudomonas aeruginosa and facilitate early medical intervention. Sensitive detection of quorum sensing (QS) molecules has the potential for early identification of Pseudomonas aeruginosa and, thereby, facilitating early medical intervention. Shang et al. (2014) developed a 1-step method to prepare thiazole termed IQS, which is a recently isolated cell–cell communication molecule, and established a highly sensitive method for detecting this molecule using boron-doped diamond and glassy carbon electrodes by cyclic voltammetry and amperometry.115 Certainly, electrochemical technique was widely acceptable and popular in detection of Pseudomonas aeruginosa and other microoragnisms, due to its preponderant feature. For example, Maciej et al. (2015) devised a selective electrochemical sensing for human serum albumin (HSA) determination using semi-covalent imprinting, and established an excellent selectivity to the myoglobin and cytochrome c interferences.116 Sofia et al. (2017) reported an electrochemical aptasensor for detection of human osteopontin (OPN), a potenrial breast cancer biomarker, by using a DNA aptamer selected by SELEX. Their results showed that the aptasensor and the standard ELISA methods quantified similar OPN levels.117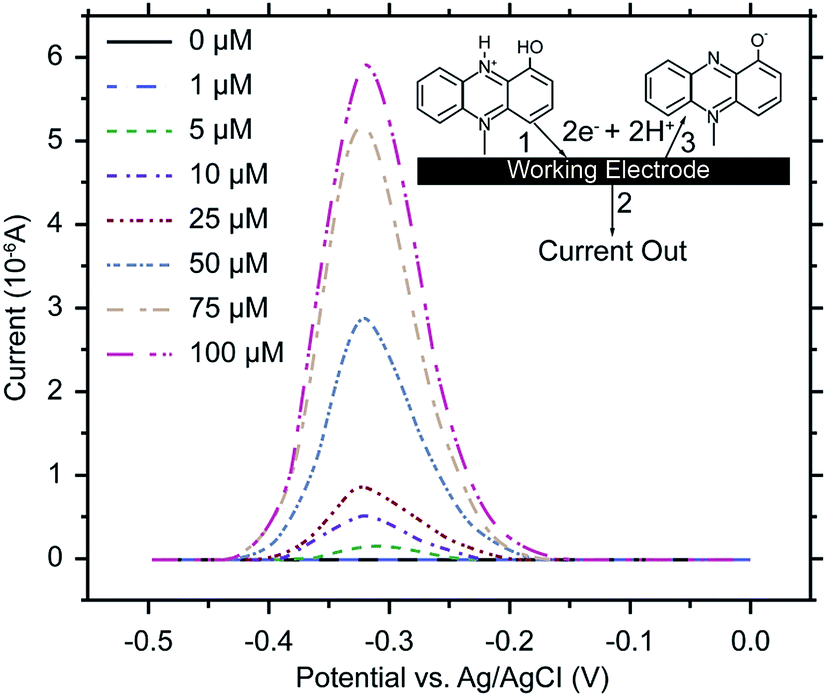 | ||
| Fig. 4 SWV of human bronchial lavage samples, with pyocyanin added. Scans were performed from 0.5 to 0 V at a frequency of 15 Hz and an amplitude voltage of 50 mV, with the current response from the blank subtracted from each sample measurement. Inset: pyocyanin reacts at the working electrode (1) releasing 2 electrons and 2 hydrogens (2) and is converted to its oxidized form (3). (Adapted from Webster, et al., 2014). Order number from the Copyright Clearance Center: 4066351485823. | ||
6. MALDI-TOF MS assays
Matrix-assisted laser desorption/ionisation time of flight mass spectrometry (MALDI-TOF MS) is a new type of soft ionization mass spectrometry, which is used to map the protein spectrum of microorganisms.118 The mass spectrometry data of clinical microorganisms are compared with the standard protein database of known microorganisms to achieve the purpose of identification.119 Because of its rapid, accurate, sensitive, automated and high throughput, MALDI-TOF MS has become an efficient microbial rapid identification technology used in clinical diagnosis,120 environmental monitoring121 and microbiological classification studies.122 Some researchers have applied this technique to the identification of Pseudomonas aeruginosa. Imperi et al. (2009) report a detailed description of the periplasmic proteome of the wild-type Pseudomonas aeruginosa strain PAO1 by 2-DE and MALDI-TOF/TOF analysis.123 In 2012, a rapid detection and identification method for Pseudomonas aeruginosa by MALDI-TOF MS was established.124 It was observed that the results of MALDI-TOF MS for the detection of Pseudomonas aeruginosa were consistent with that of automated identification systems Phoenix (BD Biosciences, USA). Recently, MALDI-TOF MS was used to accurately and quickly identify the five high-risk clones of Pseudomonas aeruginosa sequence type 111 (ST111), ST175, ST235, ST253, and ST395.125 More often, this technique was used in Pseudomonas aeruginosa drug resistance analysis such as carbapenemase (Wang et al., 2013; Johansson et al., 2014; Hrabak, 2015).126–128 MALDI-TOF MS represents an innovative technology and has a very good application prospects in the identification of Pseudomonas aeruginosa.MALDI-TOF has been developed for identification of clinical pathogens. In fact, in addition to Pseudomonas aeruginosa, many other pathogens were detected using this technique. For example, Camoez et al.