
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
First assessment on degradability of sodium p-perfluorous nonenoxybenzene sulfonate (OBS), a high volume alternative to perfluorooctane sulfonate in fire-fighting foams and oil production agents in China†
Yixiang Baoa,
Yingxi Qua,
Jun Huang *a,
Giovanni Cagnettaa,
Gang Yua and
Roland Weberb
*a,
Giovanni Cagnettaa,
Gang Yua and
Roland Weberb
aState Key Joint Laboratory of Environment Simulation and Pollution Control (SKJLESPC), Beijing Key Laboratory for Emerging Organic Contaminants Control (BKLEOC), School of Environment, POPs Research Center, Tsinghua University, Beijing, 100084, China. E-mail: huangjun@tsinghua.edu.cn; Fax: +86 10 6279 4006; Tel: +86 10 6279 2598
bPOPs Environmental Consulting, Lindenfirststr. 23, D-73527 SchwäbischGmünd, Germany
First published on 4th October 2017
Abstract
Sodium p-perfluorous nonenoxybenzene sulfonate (OBS) is widely used in China as fire-fighting foam co-formulant and oil production agent, because it is an economical alternative to perfluorooctane sulfonate (PFOS), classified as persistent organic pollutant by the Stockholm Convention. Such chemical very likely possesses toxicological and bioaccumulative characteristics that are similar to PFOS and many other perfluorinated compounds. However, in this first report we demonstrate that OBS, in spite of its non-readily biodegradability, can be decomposed by UV/H2O2 or even sole UV (254 nm) system (contrary to PFOS). More than 96% OBS is degraded in aqueous solution within 20 minutes by both methods thanks to its peculiar molecular structure. Yet, after 2 h reaction UV/H2O2 gives 99% sulfate recovery, while in UV system only 61% sulfate is detected; fluoride recovery was less than 16% for both treatments. Such results, corroborated by intermediate identification, highlight that UV/H2O2 system can effectively destroy the OBS sulfonated aromatic moiety. On contrary, it remains intact in several products of the UV treatment, raising a concern on their potential toxicity. Such results demonstrate OBS better degradability and treatability, compared to other perfluorinated chemicals, and suggest that it might be biodegraded on long term. Moreover, these findings may be of help for a more environmentally friendly eco-design of other fluorinated alternatives.
1. Introduction
Perfluorooctane sulfonate (PFOS) is an environmental pollutant and belongs to the chemical group of perfluoroalkyl substances. PFOS has been widely used for various commercial applications for more than 50 years.1 It has been found to be persistent,2 toxic and bioaccumulative3 in the environment, and has been detected worldwide in the environment, as well as in human blood and liver samples.4,5 Due to these findings, the phase-out of PFOS has been initiated since 2000. Similarly, an increasing number of other long-chain fluorinated chemicals are being phased out.6 In May 2009, PFOS, its salts and related precursor substances produced from perfluorooctane sulfonyl fluoride were listed under the Annex B of Stockholm Convention as persistent organic pollutants. Therefore, as a contracting party of Stockholm Convention, China is working on the control and phase out of these chemicals.The majority of alternatives currently available on the market are still organofluorine chemicals. As indicated in the Madrid Statement, also these alternative perfluoroalkyl substances are of concern due to their persistence (or that of their degradation products) combined with lack of detailed toxicity assessment.7 In fact, almost all these chemicals have similar toxicity and tendency to bioaccumulate in living tissues.8 Also, some short chain alternatives (<6 carbon atoms), claimed to be safer than long chain homologues, have even been found to be bioaccumulative and toxic, as well as persistent.9–12
Sodium p-perfluorous nonenoxybenzene sulfonate (OBS, C9F17OC6H4SO3Na, CAS no. 70829-87-7, Fig. 1) belongs to the group of PFASs with an aromatic moiety. As a cost-effective surfactant, it is widely used in China as co-formulant of fluoroprotein fire-fighting foams, oil production agents, and photographic materials.13 There are several manufacturers of OBS in China, with an estimated total production capacity of about 3500 t per year. Also in recent years, some efforts were put to develop synthesis method of OBS and its derivatives.14,15 With regard to its pollution potential, OBS has been recently detected in water samples from urban wastewater treatment plant and local rivers in oil-field area of North China.16 This fact raises a serious concern about its possible further spread in the environment and, consequently, on risk for humans, because OBS is very likely as toxic and persistent as the large majority of perfluoroalkyl chemicals.
 | ||
| Fig. 1 Chemical structure of OBS. | ||
A key issue to predict OBS environmental behavior is its degradability, caused by both biotic and abiotic processes. But, there is a lack of information on this aspect. Still, OBS molecule presents some weak points (i.e. a double bond, an etheric bridge, and a phenylsulfonate moiety) that may cause lower stability. A study on this aspect would be helpful not only to collect clues on possible fate of OBS into the environment, but also to estimate its treatability in water to prevent point source emissions. As a matter of fact, such studies were already conducted on other fluorocompounds. Schröder and Meesters17 have investigated the stability of several anionic and non-ionic fluorinated surfactants under conditions of advanced oxidation processes (AOPs), including ozone (O3), O3/UV, O3/H2O2, and H2O2/Fe2+. The results provided insights for an improved treatability of these pollutants. Quinete et al.18 tested both biodegradability and AOP stability of several emerging perfluorinated surfactant substitutes (i.e. fluoroaliphatic esters, the fluorosurfactant Zonyl, perfluorobutane sulfonate, and 10-(trifluoromethoxy)decane-1-sulfonate). Biodegradability tests for these all fluorosurfactants using the manometric respirometry method (Oxi-Top) yielded the same result of non-readily biodegradability. In comparison, the AOP stability of NOVEC FC-4430 and NOVEC FC-4432 (two fluorochemicals employed as coating additives) was significantly lower than that of perfluorobutane sulfonate. Also the authors found that the more stable alternatives under AOPs showed stronger recalcitrance to biological treatment. Therefore, AOP stability can be a promising indicator of biological recalcitrance or treatability of fluorosurfactants.
In our previous studies, we successfully investigated the AOP stability of two fluorosurfactants used as PFOS-alternatives, i.e. F–53B (Cl(CF2)6O(CF2)2SO3−˙K+, CAS no. 73606-19-6) and 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 FTS-K (F(CF2)6CH2CH2SO3−˙K+, CAS no. 59587-38-1). F–53B was found to be as stable as PFOS potassium under all AOPs conditions, while 6:2 FTS-K was proven to be more degradable.19,20 Following the same methodology, the stability of OBS under UV/H2O2, which has high OH radical productivity, is assessed, and main intermediates and products are also identified. In comparison, OBS degradation by UV irradiation in aqueous solution is also investigated, as well as degradation products and photochemical degradation mechanisms. These data are important for the evaluation of OBS environmental fate and to estimate the pollution potential of this large volume PFOS-alternative mainly produced and widely used in China.