(2016) described a novel MALDI-TOF-automated method to identify methicillin-resistant Staphylococcus aureus (MRSA) lineages from hospital during a 20 year period (1990–2009) and demonstrated that this tool was reliable, powerful, promising for S. aureus typing.129 Maasz et al. (2017)130 reported that MALDI-TOF MS was a cheap, reliable, sensitive and fast detection tool in mayfly and fish species identification and sex determination in bleak (Alburnus alburnus).
7. Summary and prospect
Pseudomonas aeruginosa is a prevalent, opportunistic, Gram-negative bacterium that commonly infects immunocompromised individuals; thus, many cases of hospital-acquired and community-acquired infections are caused by Pseudomonas aeruginosa. P. aeruginosa has been associated with increased mortality relative to Staphylococcus aureus or other Gram-negative in bloodstream infections.131 Previous findings have shown that as few as 10–100 bacilli are capable of colonizing the intestine of critically ill or immunocompromised patients.132 Therefore, early detection of Pseudomonas aeruginosa is particularly important. At present, culturing bacteria remains the most commonly applied method for detecting Pseudomonas aeruginosa in hospitals, but this method is time-consuming and susceptible to inconsistent results due to sample contamination. In addition, this method has low sensitivity. To address these issues, researchers have developed various assays, each with their own advantages and disadvantages. These methods may be limited by low stability and repeatability with regard to clinical applications, and most importantly, the risk of sample contamination remains unresolved. The developmental trend shows that direct detection of Pseudomonas aeruginosa in clinical samples will shorten diagnostic turnaround time and reduce the risk of contamination. In addition, the development of fully enclosed automatic detection techniques likely represents a primary direction of future developments.Conflicts of interest
The authors report no conflicts of interest.Acknowledgements
We would like to thank Editage (www.editage.com) for English language editing. This work was supported by the Scientific Research Fund of the Shenzhen Basic Research Projects under Grant number JCYJ20140901141243646; the GDPRSFS under Grant number 2012; the GDUHTP under Grant number 2011 and 2013; the Science and Technology Planning Project of Guangdong Province under Grant number 2014A020217021; and the Shenzhen Basic Research Projects under (Grant number JCYJ2016043164316142 and JCYJ20170413155047512).References
- C. S. Tran, Y. Eran, T. R. Ruch, D. M. Bryant, A. Datta, P. Brakeman, A. Kierbel, T. Wittmann, R. J. Metzger and K. E. Mostov, Cell Host Microbe, 2014, 15, 636–643 CAS.
- K. H. Turner, J. Everett, U. Trivedi, K. P. Rumbaugh and M. Whiteley, PLoS Genet., 2014, 10, e1004518 Search PubMed.
- A. Y. Koh, G. P. Priebe and G. B. Pier, Infect. Immun., 2005, 73, 2262–2272 CrossRef CAS PubMed.
- T. Ajayi, L. R. Allmond, T. Sawa and J. P. Wienerkronish, J. Clin. Microbiol., 2003, 41, 3526–3531 CrossRef CAS PubMed.
- J. Y. Fagon, J. Chastre, Y. Domart, J. L. Trouillet, J. Pierre, C. Darne and C. Gibert, Am. Rev. Respir. Dis., 1989, 139, 877–884 CrossRef CAS PubMed.
- J. Y. Fagon, J. Chastre, A. J. Hance, P. Montravers, A. Novara and C. Gibert, Am. J. Med., 1993, 94, 281–288 CrossRef CAS PubMed.
- L. V. Ivanova, T. Z. Artemova, E. K. Gipp, T. N. Maksimkina and A. V. Krasniak, Gig. Sanit., 2013, 4, 29–32 Search PubMed.
- J. Zheng, P. Ma and X. R. Huang, Food Sci., 2007, 7, 419–424 Search PubMed.
- M. Rüger, M. Ackermann and U. Reichl, BMC Microbiol., 2014, 14, 1–15 CrossRef PubMed.
- A. R. Rao, M. S. Splaingard, W. M. Gershan, P. L. Havens, A. Thill and J. T. Barbieri, Pediatr. Pulmonol., 2005, 39, 402–407 CrossRef CAS PubMed.
- H. J. Choi, M. H. Kim, M. S. Cho, B. K. Kim, J. Y. Kim, C. Kim and D. S. Park, Appl. Microbiol. Biotechnol., 2013, 97, 3643–3651 CrossRef CAS PubMed.
- E. J. Lowbury and A. G. Collins, J. Clin. Pathol., 1955, 8, 47–48 CrossRef CAS PubMed.
- V. I. Brown and E. J. Lowbury, J. Clin. Pathol., 1965, 18, 752–756 CrossRef CAS PubMed.
- G. Szita and G. Biró, Acta Vet. Hung., 1990, 38, 187–194 CAS.
- H. Kodaka, M. Iwata, S. Yumoto and F. Kashitani, J. Basic Microbiol., 2003, 43, 407–413 CrossRef CAS PubMed.
- G. Szita, V. Tabajdi, A. Fábián, G. Biró, O. Reichart and P. S. Körmöczy, Int. J. Food Microbiol., 1998, 43, 123–127 CrossRef CAS PubMed.
- K. Fonseca, J. Macdougall and T. L. Pitt, J. Clin. Pathol., 1986, 39, 220–222 CrossRef CAS PubMed.
- P. Joyanes, C. C. M. Del, L. Martínezmartínez and E. J. Perea, J. Clin. Microbiol., 2001, 39, 3247–3253 CrossRef CAS PubMed.
- G. Funke and P. Funkekissling, J. Clin. Microbiol., 2004, 42, 1466–1470 CrossRef CAS PubMed.
- L. Saiman, J. L. Burns, D. Larone, Y. Chen, E. Garber and S. Whittier, J. Clin. Microbiol., 2003, 41, 492–494 CrossRef PubMed.
- W. S. Hsieh, L. L. Sung, K. C. Tsai and H. T. Ho, APMIS, 2009, 117, 241–247 CrossRef CAS PubMed.
- M. J. Bruins, P. Bloembergen, G. J. H. M. Ruijs and M. J. H. M. Wolfhagen, J. Clin. Microbiol., 2004, 42, 7–11 CrossRef PubMed.
- E. Stefaniuk, A. Baraniak, M. Gniadkowski and W. Hryniewicz, Eur. J. Clin. Microbiol. Infect. Dis., 2003, 22, 479–485 CrossRef CAS PubMed.
- M. D. Quesada, M. Giménez, S. Molinos, G. Fernández, M. D. Sánchez, R. Rivelo, A. Ramírez, G. Banqué and V. Ausina, Clin. Microbiol. Infect., 2010, 16, 137–140 CrossRef CAS PubMed.
- C. M. O'Hara and J. M. Miller, J. Clin. Microbiol., 2002, 40, 3750–3752 CrossRef.
- I. Otto-Karg, S. Jandl, T. Müller, B. Stirzel, M. Frosch, H. Hebestreit and A. M. Abele-Horn, J. Clin. Microbiol., 2009, 47, 3283–3288 CrossRef CAS PubMed.
- J. Beuving, C. F. V. D. Donk, C. F. Linssen, P. F. Wolffs and A. Verbon, BMC Microbiol., 2011, 11, 156–162 CrossRef PubMed.