:2 FTS-K (F(CF2)6CH2CH2SO3−˙K+, CAS no. 59587-38-1). F–53B was found to be as stable as PFOS potassium under all AOPs conditions, while 6:2 FTS-K was proven to be more degradable.19,20 Following the same methodology, the stability of OBS under UV/H2O2, which has high OH radical productivity, is assessed, and main intermediates and products are also identified. In comparison, OBS degradation by UV irradiation in aqueous solution is also investigated, as well as degradation products and photochemical degradation mechanisms. These data are important for the evaluation of OBS environmental fate and to estimate the pollution potential of this large volume PFOS-alternative mainly produced and widely used in China.
2. Materials and methods
2.1 Chemicals
OBS was purchased from 3F Corporation (Shanghai, China). H2O2 (30%, Sinopharm Chemical Reagent) was used in AOP treatment tests. Tert-butanol (C4H10O, TBA, Sinopharm Chemical Reagent Co.), sorbic acid (C6H8O2, SA, AccuStandard), and 1,4-benzoquinone (C6H4O2, BQ, 99%, J&K Scientific) were used as scavengers. Trifluoroacetic acid (TFA, LC-MS grade, CNW Technologies), phenol sulfonic acid (C6H5O4SNa, PSA, >98.0%, TCI (Shanghai) Development Co.), methanol (CH3OH, HPLC grade, Avantor Performance Materials), sodium dihydrogen phosphate (NaH2PO4·2H2O, Modern Oriental (Beijing) Technology Development Co.), and ammonium acetate (NH4Ac, Sinopharm Chemical Reagent Co.) were used for identification/quantification of the degradation products. Aqueous solutions were prepared using ultrapure water (18.2 MΩ cm, Millipore, USA).2.2 Experimental methods
The readily-biodegradability of OBS was assessed by the Closed Bottle Test (CBT) method according to the guidelines for testing of chemicals (method 301 D) of the Organization for Economic Cooperation and Development (OECD) (the detailed procedure is described in ESI, Section S1†). The secondary effluent from a municipal wastewater treatment plant was adopted as microbial inoculum in all tests. Duplicate measurements for OBS, reference substance and blank were performed.The OBS solution used in all abiotic degradation experiments (namely, under AOP conditions) were prepared with concentration of 0.16 mM in ultrapure water. The UV irradiation experiment was performed in quartz glass tube containing 300 mL OBS solution in a photochemical reactor (RPR200, Southern New England Ultraviolet (Rayonet) Co., USA). The quartz tube was placed in the middle of the reactor (25 cm diameter) and surrounded by 4 of UV lamps with wavelength of 253.7 nm (with approximate intensity of 1.6 × 1016 photons s−1 cm−3) at room temperature (around 20 °C). The photochemical reactor was operated 5 minutes before the OBS solution was poured into the quartz tube. The UV/H2O2 experiments were carried out by adding different volumes of 30% H2O2 solution in 300 mL OBS solution under the aforementioned UV irradiation conditions. Reaction medium was magnetically stirred at 200 rpm. Samples (3 mL) were withdrawn from the reaction solution at specified times.
2.3 Instrumental analysis
Fluorides and sulfates were quantified by ion chromatography (Dionex ICS 2000, USA). OBS was determined by high-performance liquid chromatography (HPLC, LC-20AT, Shimadzu, Japan) equipped with UV/Vis detector at 275 nm (SPD-20A, Shimadzu, Japan). The mobile phase was methanol:0.02 M dihydrogen phosphate buffer solution (8:2, v/v) with a constant elution rate of 1 mL min−1. Intermediates and by-products were identified by liquid chromatography (LC, DIONEX UltiMate 3000, USA) equipped with XBridge C18 column (3.0 × 150 mm, 3.5 μm, Waters), followed by a tandem of two time of flight mass spectrometers (TOFMS, micrOTOF-Q III BRUKER, Germany) The LC column temperature was 30 °C, flow rate of the mobile phase (10 mM NH4Ac aqueous solution (phase A) and methanol (phase B)) was kept at 0.3 mL min−1. The linear gradient was set to 1:9 A:B from 0 to 15 min, 9:1 A:B from 15 to 25 min and 1:9 A:B from 25 to 32 min. The TOFMS was operated in electrospray ionization (ESI) negative ion mode, with spray voltage of 3.5 kV and mass scan range of 100–1000. The mass accuracy was corrected by infusion of sodium formate aqueous solution. LC (DIONEX UltiMate 3000, USA) with tandem of 2-stage mass spectrometer (MS/MS, API 3200, AB SCIEX, USA) was used to quantify the TFA and PSA generated during the degradation of OBS. The same conditions of LC were used for the (semi)quantification of the products. Negative electrospray ionization mode with multiple reaction monitoring (MRM) was used for MS/MS. Ionization voltage was 4.5 kV and temperature of 450 °C. The pressures for curtain gas, collision gas, ion source gas 1 and ion source gas 2 were set to 10, 5, 60, and 40 psi, respectively. Optimized parameters for quantification of PSA and TFA are summarized in Table S1.† A UV-vis spectrophotometer (DR 5000, Hatch, USA) was used to detect the absorption band of the various solutions. Electron spin resonance spectrometer (ESR, JES-FA200, JEOL, Japan) equipped with an USHIO Optical MoluleX as light source was used to detect radicals generated during the reaction.