- A. Endimiani, F. Luzzaro, A. Tamborini, G. Lombardi, V. Elia, R. Belloni and A. Toniolo, New Microbiol., 2002, 25, 323–329 CAS.
- G. L. Bratthauer, Methods Mol. Biol., 2010, 588, 243–255 CAS.
- A. Schalhorn and W. Wilmanns, Onkologie, 1980, 3, 193–196 CAS.
- Y. Xu, T. Valerie, S. Crystal, D. P. Kathleen, A. Laura, S. Keirsten, M. A. Perricone and S. M. Richards, J. Transl. Med., 2008, 6, 61 CrossRef PubMed.
- G. H. Shand, S. S. Pedersen, R. Tilling, M. R. Brown and N. Høiby, J. Med. Microbiol., 1988, 27, 169–177 CrossRef CAS PubMed.
- N. Shibuya, S. Kohno, Y. Shigeno, K. Yamaguchi, A. Saito, K. Hara, C. Mochida and K. Sugahara, Kansenshogaku Zasshi, 1987, 61, 1406–1414 CrossRef CAS PubMed.
- D. K. Wagner, M. T. Yach and M. W. Rytel, Infection, 1986, 14, 105–107 CrossRef CAS PubMed.
- R. Kohler and A. White, J. Infect. Dis., 1977, 136, 112–116 CrossRef CAS PubMed.
- Y. Ueda, Y. Sanai and J. Y. Homma, Am. J. Vet. Res., 1982, 43, 55–60 CAS.
- M. Granström, B. Wretlind, B. Markman, O. R. Pavlovskis and M. L. Vasil, Eur. J. Clin. Microbiol., 1985, 4, 197–200 Search PubMed.
- A. Fomsgaard, B. Dinesen, G. H. Shand, T. Pressler and N. Høiby, J. Clin. Microbiol., 1989, 27, 1222–1229 CAS.
- D. Dogru, S. Pekcan, E. Yalcin and B. Sener, Turk. J. Pediatr., 2013, 55, 50–57 Search PubMed.
- L. Beutin, S. Zimmermann and K. Gleier, Infection, 1996, 24, 267–268 CrossRef CAS PubMed.
- C. Bisseye, J. M. N. Nodong, A. M. Matoumba and C. Bengone, Asian Pac. J. Trop. Biomed., 2017, 7, 805–808 CrossRef.
- B. Gake, M. A. Vernet, I. Leparc-Goffart, J. F. Drexler, E. A. Gould, P. Gallian and X. de Lamballerie, Braz. J. Infect. Dis., 2017, 21, 481–483 CrossRef PubMed.
- O. A. Hamid, S. Elfedawy, S. K. Mohamed and H. Mosaad, Eur. Arch. Otorhinolaryngol., 2009, 266, 1569–1573 CrossRef PubMed.
- A. Pârishamelin, M. Debruyne and S. Fustec-Isarboure, Ann. Pharm. Fr., 1999, 57, 68–75 Search PubMed.
- J. C. Chin, B. Pang and M. Carrigan, Vet. Microbiol., 1991, 26, 291–299 CrossRef CAS PubMed.
- E. Ayala-Sulca and E. Miranda-Ulloa, Rev. Peru. Med. Exp. Salud Publica, 2015, 32, 485–491 CrossRef PubMed.
- E. Caballero, M. E. Drobnic, M. T. Pérez, J. M. Manresa, A. Ferrer and R. Orriols, Thorax, 2001, 56, 669–674 CrossRef CAS PubMed.
- A. M. Weisner, H. Chart, A. Bush, J. C. Davies and T. L. Pitt, J. Med. Microbiol., 2007, 56, 670–674 CrossRef CAS PubMed.
- L. F. Pemberton, Methods Mol. Biol., 2014, 1205, 79–90 CAS.
- H. Miyazaki, Y. Yoshimatsu, Y. Akatsu, K. Mishima, M. Fukayama, T. Watabe and K. Miyazono, Cancer Sci., 2014, 105, 1116–1123 CrossRef CAS PubMed.
- D. R. Parks, R. R. Hardy and L. A. Herzenberg, Immunol. Today, 1983, 4, 145–150 CrossRef CAS PubMed.