3. Results and discussion
3.1 Biotic degradation
The biochemical oxygen demand (BOD) evaluated by 28 day tests (referred to that of the blank) of OBS and a reference substrate (sodium benzoate) is shown in Table S2.† The biodegradation percentage of OBS (calculated as ratio of BOD to theoretical oxygen demand (ThOD, 0.599 mg mg−1 for OBS), and shown in Table S3†) demonstrated that OBS caused irrelevant oxygen depletion during the test. On contrary, the reference substance was significantly biodegraded in the same period. These findings strongly suggest that the fluorinated compound was not involved in any short term biological process. In other words, it is non-readily biodegraded. It clearly implies a significant recalcitrance of OBS to biodegradation and its potential persistence in the natural environment. Hence, the present study mainly focuses on abiotic degradation, which can provide clues also on long term biological degradation.3.2 Abiotic degradation
Unlike PFOS or other perfluoroalkyl substances, which are highly resistant to AOPs, OBS can be readily decomposed in both UV and UV/H2O2 systems (Fig. 2a). More than 96% OBS parent molecules were degraded under the tested conditions within 20 min. Early degradation stage demonstrates in a straightforward manner that 10 mM H2O2 dosage (62.5 H2O2/OBS molar ratio) induces the fastest OBS depletion rate (Fig. 2b). Higher dosages, however, determine a decrease in degradation rate, caused by self-consumption of generated OH radicals (eqn (1)). OBS was still detectable after 120 min under UV irradiation. The degradation processes fitted well with pseudo-first order kinetics (Fig. S1†). The test series confirmed that the optimal dose for H2O2 is 10 mM. In this case, the related fitting calculation showed a degradation rate constant of k = 0.2597 ± 0.0074 min−1, which is 1.6 times higher than that in the UV system without H2O2 (k = 0.1695 ± 0.0127 min−1).| H2O2 + 2OH˙ = 2H2O + O2 | (1) |
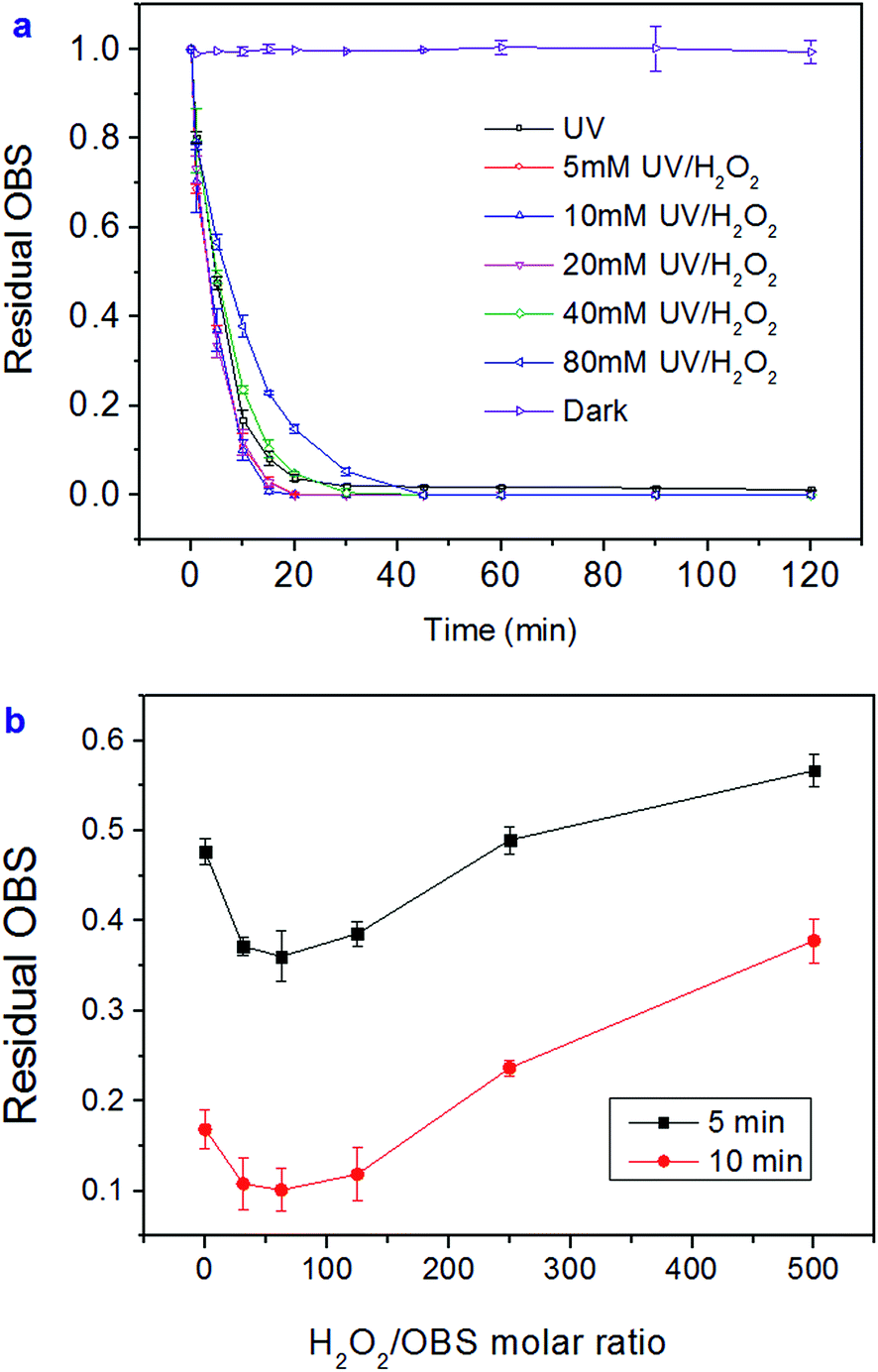 | ||
| Fig. 2 Residual OBS in UV and UV/H2O2 system correlated with irradiation time (a) and H2O2/OBS molar ratio for 5 and 10 min irradiation (b). | ||
Fluorine ion recovery during the degradation of OBS is important to determine the mineralization degree of its perfluorinated moiety. Under sole UV light, 14.8% of total fluorine content of the OBS molecule was found in solution as fluorides, while in UV/H2O2 system fluoride recovery was 16.1%. This shows that the perfluorinated chain is only partly degraded. On the other hand, release of sulfates in the reaction medium indicates that the aromatic moiety undergoes degradation. In fact, more than 99% and 61% sulfate is found in solution for UV/H2O2 and UV tests, respectively. Such remarkable difference indicates that the benzene sulfonate moiety is effectively destroyed by UV/H2O2, but only partially mineralized under UV irradiation (Fig. 3).
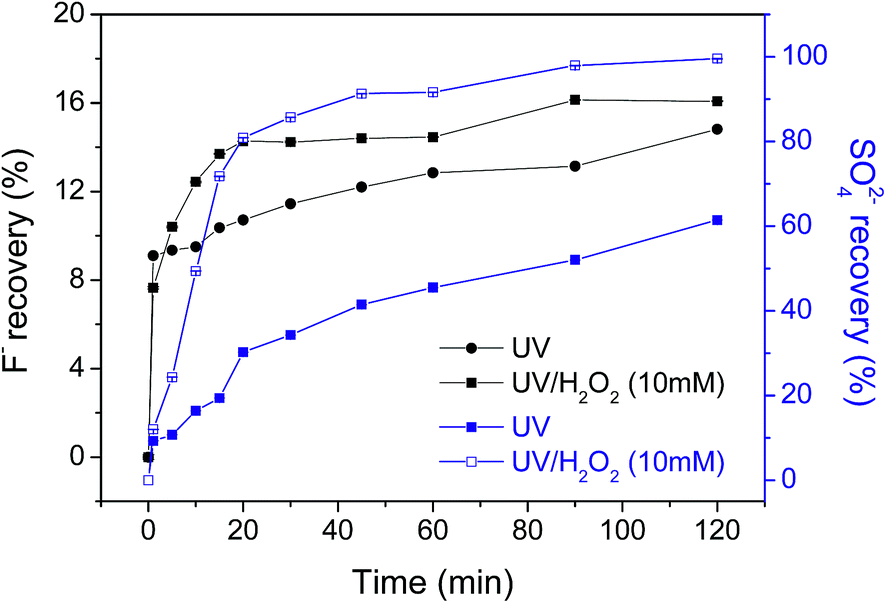 | ||
| Fig. 3 Recovery ratio of fluorine ion and sulfate ion for OBS in UV and UV/H2O2 system (error is ≤0.05 for all data). | ||
3.3 Degradation products and proposed pathway of OBS degradation in UV/H2O2 system
OBS degradation intermediates were identified by liquid chromatography with MS detector (LC-TOFMS). A base peak chromatogram (BPC-All MS) model was adopted to determine the products with prominent peak intensity. The blank sample (ultrapure water) was used to exclude unknown substances from the base peak chromatogram. In total, 7 stable degradation products were detected after 2 h reaction. The compound spectra and the assigned corresponding structures are showed in Fig. S2† and hereafter identified by their chemical formula: C10H2F15O5− (m/z = 486.9), C7HF14O− (m/z = 366.9), C7HF12O3− (m/z = 360.9), C4F7O2− (m/z = 212.9), C4HF6O3− (m/z = 210.9), C3HF6O2− (m/z = 182.9), and C2F3O2− (i.e. TFA, m/z = 112.9). In addition, 6 intermediates were detected (Fig. S2†): C15H6F17O7S− (m/z = 652.9), C15H4F17O5S− (m/z = 618.9), C15H6F15O6S− (m/z = 598.9), C15H6F15O7S− (m/z = 614.9), C14H5F14O7S− (m/z = 582.5), and C9F17O− (m/z = 446.9).Based on the identified products above, a possible degradation pathway of OBS under UV/H2O2 (dominated by hydroxyl radical) is proposed (Fig. 4). It is well-known that OH radical reacts with organic compounds in aqueous solution by 3 mechanisms: OH addition on aromatic or unsaturated groups, which result in radical adducts; hydrogen atom transfer; and single electron transfer.21,22 For OBS, the double bond and benzene ring are the most likely attacked sites by OH radicals. Hence, in UV/H2O2 system the OH radical can react with OBS through four possible routes: (I) OH addition on double bond to from an epoxy structure, (II) OH addition on benzene ring to form OH adducts, (III) electrophilic attack of C–F bond at position 3 and 4 (C atoms are arbitrarily labelled, as shown in Fig. 4), (IV) electrophilic attack of O at position 6. For the addition reaction through Route I and Route II, the OH radical is added to the double bond, thus forming of a dihydroxy-alcohol. The addition reaction by OH addition on the benzene ring results in formation of two adducts, namely C15H6F17O7S− and C15H4F17O5S−. By elimination of a H2O molecule on C3 and C4 of C15H6F17O7S−, an epoxy group is formed (C15H4F17O5S−). If the CF3 group at position 5 is substituted by OH, then C14H5F14O7S− is formed. In the second step, the aromatic group of C14H5F14O7S− is rather quickly decomposed to form the stable C7HF14O− product, whose concentration increases continuously along 2 h reaction. The alcoholic group on C2 is further oxidized into ketone with simultaneous replacement of F at C3 and C4 by OH radicals to form C7HF12O3−.
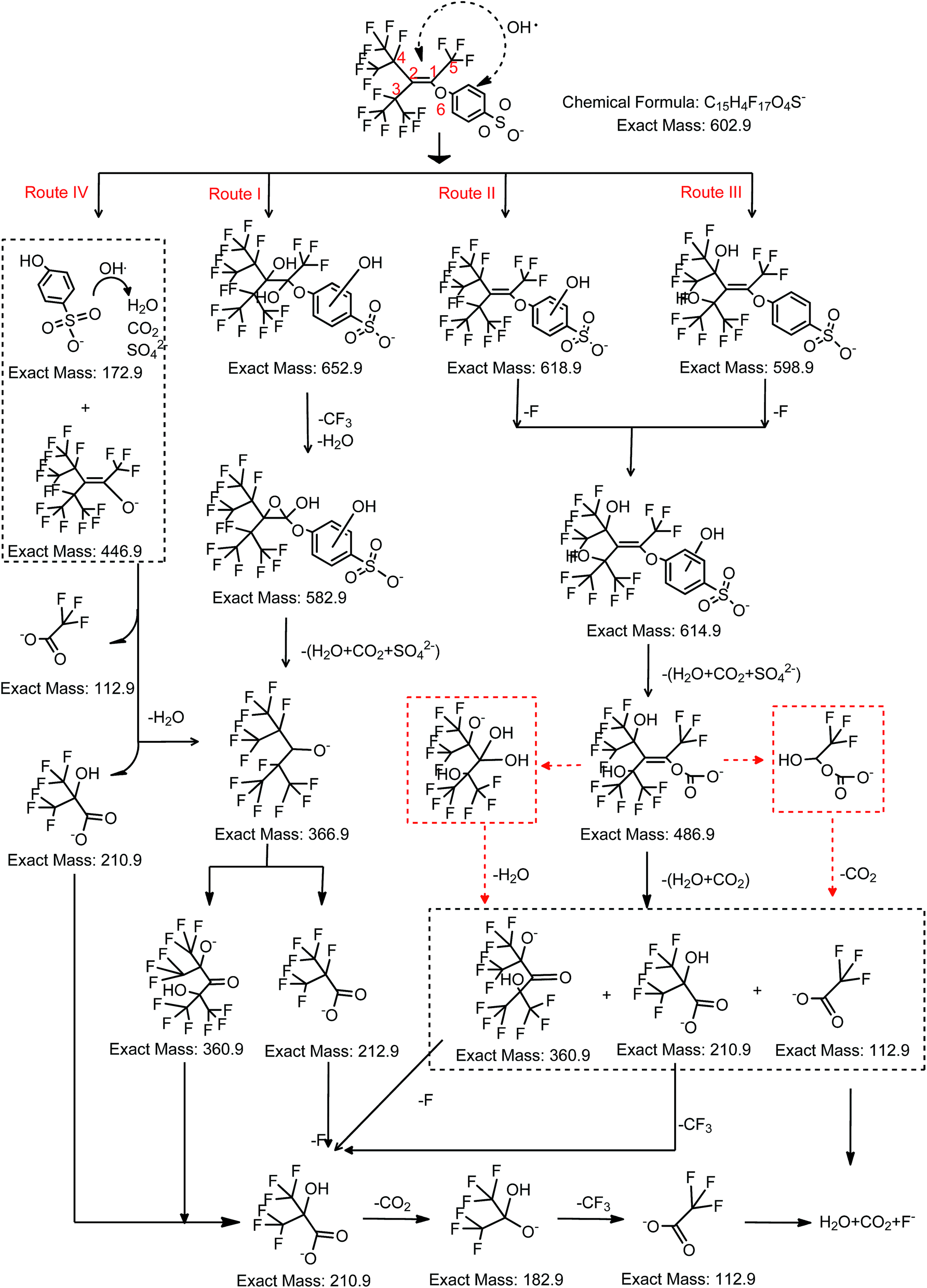 | ||
| Fig. 4 Proposed degradation pathway of OBS under UV/H2O2. | ||
Concerning the degradation of C7HF14O−, the C–C bond is broken up at position 2 to form C4F7O2−. Consequently, the two C–C adjacent bonds (i.e. C2–C3 and C2–C4) are weakened due to the branched CF3 group.23,24 Jin and Zhang also found that branched PFOS shows higher degradability respect to the linear one.25 From this perspective, the branched structures in OBS increase its degradability respect to linear perfluoroalkyl compounds.