- C. Chen, J. Peng, H. S. Xia, G. F. Yang, Q. S. Wu, L. D. Chen, L. B. Zeng, Z. L. Zhang, D. W. Pang and Y. Li, Biomaterials, 2009, 30, 2912–2918 CrossRef CAS PubMed.
- X. Yang, Y. Zhuo, S. Zhu, Y. Luo, Y. Feng and Y. Xu, Biosens. Bioelectron., 2015, 64, 345–351 CrossRef CAS PubMed.
- L. Zhang, Z. Wang, Z. Lu, K. Xia, Y. Deng, S. Li, C. Zhang, Y. Huang and N. He, J. Nanosci. Nanotechnol., 2014, 14, 4710–4713 CrossRef CAS PubMed.
- A. M. Oliveira and C. A. French, Acta Cytol., 2005, 49, 587–594 CrossRef PubMed.
- H. W. Roberts, J. F. Donati-Bourne, V. L. Wilson and J. C. Wilton, J. Invest. Surg., 2013, 26, 283–293 CrossRef PubMed.
- M. A. Pfaller, M. Barrett, F. P. Koontz, R. P. Wenzel, M. D. Cunningham, N. Rollins and R. P. Darveau, J. Clin. Microbiol., 1989, 27, 558–560 CAS.
- Y. Tang, Chin. J. Tuberc. Respir. Dis., 1992, 15, 8–10 CAS.
- D. Baud, J. Zufferey, P. Hohlfeld and G. Greub, Diagn. Microbiol. Infect. Dis., 2014, 78, 217–219 CrossRef CAS PubMed.
- H. Gotfred-Rasmussen, M. Lund, H. L. Enemark, M. Erlandsen and E. Petersen, Diagn. Microbiol. Infect. Dis., 2016, 84, 187–190 CrossRef CAS PubMed.
- H. Seliger, Trends Biotechnol., 1990, 8, 335 CrossRef.
- A. C. Cheng, J. Microbiol., Immunol. Infect., 2015, 48, S19 Search PubMed.
- D. Bayley and A. Teng, J. Dent. Res., 2002, 81, A195 Search PubMed.
- Y. Li, D. E. Cooke, E. Jacobsen and D. L. T. Van, J. Microbiol. Methods, 2013, 92, 316–322 CrossRef CAS PubMed.
- N. M. Kingsford and H. W. Raadsma, Vet. Microbiol., 1995, 47, 61–70 CrossRef CAS PubMed.
- A. Hummel and G. Unger, Zentralbl. Hyg. Umweltmed., 1998, 201, 349–355 CAS.
- P. Deschaght, S. V. Daele, F. D. Baets and M. Vaneechoutte, J. Cystic Fibrosis, 2011, 10, 293–297 CrossRef CAS PubMed.
- D. De Vos, A. J. Lim, J. P. Pirnay, M. Struelens, C. Vandenvelde, L. Duinslaeger, A. Vanderkelen and P. Cornelis, J. Clin. Microbiol., 1997, 35, 1295–1299 CAS.
- R. Lavenir, D. Jocktane, F. Laurent, S. Nazaret and B. Cournoyer, J. Microbiol. Methods, 2007, 70, 20–29 CrossRef CAS PubMed.
- S. N. Anuj, D. M. Whiley, T. J. Kidd, K. A. Ramsay, S. C. Bell, M. W. Syrmis, K. Grimwood, C. E. Wainwright, M. D. Nissen and T. P. Sloots, Clin. Microbiol. Infect., 2011, 17, 1403–1408 CrossRef CAS PubMed.
- X. Qin, J. Emerson, J. Stapp, L. Stapp, P. Abe and J. L. Burns, J. Clin. Microbiol., 2003, 41, 4312–4317 CrossRef CAS PubMed.
- S. F. L. Da, J. E. Levi, C. N. Oda Bento, S. R. S. Da and T. Rozov, J. Med. Microbiol., 1999, 48, 357–361 CrossRef PubMed.
- S. N. Anuj, D. M. Whiley, T. J. Kidd, S. C. Bell, C. E. Wainwright, M. D. Nissen and T. P. Sloots, Diagn. Microbiol. Infect. Dis., 2009, 63, 127–131 CrossRef CAS PubMed.