The C7HF12O3− is split into C4HF6O3− and C3HF6O2− by attack to the ketonic group; the carboxyl group on C4HF6O3− can also be cleaved to generate C3HF6O2− with further oxidation. On the other hand, the F atom on C4 of C4F7O2− (generated from C7HF14O−) can also be replaced by OH. The further mineralization occurs by cleavage of CF3 group on C3 to give C2F3O2− (i.e. TFA). Although TFA is considered to be relatively stable in UV/H2O2 system,20 we found its mineralization after prolonged time.
OH radical can difficultly break C–F bonds owing to the high energy demand. However, for the branched fluorinated structure of OBS, there are two strong electron withdrawing CF3 groups on both C3 and C4 atoms, which reduce the electron density and consequently weaken the bond energy of C–F on C3 and C4. This facilitates the substitution of F atom by OH on both carbons. Therefore, we hypothesize that the electrophilic substitution of F at C3 and C4 happens at the second step of Route II to form C15H6F15O7S− and also at the beginning of Route III to form C15H6F15O6S−. The further degradation proceeds by attack to the benzene ring, thus the C15H6F15O7S− produced from the second step of Route II and Route III is decomposed to C10H2F15O5−. The resulting C10H2F15O5− is split into C7HF12O3− (or C3HF6O2−) and TFA by cleavage of the C1 = C2 bond. Then, the following degradation pathway of C7HF12O3− (or C4HF6O3−) and TFA is the same of Route I.
In Route IV, the electrophilic attack takes place at O6 atom (Fig. 4), and OBS is split into C9F17O− (m/z = 446.9) and PSA. No increase of PSA was observed during the reaction due to its fast degradation kinetic in UV/H2O2 system (Fig. 5). This was totally different from that in UV system (see Section 3.4). For the further degradation of C9F17O−, it follows the same procedure like C10H2F15O5− by breakage of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, and the resulting C7HF12O3− (or C4HF6O3−) and TFA can be decomposed stepwise in the same way above-mentioned.
C bond, and the resulting C7HF12O3− (or C4HF6O3−) and TFA can be decomposed stepwise in the same way above-mentioned.
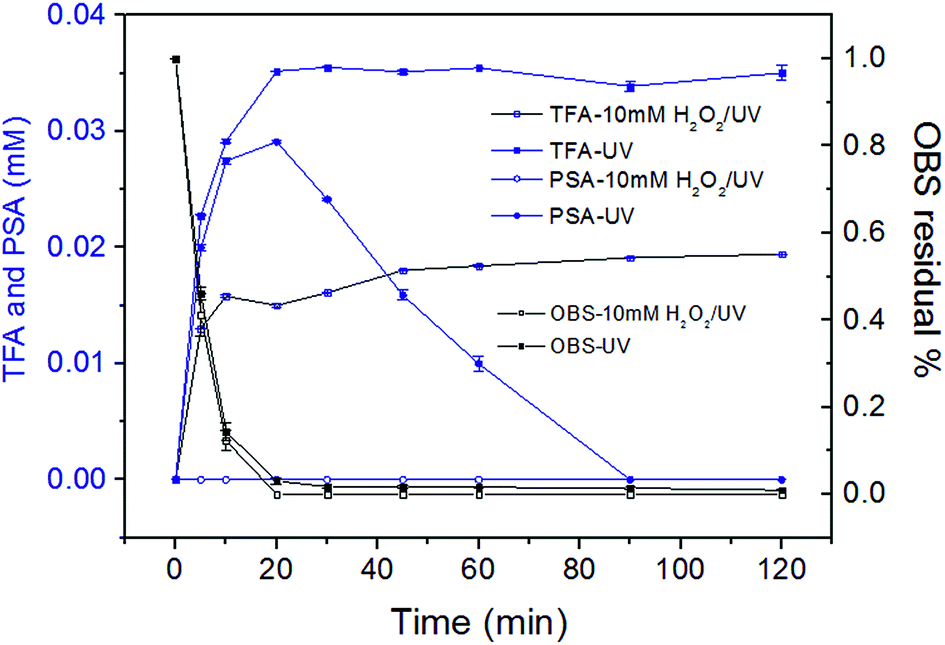 | ||
| Fig. 5 Variation of PSA and TFA in UV and UV/H2O2 system during 2 h reaction. | ||
3.4 Degradation products and proposed pathway of OBS in UV system
As constituent of sunlight, ultraviolet light plays an important role for transformation of organic pollutants in the environment. The sunlight that reaches the earth has its maximum wavelength at 365 nm.26 Therefore, the photochemical behavior of OBS was firstly tested under 365 nm UV. However, no significant elimination was found under such light (Fig. S3†). This indicates that the decomposition of OBS in the natural environment by sunlight is conceivably quite slow. Conversely, as showed in Fig. 2a, we found that OBS can be easily decomposed under 254 nm UV, through the mechanism described hereafter.OBS molecule possesses an aromatic ring that acts as a unit to initiate photosensitized reactions with reactive oxidative species (ROS), which may be present in the reaction medium.27,28 In order to identify such ROS under UV (254 nm) light, electron spin resonance analysis was performed. As showed in Fig. 6, singlet state oxygen (1O2) was generated in OBS aqueous solution by irradiation, but no appreciable OH radical and superoxide anion signal (O2˙−) were found. The generation of singlet oxygen decreases gradually due to discontinuous introduction of oxygen into the system. To further corroborate identification of the dominant ROS, inhibition experiment was also conducted. Only quenching of 1O2 and OBS excited triplet state (OBS*) by sorbic acid addition could substantially lower the removal efficiency (Fig. S4†). These results highlight that, in the UV system, two mechanisms are responsible for the OBS degradation: (1) direct photolysis, where OBS is firstly excited to the triplet state (T1) by UV light and, then, split at the weaker bond (while the excited electrons return to the ground state); (2) oxidation of OBS by 1O2. The simplified reaction scheme of the overall degradation process is described by eqn (2)–(5).29–31
 | (2) |
| OBS*(T1) → products. | (3) |
| OBS*(T1) + 3O2 → OBS + 1O2. | (4) |
| OBS + 1O2 → products. | (5) |
 | ||
| Fig. 6 ESR spectra of TEMP–1O2 and DMPO–OH˙ in OBS aqueous solution and DMPO–O2˙− in OBS methanol solution. | ||
For the sake of clarity, the two mechanisms (viz. direct photolysis and oxidation by 1O2) are later proposed separately.
Some of the products and intermediates of OBS degradation in UV system are the same as those detected in the UV/H2O2 system. Intermediates are C15H4F17O5S− (m/z = 618.9) and C15H6F15O6S− (m/z = 598.9), which were eliminated stepwise within the 2 h treatment. C7HF14O− (m/z = 366.9), C7HF12O3− (m/z = 360.9), C4F7O2− (m/z = 212.9), C4HF6O3− (m/z = 210.9), C3HF6O2− (m/z = 182.9) and C2F3O2− (m/z = 112.9) were formed as stable degradation products. In addition, several new products were found, that is, C15H4F17O5S− (m/z = 618.9) and its isomer (Fig. 7 and 8), C14H5F14O7S− (m/z = 582.9), C14H6F13O7S− (m/z = 564.9), C7H5O11S− (m/z = 296.9), C7H5O10S− (m/z = 280.9), C6H5O8S− (m/z = 236.9), and C6H5O4S− (m/z = 172.9). Their extracted compound spectra and the corresponding structures were shown in Fig. S5.†
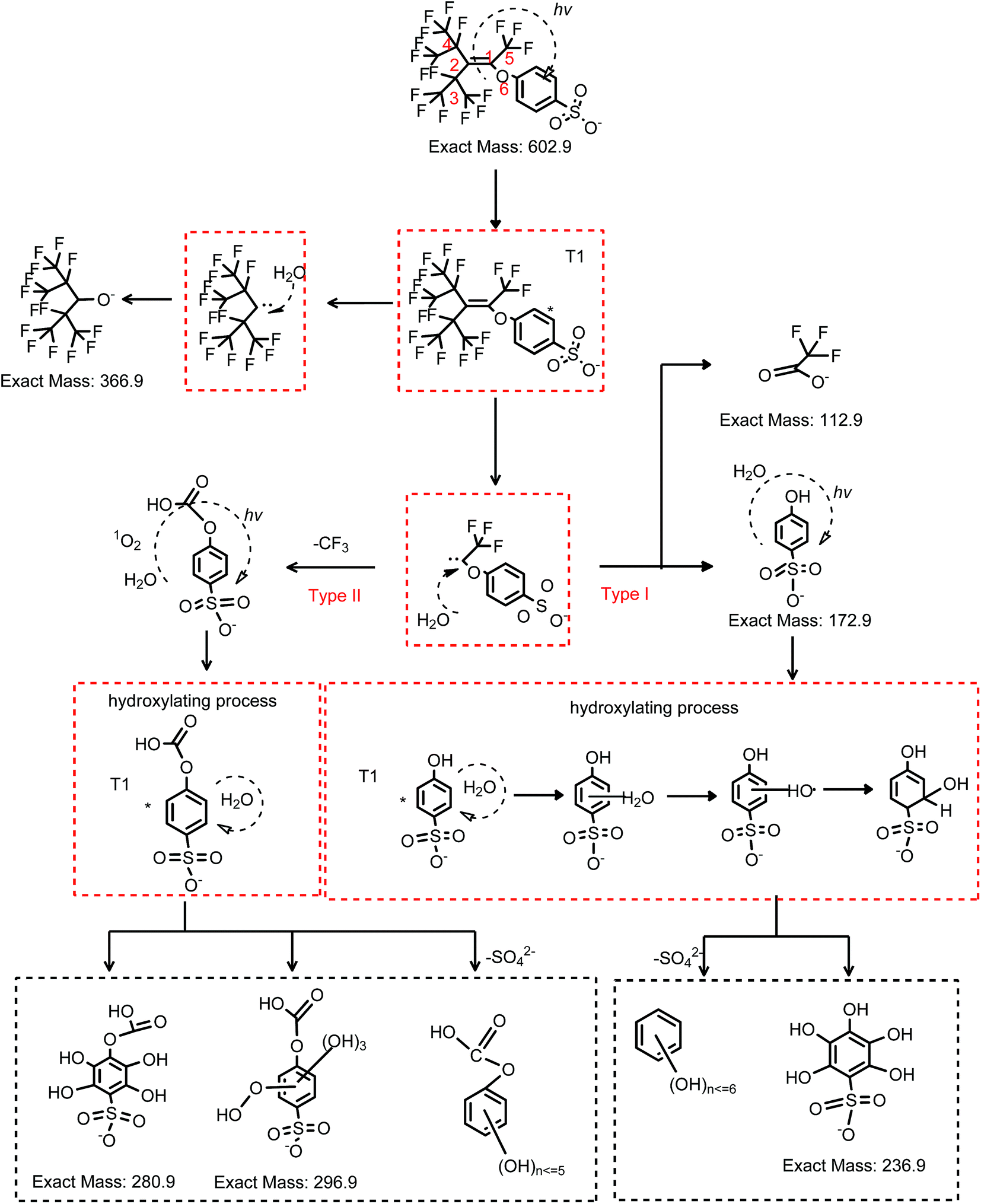 | ||
| Fig. 7 Proposed degradation pathway of OBS under UV dominated by direct photolysis. | ||
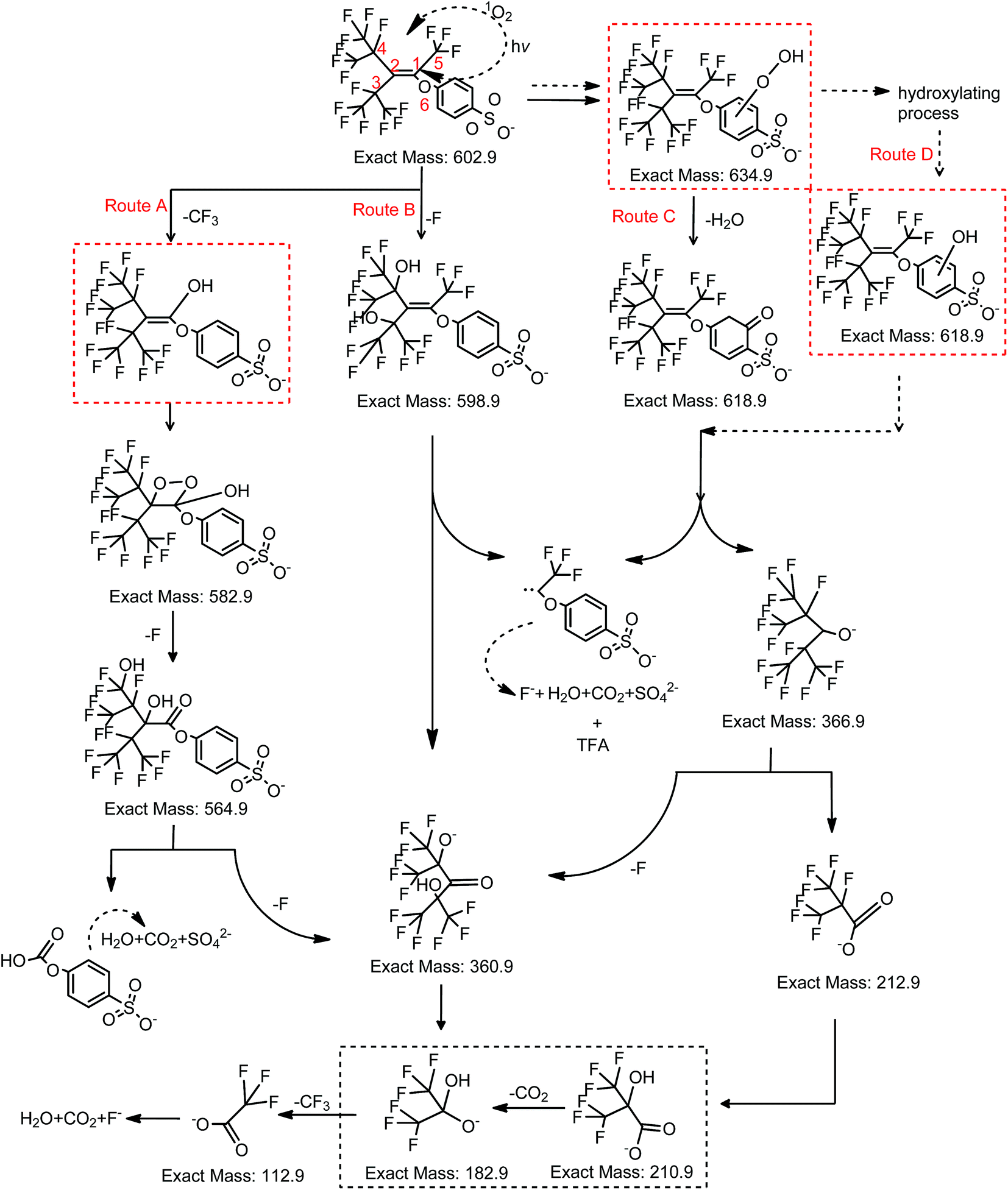 | ||
| Fig. 8 Proposed degradation pathway of OBS under UV dominated by 1O2. | ||
3.5 Comparison of degradation mechanism under UV and UV/H2O2
The aromatic group, the double bond and CF3 groups in the branched perfluorinated moiety make OBS easier to be degraded compared to PFOS. Still, the defluorination process is not complete, and stable degradation products are formed in both systems. Therefore, longer reaction time and combination of other techniques (e.g. catalytic oxidation) would obtain a satisfactory degradation and possibly entire mineralization.In UV system, the generation of TFA and PSA reached the maxima simultaneously until 20 min when >96% OBS was decomposed (Fig. 5). TFA was levelled off at prolonged time, while PSA was further degraded (as described in Section 3.6). OBS decomposition rate by UV was sensibly lower than that in UV/H2O2. This is reasonably due, in the first instance, to the 1O2 low concentration (compared to OH radicals generated from 10 mM H2O2), determined by low O2 amount in the system (no forced aeration was adopted in the experiments). Secondly, the complex degradation products in UV system could quench OBS*, thus slowing down the elimination of OBS. Finally, the acidic environment (pH = 3.3 after reaction) probably inhibited OBS photolysis.36