- M. Motoshima, K. Yanagihara, K. Fukushima, J. Matsuda, K. Sugahara, Y. Hirakata, Y. Yamada, S. Kohno and S. Kamihira, Diagn. Microbiol. Infect. Dis., 2007, 58, 53–58 CrossRef CAS PubMed.
- R. I. Jaffe, J. D. Lane and C. W. Bates, J. Clin. Lab. Anal., 2001, 15, 131–137 CrossRef CAS PubMed.
- G. J. Al-Ahmad and R. Z. Roodsari, Ann. Burns Fire Disasters, 2016, 29, 264–267 Search PubMed.
- B. S. Pritt, P. S. Mead, D. K. H. Johnson, D. F. Neitzel, L. B. Respicio-Kingry, J. P. Davis, E. Schiffman, L. M. Sloan, M. E. Schriefer and A. J. Replogle, Lancet Infect. Dis., 2016, 16, 556–564 CrossRef PubMed.
- Y. Tang, J. Zou, C. Ma, Z. Ali, Z. Li, X. Li, N. Ma, X. Mou, Y. Deng, L. Zhang, K. Li, M. Lu, H. Yang and N. He, Theranostics, 2013, 3, 85–92 CrossRef CAS PubMed.
- Y. Tang, Z. Li, N. He, L. Zhang, C. Ma, X. Li, C. Li, Z. Wang, Y. Deng and N. He, J. Biomed. Nanotechnol., 2013, 9, 312–317 CrossRef CAS PubMed.
- Y. Tang, Z. Ali, J. Zou, K. Yang, X. Mou, Z. Li, Y. Deng, Z. Lu, C. Ma and M. A. Shah, J. Nanosci. Nanotechnol., 2014, 14, 4886–4890 CrossRef CAS PubMed.
- G. Johnson, T. Nolan and S. A. Bustin, Methods Mol. Biol., 2013, 943, 1–16 CAS.
- F. L. Gall, R. L. Berre, S. Rosec, J. Hardy, S. Gouriou, S. Boisramé-Gastrin, S. Vallet, G. Rault, C. Payan and G. Héry-Arnaud, BMC Microbiol., 2013, 13, 143 CrossRef PubMed.
- N. Miyashita, Y. Kawai, T. Tanaka, H. Akaike, H. Teranishi, T. Wakabayashi, T. Nakano, K. Ouchi and N. Okimoto, J. Infect. Chemother., 2015, 21, 473–475 CrossRef CAS PubMed.
- I. Diawara, K. Katfy, K. Zerouali, H. Belabbes and N. Elmdaghri, J. Infect. Dev. Ctries, 2016, 10, 53–61 CrossRef PubMed.
- T. Schwartz, H. Volkmann, S. Kirchen, W. Kohnen, K. Schönhölz, B. Jansen and U. Obst, FEMS Microbiol. Ecol., 2006, 57, 158–167 CrossRef CAS PubMed.
- M. M. Feizabadi, A. Majnooni, B. Nomanpour, B. Fatolahzadeh, N. Raji, S. Delfani, M. Habibi, S. Asadi and M. Parvin, Infect., Genet. Evol., 2010, 10, 1247–1251 CrossRef CAS PubMed.
- C. S. Lee, K. Wetzel, T. Buckley, D. Wozniak and J. Lee, J. Appl. Microbiol., 2011, 111, 893–903 CrossRef CAS PubMed.
- J. B. Mahony, G. Blackhouse, J. Babwah, M. Smieja, S. Buracond, S. Chong, W. Ciccotelli, T. O'Shea, D. Alnakhli and M. Griffithsturner, J. Clin. Microbiol., 2009, 47, 2812–2817 CrossRef PubMed.
- X. C. Li, J. Z. Wang and Y. H. Liu, Chin. J. Contemp. Pediatr., 2012, 14, 406–408 Search PubMed.
- D. L. R. Gomes, C. A. S. Martins, L. M. D. Faria, L. S. Santos, C. S. Santos, P. S. Sabbadini, M. C. Souza, G. B. Alves, A. C. P. Rosa and P. E. Nagao, J. Med. Microbiol., 2009, 58, 1419–1427 CrossRef CAS PubMed.