In the UV/H2O2 system, no detectable amount of PSA was generated during reaction (Fig. 5). So the PSA was decomposed effectively to entirely release sulfate ions. The degradation of PSA in both systems were tested (Fig. S7†). The results showed that almost 100% of sulfate ion was recovered in UV/H2O2 system and much less in UV system. With regard to the second degradation product that was investigated in detail, i.e. TFA, its generation did not reach the theoretical maximum when OBS was totally decomposed, but it kept gradually increasing until 60 min, and then its concentration remained stable. The reason is that after 60 min reaction, production and degradation rates of TFA were in equilibrium. The concentration of TFA after 2 h in UV/H2O2 system was considerably lower than that in UV system. This is probably due to partial mineralization of CF3 group of aromatic moiety linked to C1 that could not generate TFA.
The defluorination efficiency in UV/H2O2 system was not much higher compared to the UV treatment indicating that a similar amount of fluorinated degradation products still existed. However, PSA and any aromatic products, as well as TFA, showed considerable lower concentrations at the end of the treatment compared to the sole UV degradation. In terms of safety of a possible AOP treatment process, the aromatic compounds might be more toxic, so their higher concentration in water under UV light should be taken into account.37 TFA also possesses some toxicity (for instance, its oral LC50 for rat is 200 mg kg−1 bw).38 Hence, the UV/H2O2 is a safer treatment for degrading OBS.
4. Conclusions
OBS, a widely used polyfluorinated PFOS-alternative in China, was for the first time assessed for biotic and abiotic degradability. Closed bottle tests showed a strong recalcitrance of OBS to any biological transformation in the short period (28 d). Hence, such surfactant, like all the other PFASs, is a non-readily biodegradable chemical. Nonetheless, our investigation on abiotic degradability under AOP conditions highlighted that, differently from the majority of fluorinated compounds, OBS is degradable in substantial manner.OBS could be decomposed under UV (254 nm) and even better in UV/H2O2. However, under both conditions complex by-products were formed and less than 20% of fluorine was mineralized. The UV/H2O2 showed a more promising degradation performance, which might be combined with other technologies to detoxify OBS effectively. In addition, OBS was not degraded under 365 nm UV irradiation (main UV component of sunlight reaching the earth). Therefore, the photolysis of OBS in natural environment might be rather slow. On contrary, the efficient degradation in UV/H2O2 suggests that OBS may be affected by long term biological transformations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the National Natural Science Fund of China (No.21477060) and Tsinghua University Initiative Scientific Research Program (20131089251).References
- V. Ciccotelli, M. C. Abete and S. Squadrone, Food Control, 2016, 59, 46–52 CrossRef CAS.
- T. Frömel and T. P. Knepper, Rev. Environ. Contam. Toxicol., 2010, 208, 161–177 Search PubMed.
- T. M. Boudreau, C. J. Wilson, W. J. Cheong, P. K. Sibley, S. A. Mabury, D. C. G. Muir and K. R. Solomon, Environ. Toxicol. Chem., 2003, 22, 2739–2745 CrossRef CAS PubMed.
- B. O. Clarke and S. R. Smith, Environ. Int., 2011, 37, 226–247 CrossRef CAS PubMed.
- J. P. Giesy and K. Kannan, Environ. Sci. Technol., 2001, 35, 1339–1342 CrossRef CAS PubMed.
- S. K. Ritter, Chem. Eng. News, 2010, 88, 12–17 Search PubMed.
- A. Blum, S. A. Balan, M. Scheringer, X. Trier, G. Goldenman, I. T. Cousins, M. Diamond, T. Fletcher, C. Higgins, A. E. Lindeman, G. Peaslee, P. Voogt, Z. Wang and R. Weber, Environ. Health Perspect., 2015, 123, A107–A111 CrossRef CAS PubMed.
- Z. Wang, J. C. DeWitt, C. P. Higgins and I. T. Cousins, Environ. Sci. Technol., 2017, 51, 2508–2518 CrossRef CAS PubMed.
- L. Zhao, P. K. Mccausland and P. W. Folsom, Chemosphere, 2013, 92, 464–470 CrossRef CAS PubMed.
- J. C. W. Lam, J. Lyu and K. Y. Kwok, Environ. Sci. Technol., 2016, 50, 6728–6734 CrossRef CAS PubMed.
- Q. Zhang, W. Liu and Q. Niu, Toxicol. Res., 2014, 5, 539–546 RSC.
- H. Viberg, I. Lee and P. Eriksson, Toxicology, 2013, 304, 185–191 CrossRef CAS PubMed.
- http://www.sh3f.com/product_view.asp?id=34%26mc=e851a03267f01c172e9c1e204ea16525f766fa44e1ed0ff6.
- L. Chen, H. Shi, H. Wu and J. Xiang, Colloids Surf., A, 2011, 384, 331–336 CrossRef CAS.
- H. K. Wu, J. Q. Zhong, H. M. Shen and H. X. Shi, J. Fluorine Chem., 2013, 156, 5–8 CrossRef CAS.
- C. Li, Master Dissertation, Qingdao Technological University, 2016.
- H. F. Schröder and R. J. W. Meesters, J. Chromatogr. A, 2005, 1082, 110–119 CrossRef.
- N. Quinete, F. Orata, A. Maes, M. Gehron, K. H. Bauer, I. Moreira and R. D. Wilken, Arch. Environ. Contam. Toxicol., 2010, 59, 20–30 CrossRef CAS PubMed.
- S. Wang, J. Huang, Y. Yang, Y. Hui, Y. Ge, T. Larssen, G. Yu, S. Deng, B. Wang and C. Harman, Environ. Sci. Technol., 2013, 47, 10163–10170 CrossRef CAS PubMed.
- X. Yang, J. Huang, K. Zhang, G. Yu, S. Deng and B. Wang, Environ. Sci. Pollut. Res., 2014, 21, 4634–4642 CrossRef CAS PubMed.
- G. H. Naik, K. I. Priyadarsini, A. K. Maity and H. Mohan, J. Phys. Chem. A, 2005, 109, 2062–2068 CrossRef CAS PubMed.
- A. Galano and J. R. Alvarez-Idaboy, Org. Lett., 2009, 11, 5114–5117 CrossRef CAS PubMed.
- V. Tortelli, C. Tonelli and C. Corvaja, J. Fluorine Chem., 1993, 60, 165–174 CrossRef CAS.
- C. Tonelli and V. Tortelli, J. Fluorine Chem., 1994, 67, 125–128 CrossRef CAS.
- L. Jin and P. Zhang, Chem. Eng. J., 2015, 280, 241–247 CrossRef CAS.
- X. Zhao, T. Toyooka and Y. Ibuki, Sci. Total Environ., 2013, 458–460, 54–62 CrossRef CAS PubMed.
- M. S. Baptista, J. Cadet, D. P. Mascio, A. A. Ghogare, A. Greer, M. R. Hamblin, C. Lorente, S. C. Nunez, M. S. Ribeiro, A. H. Thomas, M. Vignoni and T. M. Yoshimura, Photochem. Photobiol., 2017, 93, 912–919 CrossRef CAS PubMed.
- M. C. Derosa and R. J. Crutchley, Coordin. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- C. S. Foote, Acc. Chem. Res., 1968, 1, 104–110 CrossRef CAS.
- K. Li and J. L. Ferry, Environ. Sci. Technol., 2003, 37, 4894–4900 CrossRef.
- Y. Chen, C. Hu, J. Qu and M. Yang, J. Photochem. Photobiol., A, 2008, 197, 81–87 CrossRef CAS.
- A. Pochon, P. P. Vaughan, D. Q. Gan, P. Vath, N. V. Blough and D. E. Falvey, J. Phys. Chem. A, 2002, 106, 2889–2894 CrossRef CAS.
- M. Kimura and Y. Ogata, Bull. Chem. Soc. Jpn., 2006, 56, 471–473 CrossRef.
- S. Mazur and C. S. Foote, J. Am. Chem. Soc., 2002, 92, 3225–3226 CrossRef.
- X. Tian, P. Gao, Y. Nie, C. Yang, Z. Zhou, Y. Li and Y. Wang, Chem. Commun., 2017, 53, 6589–6592 RSC.
- J. S. Miller, Rose bengal-sensitized photooxidation of 2-chlorophenol in water using solar simulated light, Water Res., 2005, 39, 412–422 CrossRef CAS PubMed.
- S. Weng, J. Y. Yang, Y. H. Li and E. R. Blatchley 3rd, Sci. Total Environ., 2017, 599–600, 94–97 CrossRef CAS PubMed.
- US National Library of Medicine, trifluoroacetic acid, 76-05-1, animal toxicity studies, https://toxnet.nlm.nih.gov/cgi-bin/sis/search2/f?./temp/%7EeE5qWO:1.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09728j |
| This journal is © The Royal Society of Chemistry 2017 |