- S. F. L. Da, A. F. Tateno, L. F. Velloso, J. E. Levi, S. Fernandes, C. N. Bento, J. C. Rodrigues and S. R. Ramos, Pediatr. Pulmonol., 2004, 37, 537–547 CrossRef PubMed.
- J. L. Fothergill, A. L. Upton, T. L. Pitt, C. A. Hart and C. Winstanley, J. Cystic Fibrosis, 2008, 7, 258–261 CrossRef CAS PubMed.
- K. L. Thong, M. Y. Lai, C. S. J. Teh and K. H. Chua, Trop Biomed., 2011, 28, 21–31 CAS.
- M. Salman, A. Ali and A. Haque, Pak. J. Med. Sci., 2013, 29, 957–961 Search PubMed.
- H. Aghamollaei, M. M. Moghaddam, H. Kooshki, M. Heiat, R. Mirnejad and N. S. Barzi, J. Infect. Public Health, 2015, 18, 314–322 CrossRef PubMed.
- L. Jiang, H. Ren, H. Zhou, S. Zhao, B. Hou, J. Yan, T. Qin and Y. Chen, Biomed. Environ. Sci., 2017, 30, 549–561 Search PubMed.
- A. N. A. Tayoun, P. R. Burchard, A. M. Caliendo, A. Scherer and G. J. Tsongalis, Exp. Mol. Pathol., 2015, 98, 214–218 CrossRef PubMed.
- K. Dhama, K. Karthik, S. Chakraborty, R. Tiwari, S. Kapoor, A. Kumar and P. Thomas, Pak. J. Biol. Sci., 2014, 17, 151–166 CrossRef CAS PubMed.
- M. Goto, K. Shimada, A. Sato, E. Takahashi, T. Fukasawa, T. Takahashi, S. Ohka, T. Taniguchi, E. Honda and A. Nomoto, J. Microbiol. Methods, 2010, 81, 247–252 CrossRef CAS PubMed.
- M. L. Liu, Y. Xia, X. Z. Wu, J. Q. Huang and X. G. Guo, AMB Express, 2017, 7, 48 CrossRef PubMed.
- B. J. Parcell and G. V. Orange, J. Microbiol. Methods, 2013, 95, 253–255 CrossRef CAS PubMed.
- C. Almeida, J. M. Sousa, R. Rocha, L. Cerqueira, S. Fanning, N. F. Azevedo and M. J. Vieira, Appl. Environ. Microbiol., 2013, 79, 6293–6300 CrossRef CAS PubMed.
- M. Søgaard, H. Stender and H. C. Schønheyder, J. Clin. Microbiol., 2005, 43, 1947–1949 CrossRef PubMed.
- A. Y. Peleg, Y. Tilahun, M. J. Fiandaca, E. M. C. D'Agata, L. Venkataraman, R. C. Moellering Jr and G. M. Eliopoulos, J. Clin. Microbiol., 2009, 47, 830–832 CrossRef CAS PubMed.
- P. Ruff, R. B. Pai and F. Storici, ISRN Mol. Biol., 2012, 939083, 1–9 Search PubMed.
- M. Labib, A. S. Zamay, O. S. Kolovskaya, I. T. Reshetneva, G. S. Zamay, R. J. Kibbee, S. A. Sattar, T. N. Zamay and M. V. Berezovski, Anal. Chem., 2012, 84, 8114–8117 CrossRef CAS PubMed.
- M. Valencia-Burton and N. E. Broude, Curr. Protoc. Cell Biol., 2007, 37, 1–20 Search PubMed.
- K. Wang, Y. Zeng, X. Yang, W. Li and X. Lan, Eur. J. Clin. Microbiol. Infect. Dis., 2011, 30, 273–278 CrossRef PubMed.
- P. Kurupati, G. Kumarasinghe and P. C. Laa, Mol. Cell. Probes, 2005, 19, 417–421 CrossRef CAS PubMed.
- F. A. Medeiros, L. I. Gomes, E. Oliveira, C. S. de Souza, M. V. Mourão, G. F. Cota, L. H. Marques, M. Carneiro and A. Rabello, J. Trop. Med., 2017, 7364854, 1–10 Search PubMed.
- C. F. Becker, N. J. Watmough and S. J. Elliott, Biochemistry, 2009, 48, 87–95 CrossRef CAS PubMed.
- C. Liu, G. M. Zeng, L. Tang, Y. Zhang, Y. P. Li, Y. Y. Liu, Z. Li, M. S. Wu and J. Luo, Enzyme Microb. Technol., 2011, 49, 266–271 CrossRef CAS PubMed.
- T. A. Webster, H. J. Sismaet, J. L. Conte, I. P. Chan and E. D. Goluch, Biosens. Bioelectron., 2014, 60, 265–270 CrossRef CAS PubMed.
- H. J. Sismaet, T. A. Webster and E. D. Goluch, Analyst, 2014, 139, 4241–4246 RSC.
- F. Shang, E. Ó. Muimhneacháin, F. J. Reen, A. Buzid, F. O'Gara, J. H. Luong, J. D. Glennon and G. P. Mcglacken, Bioorg. Med. Chem. Lett., 2014, 24, 4703–4707 CrossRef CAS PubMed.
- M. Cieplak, K. Szwabinska, M. Sosnowska, B. K. Chandra, P. Borowicz, K. Noworyta, F. D'Souza and W. Kutner, Biosens. Bioelectron., 2015, 74, 960–966 CrossRef CAS PubMed.
- S. G. Meirinho, L. G. Dias, A. M. Peres and L. R. Rodrigues, Anal. Chim. Acta, 2017, 987, 25–37 CrossRef CAS PubMed.
- P. Seng, M. Drancourt, F. Gouriet, B. L. Scola, P. E. Fournier, J. M. Rolain and D. Raoult, Clin. Infect. Dis., 2009, 49, 543–551 CrossRef CAS PubMed.
- C. Benagli, V. Rossi, M. Dolina, M. Tonolla and O. Petrinil, PLoS One, 2011, 6, e16424 CAS.
- A. R. Mcmullen, M. A. Wallace, D. H. Pincus, K. Wilkey and C. A. Burnham, J. Clin. Microbiol., 2016, 54, 2068–2073 CrossRef CAS PubMed.
- P. Khot and C. Farrance, Am. Pharm. Rev., 2016, 19 Search PubMed.
- A. Wieser, L. Schneider, J. Jung and S. Schubert, Appl. Microbiol. Biotechnol., 2012, 93, 965–974 CrossRef CAS PubMed.
- F. Imperi, F. Ciccosanti, A. B. Perdomo, F. Tiburzi, C. Mancone, T. Alonzi, P. Ascenzi, M. Piacentini, P. Visca and G. M. Fimia, Proteomics, 2009, 9, 1901–1915 CrossRef CAS PubMed.
- T. G. Xie, Y. D. Wang, Q. Y. Zheng and X. Zhao, Chin. J. Microecol., 2012, 24, 938–940 CAS.
- N. Cabrolier, M. Sauget, X. Bertrand and D. Hocquet, J. Clin. Microbiol., 2015, 53, 1395–1398 CrossRef CAS PubMed.
- L. Wang, Y. Fan, M. Wang and X. Lu, Zhonghua Yixue Zazhi, 2013, 93, 2079–2081 CAS.
- A. Johansson, J. Ekelof, C. G. Giske and M. Sundqvist, BMC Microbiol., 2014, 14, 1–8 CrossRef PubMed.
- J. Hrabak, Methods Mol. Biol., 2015, 1237, 91–96 CAS.
- M. Camoez, J. M. Sierra, M. A. Dominguez, M. Ferrer-Navarro, J. Vila and I. Roca, Clin. Microbiol. Infect., 2016, 22, 161.e1–161.e7 CrossRef CAS PubMed.
- G. Maasz, P. Takács, P. Boda, G. Varbiro and Z. Pirger, Sci. Total Environ., 2017, 601–602, 317–325 CrossRef CAS PubMed.
- J. T. Thaden, Y. Li, F. Ruffin, S. A. Maskarinec, J. M. Hill-Rorie, L. C. Wanda, S. D. Reed and V. G. Fowler Jr, Antimicrob. Agents Chemother., 2017, 61, e01709–e01716 CAS.
- D. Van der Waaij, J. Antimicrob. Chemother., 1982, 10, 263–270 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |