
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development and application of immobilized surfactant in mass spectrometry-based proteomics†
Qiong Wu ab,
Fenglong Jiaob,
Fangyuan Gaob,
Chaoshuang Xiab,
Yayao Lvb,
Qian Yub,
Yangjun Zhang*b and
Xiaohong Qian*ab
ab,
Fenglong Jiaob,
Fangyuan Gaob,
Chaoshuang Xiab,
Yayao Lvb,
Qian Yub,
Yangjun Zhang*b and
Xiaohong Qian*ab
aSchool of Pharmacy, Guangdong Pharmaceutical University, Guangzhou 510006, Guangdong, China. E-mail: 13911734119@163.com
bState Key Laboratory of Proteomics, National Center for Protein Science Beijing, Beijing Institute of Radiation Medicine, Beijing 102206, China. E-mail: 13683167093@163.com
First published on 14th September 2017
Abstract
In “bottom-up” proteomics, proteins are first digested into peptides and then characterized by chromatographic separation and mass spectrometry analysis, in which proteolysis has an appreciable influence on the repeatability and reliability of analytical results. To improve recovery and enzymatic efficiency, proteins are often denatured and solubilized with chemical agents such as sodium dodecyl sulphate (SDS), urea (UA), etc. However, surfactant like SDS is difficult to remove from the reacting system and may interfere with protease activity, chromatographic separation of peptides and mass spectrometry analysis. To this end, we have prepared an immobilized surfactant which can preserve the solubilization and denaturation abilities of a surfactant and has the advantage of being easily separated from samples. The denaturation and solubilization capabilities of the newly developed immobilized surfactant were evaluated by comparing the enzymatic hydrolysis efficiencies in the pretreatment of protein samples treated with immobilized surfactant and other common methods. The results showed that the average protein sequence coverage was 42.1% (digested for 1 h), which was 76.89% higher than that of a control experiment (23.8%) in the digestion of a standard protein (BSA) and reached the same denaturation ability as that of heating at 95 °C. Moreover, the immobilized surfactant was further tested in the extraction and digestion of the total protein of HeLa cells. The number of identified proteins resulted in similar levels to those exploited with 0.1% SDS and 8 M UA, both of which are frequently used in lysis buffer, indicating that the immobilized surfactant had a good ability to denature and solubilize proteins. In brief, the immobilized surfactant avoids the shortcomings of UA and SDS and can be utilized in the pretreatment of complex biological samples.
1. Introduction
Proteomics is the large-scale study of proteins, particularly their amino acid sequences, post-translational modifications, structures and functions, expressed from a genome, organism, tissue, or cell type.1–3 Genes are carriers of genetic information, and proteins are carriers of life activity. The human genome project, which began in the 1990s, has made great advancements. Life science has entered the post-genome era, and the focus has shifted from revealing the genetic information to exposing the laws of life activity at the molecular level, in which proteomics is one of the most important components.4–6 The main research strategies of proteomics include “bottom-up”, “middle-down” and “top-down”.7–9 In bottom-up proteomics, also called “shotgun” proteomics,10 proteins are first digested into peptides, followed by reversed-phase liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. The resulting MS data are searched against a protein sequence database for peptide identification; then, the corresponding proteins can be inferred.11–13 The “bottom-up” proteomic strategy is widely used in protein expression profiles, post-translational modifications, protein interactions and proteome quantification.14–16 However, there are still many difficulties involved when utilizing this strategy, such as obtaining deep coverage, reliability, and repeatability due to the variable physical and chemical properties resulting from differences in amino acid sequences, the varying abundance of different proteins, and the complexity of post-translational modifications, spatial structure and protein–protein interactions.17,18 Therefore, it is necessary to develop new technologies to improve proteomics methods.19–22The pretreatment conditions of protein samples have a significant effect on the composition, chromatographic separation and mass spectrometry analysis of peptide mixtures.23 Improving recovery and enzymatic efficiency is of great importance for the reproducibility and reliability of proteomic analysis.24 Surfactants are commonly used reagents in sample pretreatment and are applied to denature and solubilize proteins, thereby increasing the enzymatic efficiency of the digestion enzymes acting upon the protein sample. Sodium dodecyl sulfate (SDS) is an effective and popularly used surfactant.25,26 However, SDS has many disadvantages, including reducing protease activity, thereby affecting enzymatic efficiency, interfering with chromatographic separation and suppressing peptide mass spectrometry signals. Therefore, it often be removed from the system before enzymatic digestion and LC-MS/MS analysis. Conventional sample purification methods, such as precipitation, extraction, ion exchange and dialysis cannot completely remove the SDS in samples, and the removal process can cause an excessive loss of proteins and peptides. New reagents have been utilized to improve the solubilization process, such as sodium deoxycholate (SDC)27 and ionic liquids,28 which are easier to remove and have better mass spectrometric compatibility. In addition, the filter-aided sample preparation (FASP) method29 and desalting columns30 are also used for removal of SDS. However, these methods suffer from cumbersome and time-consuming steps, and need to be further improved.
In recent years, magnetic nanoparticles have been widely used as a new biofunctional material31 due to their excellent biocompatibility, low toxicity and large surface area.32,33 The easy preparation and rapid magnetic response make magnetic nanoparticles an ideal carrier material for use in fluorescence imaging, drug delivery and proteomic research.34–36
In this study, we prepared surface-active surfactant immobilized magnetic nanoparticles that preserve the solubilization and denaturation abilities of a surfactant and have the advantage of easy magnetic separation. Thus, no surfactant remains in the sample during proteolysis, which has no negative effect on protein hydrolysis, chromatographic separation or mass spectrometry analysis.
2. Materials and methods
2.1 Reagents
Ferric chloride hexahydrate (FeCl3·6H2O), ferrous chloride tetrahydrate (FeCl2·4H2O), trimethoxy (octadecyl) silane (TMOS, technical grade), tetraethyl orthosilicate (Si(OC2H5)4), ammonium hydroxide (NH4OH), bovine serum albumin (BSA), urea (UA), Tris–HCl (1 M, pH 8.0), dithiothreitol (DTT), and iodoacetamide (IAA) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Protease inhibitor cocktail tablets were purchased from Roche (Manheim, Germany). Trypsin (from bovine pancreas) was purchased from Promega (Madison, WI, USA). Hydrochloric acid (HCl), ethanol (CH4O) and isopropanol (C3H8O) were obtained from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). All other regents were of analytical grade.2.2 Syntheses of Fe3O4 nanoparticles and Fe3O4@TMOS material
First, 1.46 g ferric chloride hexahydrate and 0.5 g ferrous chloride tetrahydrate were dissolved in 6.3 mL of 0.4 M chlorine. Next, 62.5 mL of 0.7 M ammonia was added to a round bottom flask and stirred vigorously. The hydrochloric acid mixture above was added to the round bottom flask rapidly and stirred for 1 h at room temperature. The precipitation was separated by means of a magnet, washed three times with ethanol and dried under vacuum at 60 °C, by which we obtained ferroferric oxide (Fe3O4) nanoparticles.Fifty milligrams Fe3O4 nanoparticles, 22.857 mL ethanol, 5.714 mL water and 0.214 mL ammonia were added together and sonicated for 30 min. The mixture was then transferred to a 100 mL round bottom flask, and 0.143 mL tetraethyl orthosilicate was added dropwise and then stirred for 6 h at room temperature. The precipitate was washed three times with isopropanol.
The resulting material was dispersed in 8.571 mL isopropanol, 259 μL TMOS was added dropwise under N2 atmosphere, and the mixture was stirred for 24 h at room temperature. The precipitate Fe3O4@TMOS, the final product, was washed three times with isopropanol and stored in 3 mL (Fe3O4@TMOS with a final concentration of 15 mg mL−1) isopropanol.
2.3 Pretreatment of proteins
Tryptic digestion of BSA: 1 mg BSA was dissolved in 10 mL 50 mmol NH4HCO3, with a final concentration of 0.1 μg μL−1. Four aliquots of 100 μL BSA solution were denatured separately as described in Table S1.† Trypsin was added to the BSA solutions (enzyme to protein weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100), and incubated at 37 °C for 1 h. The solutions were then analysed by MALDI-TOF mass spectrometry by first depositing 1 μL digested peptide solution onto a MALDI plate, followed by 1 μL CHCA matrix aqueous solution.
:100), and incubated at 37 °C for 1 h. The solutions were then analysed by MALDI-TOF mass spectrometry by first depositing 1 μL digested peptide solution onto a MALDI plate, followed by 1 μL CHCA matrix aqueous solution.
Extraction and trypsin digestion of HeLa proteins: four plates (25 mm × 150 mm, 2 × 107 cells per plate) of HeLa cells were separately centrifuged at 500 × g for 3 min in four different 1.5 mL EP tubes, and the supernatant was discarded. One millilitre of the different lysis buffers, as shown in Table S2,† was added to each of four EP tubes along with protease inhibitor. One hundred micrograms of the resulting protein from each method was then reduced with 10 mM DTT at 56 °C for 1 h and alkylated with 50 mM IAA at room temperature in the dark for 1 h. Trypsin was then added to the protein solution at a ratio of 1:50 and incubated at 37 °C for 16 h. The digested peptides of HeLa proteins were desalted using a self-packed C18 desalination column. Of the resulting sample, 1 μg peptides from each lysis method were taken for further HPLC-MS/MS analysis.
2.4 Mass spectrometry analysis
MALDI-TOF MS analysis: the MALDI-TOF mass analysis was performed on an ultrafleXtreme time-of-flight mass spectrometer (Bruker, Germany) in a positive-ion reflector mode with an available accelerating voltage of 20 kV. The nitrogen laser was 337 nm, and the laser energy was 80%. The mass range of the mass spectra was between m/z 700 and 3500.Nano LC-MS/MS analysis: the high-performance liquid chromatograph used was UltiMate 3000 RSLCnano (Thermo Fisher Scientific, USA). The chromatographic column was self-packed (150 μm × 12 cm, New Objective, USA) with a particle size of 1.9 μm (Dr Maisch Gmbh, Germany). The digested peptide mixture was dissolved with mobile phase A (0.1% FA aqueous solution), the loading volume was 5 μL, and the flow rate was 600 nL min−1. A 90 minute gradient was established with mobile phase A and mobile phase B (0.08% FA-80% ACN aqueous solution). The elution gradient was composed of 0 min of 5% B, 0–8 min of 5% B-8% B, 8–24 min of 8% B-13% B, 24–60 min of 13% B-28% B, 60–79 min of 28% B-40% B, 79–80 min of 40% B-95% B, 80–86 min of 95% B-6% B, and 86–90 min of 6% B. The eluted peptides were passed through an electrospray ionization source (ESI) directly into the mass spectrometer. The mass spectrometer (Q Exactive HF, Thermo Fisher Scientific, USA) was operated in positive mode with a spray voltage of 2 kV. The full MS scan was acquired from m/z 300 to 4000 with a resolution of 120000 and an MS AGC of 2e6. The MS/MS scan was acquired from m/z 300 to 1400 with a resolution of 15000, MS2 AGC of 2e4 and MS2 maximum ion implantation time of 19 ms.
2.5 Data analysis
MALDI-TOF spectra were analysed by flexAnanlysis software (version 3.3). Peptide mass fingerprints were searched via Mascot (version 2.3.01, Matrix Science). The Mascot parameters were set as follows: the enzyme was set as trypsin, and two missed cleavages at most were allowed; fixed modification was carbamidomethylation (Cys), and variable modification was methionine oxidation (Met); and the mass tolerance was set as 0.2 Da.The raw data of MS spectra was searched with Mascot built-in Proteome Discoverer software (version 2.1 SP2, Thermo Scientific). The Mascot parameters were set as follows: the enzyme was set as trypsin, and two missed cleavages at most were allowed; fixed modification was carbamidomethylation (Cys), and variable modification was methionine oxidation (Met); the precursor tolerance was 10 ppm; the fragment tolerance was 20 mmu; and the false discovery rate (FDR) was set as 1%. The database used was the human protein database (download from http://www.uniprot.org/uniprot/?query=homo%20protein%26fil=%26sort=score on April 18th, 2017).
3. Results and discussion
3.1 Preparation and characterization of the immobilized surfactant
The preparation process of the immobilized surfactant is shown in Scheme 1, in which Fe3O4 nanoparticles with a diameter of approximately 10 nm were first prepared and then coated with tetraethyl orthosilicate. Finally, the long carbon chains were fixed by Si–O to the surface of the magnetic nanoparticles. The denaturation mechanism of Fe3O4@TMOS was similar to that of a surfactant, mainly through the long carbon chains interacting with proteins and other ends interacting with water to denature and solubilize proteins. The large nanoscale hydrophilic core of Fe3O4@TMOS remained outside the proteins which was analogous to the hydrophilic side of a surfactant, but unlike the surfactant, Fe3O4@TMOS can be easily separated from samples under magnetic filed. The frequently used Fe3O4 nanoparticles have a larger diameter (approximately 250 nm), such as Fe3O4 nanoparticles prepared by Hu, etal.37 which carried immobilized enzymes. As proteins in aqueous solution are usually 5–10 nm in diameter, Fe3O4 nanoparticles with larger sizes cannot easily make full contact with proteins. Therefore, a coprecipitation method was used to prepare smaller and more uniform Fe3O4 nanoparticles. In addition, the magnetic nanoparticles had numerous hydroxyl groups after coating with SiO2 this endows good hydrophilicity for the hydrophilic side. The long carbon chain with hydrophobicity, as the hydrophobic side, can insert into proteins and induce protein denaturation and solubilization.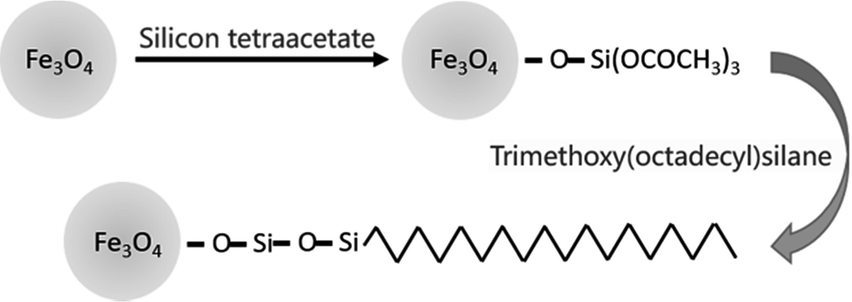 | ||
| Scheme 1 The preparation process of Fe3O4@TMOS. | ||
The morphology of Fe3O4 and Fe3O4@TMOS nanoparticles was characterized by transmission electron microscopy (TEM, FEI, USA). As shown in Fig. 1, the Fe3O4 nanoparticles have an average diameter of 10 nm. After modification, the edges of the Fe3O4@TMOS nanoparticles were obviously blurred, and the size was also marginally increased (Fig. 1e). Since Fe3O4@TMOS had a magnetic response and a small particle size, there was a tendency for it to agglomerate. However, it can be uniformly dispersed in the solution by ultrasound before use (Fig. 2a).
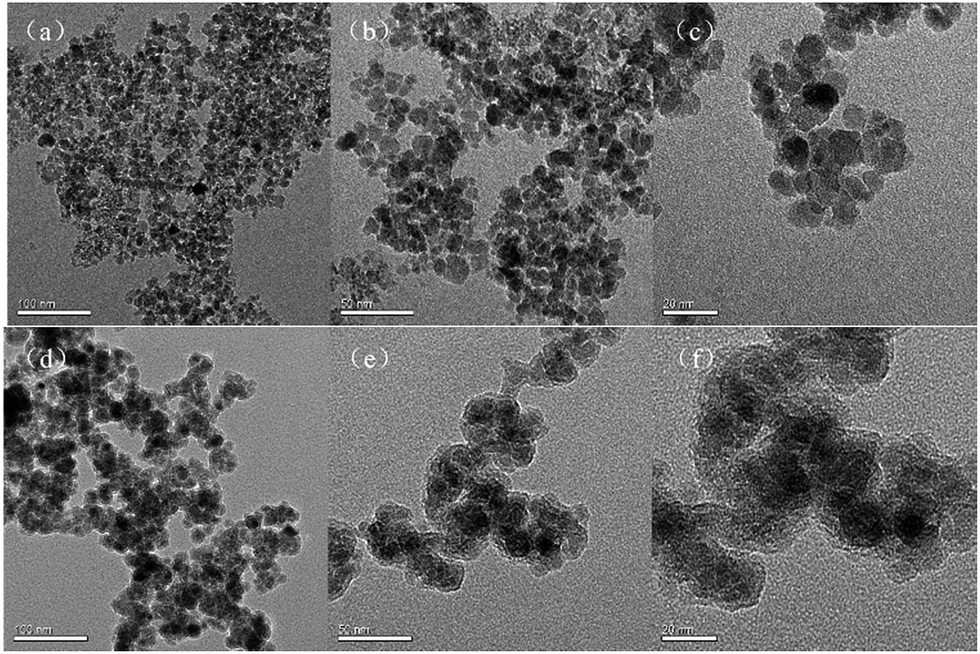 | ||
| Fig. 1 TEM of Fe3O4 magnetite nanoparticle (a, b, c) and Fe3O4@TMOS (d, e, f). | ||
 | ||
| Fig. 2 (a) Fe3O4@TMOS evenly dispersed in isopropanol; (b) the separation of Fe3O4@TMOS and the solvent under the action of an applied magnetic field. | ||
According to elemental analysis of the Fe3O4 and Fe3O4@TMOS nanoparticles (Table 1), after modification with TMOS, the carbon content increased from 0.81% to 3.44%, which indicates that TMOS was successfully immobilized on the surface of Fe3O4 nanoparticles. Fourier transform infrared spectroscopy (FT-IR, Nicolet, USA) was used to further characterize the chemical constituents of these two types of nanoparticles. As shown in Fig. 3, TMOS showed strong peaks at 2920 cm−1 and 2860 cm−1, which are the absorption peaks of C–H stretching vibrations (σC–H). These peaks were not observed in the FT-IR spectra of Fe3O4 nanoparticles, while after immobilization of TMOS on the surface, these peaks could again be clearly identified. The results suggest that TMOS was successfully immobilized on the surface of the nanoparticles. The amount of TMOS bound to the surface of Fe3O4@TMOS was measured by thermogravimetric analysis (TGA, TA instrument, USA). As shown in Fig. 4, the heat loss of Fe3O4 nanoparticles was approximately 8%, which was due to the volatilization of the crystallization water, consistent with the results of elemental analysis, which showed that Fe3O4 nanoparticles contained 1.373% elemental hydrogen. In contrast, the weight loss of Fe3O4@TMOS was up to 17%, which was due to the volatilization of crystallization water and carbon chains on the surface of Fe3O4@TMOS under high temperature conditions. The amount of TMOS bound to the surface of Fe3O4 was found to be 160 μg mg−1.
| Magnetite nanoparticles | mg | N% | C% | H% | S% |
|---|---|---|---|---|---|
| Fe3O4 | 5.338 | 0.02 | 0.81 | 1.373 | 0.506 |
| Fe3O4@TMOS | 5.352 | 0.29 | 3.44 | 1.176 | 0.269 |
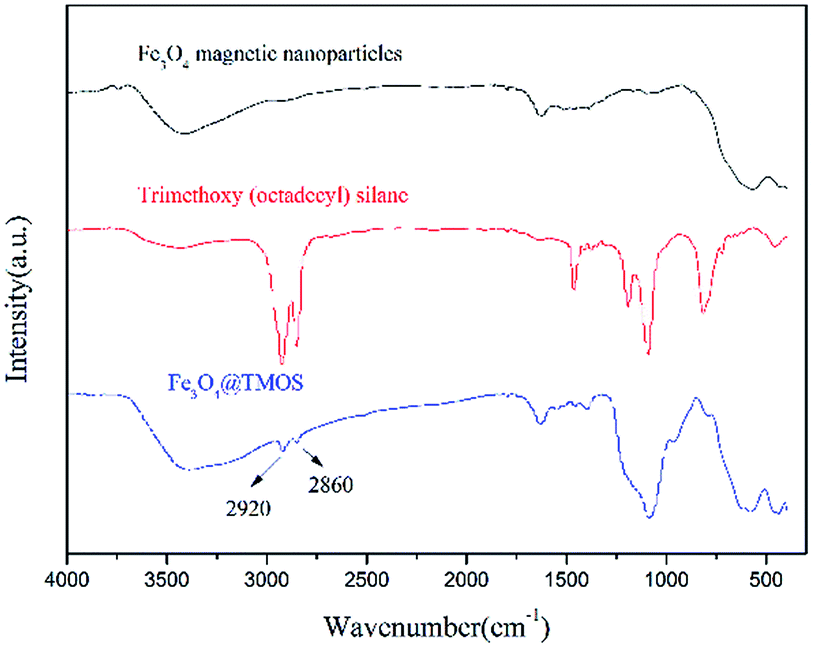 | ||
| Fig. 3 FT-IR spectra of Fe3O4 magnetite nanoparticles, trimethoxy(octadecyl)silane and Fe3O4@TMOS. | ||
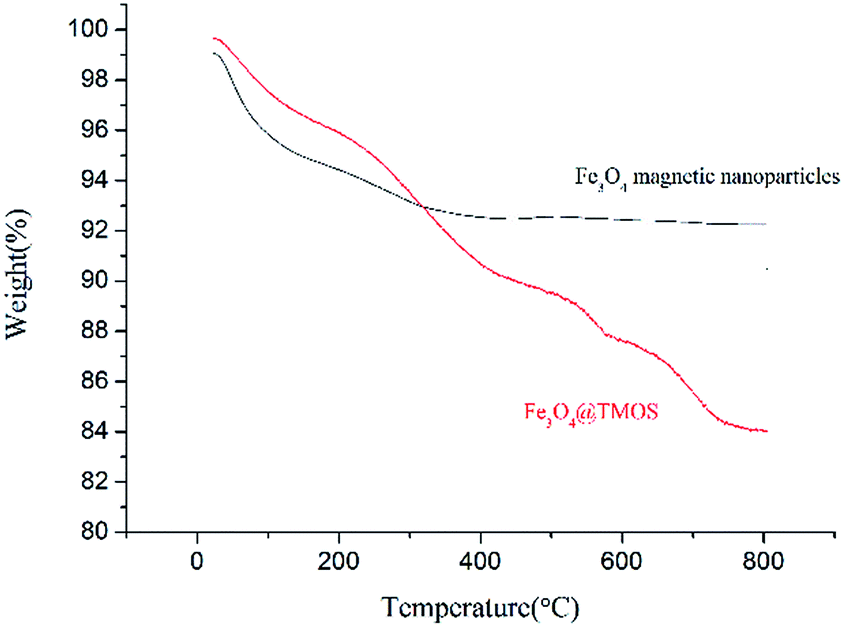 | ||
| Fig. 4 TGA profiles of Fe3O4 magnetite nanoparticles and Fe3O4@TMOS. | ||
As shown in Fig. S1,† the magnetic field lines are uniformly symmetrical in the magnetic hysteresis loop (Quantum Design, USA), indicating that the prepared Fe3O4 nanoparticles had superparamagnetism. After modification of the carbon chain, the magnetic properties of Fe3O4@TMOS were not significantly reduced, indicating that Fe3O4@TMOS had constant magnetic properties and could be easily separated under an applied magnetic field (Fig. 2a and b).
3.2 Protein denaturation ability of the immobilized surfactant on standard protein BSA
SDS is difficult to be removed from samples and interferes with the ionization behaviour of peptides when analysed by mass spectrometry. Fig. 5 shows that the MALDI-TOF mass spectra of standard protein BSA digests cleaved by trypsin for 1 h under the following conditions: (Fig. 5a) undenatured, (Fig. 5b) denatured with SDS followed by removal of SDS using the FASP method, (Fig. 5c) denatured with SDS without SDS removal, (Fig. 5d) denatured at 95 °C and (Fig. 5e) denatured with Fe3O4@TMOS. Compared with Fig. 5a, the normal peptide peaks were absent and a large number of impurity peaks were apparent in the sample denatured with SDS (Fig. 5c). The result indicated that SDS had a considerable interference with mass spectrometry analysis and could not be removed by the conventional FASP method (Fig. 5b). Thermal denaturation, a frequently used denaturation method (also a completely denatured method), (Fig. 5d), showed that the number of peptide peaks increased compared with undenatured sample (Fig. 5a). Using the Fe3O4@TMOS denaturation method, the number of peptide peaks was significantly increased, and no impurity peaks appeared (Fig. 5d). The results indicate that Fe3O4@TMOS can denature BSA effectively and can be completely separated by an external magnetic field and these nanoparticles do not interfere with the mass spectrometry analysis.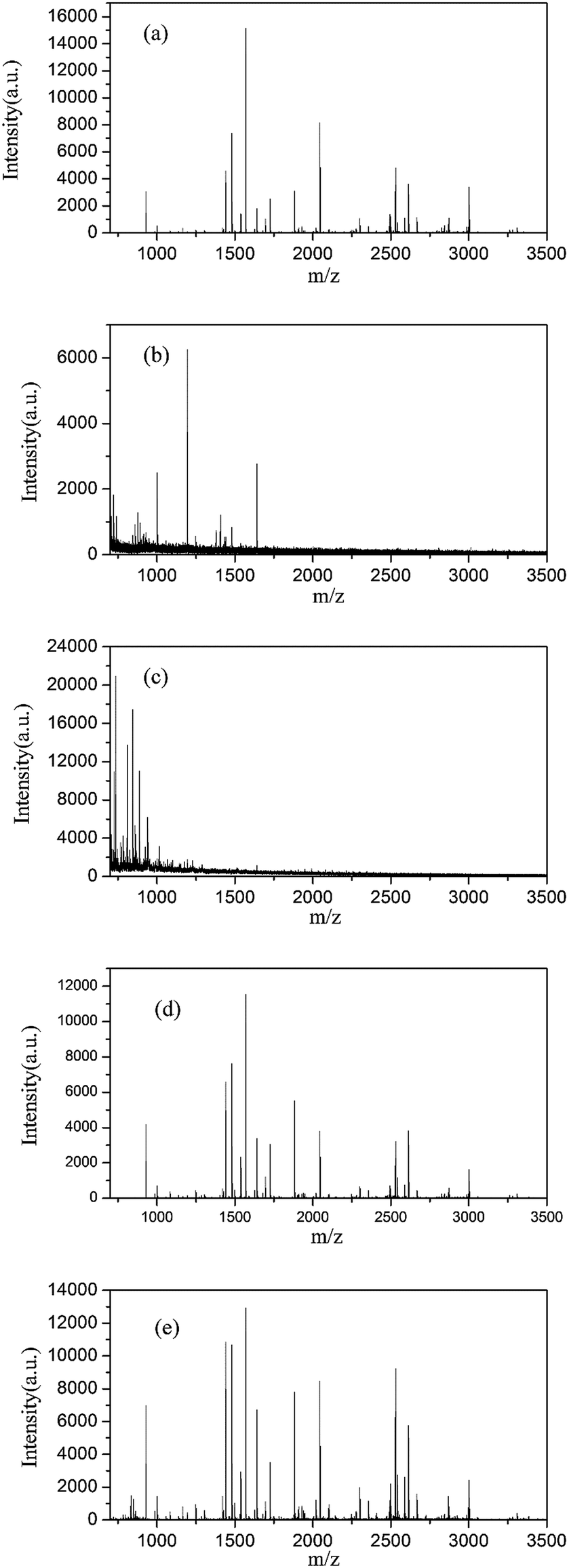 | ||
| Fig. 5 MALDI-TOFMS spectra of BSA digests (1 h) under conditions of undenatured (a), denatured with SDS followed by SDS removal (b), denatured with SDS without removal (c), denatured with 95 °C (d) and denatured with Fe3O4@TMOS (e). | ||
The amino acid sequence coverages of BSA digested under different denaturing conditions were examined. As shown in Table S3–S5† and Fig. S2–S4,† the 41% amino acid sequence coverage of BSA digestion after denaturing by calefaction at 95 °C indicated that the BSA was completely denatured. Coverage had been greatly improved compared to the results of undenatured BSA of 23.8%. In addition, no peptides can be identified in BSA digests denatured by SDS because of the interference by SDS in mass spectrometry analysis. As shown in Fig. 6, the average amino acid sequence coverage of BSA digested after denaturation by Fe3O4@TMOS for three times was 42.1%, which was much higher than that of the undenatured BSA digest (increased by 76.89%) and reached the same enzymatic efficiency of calefaction at 95 °C (Fig. 6). Therefore, Fe3O4@TMOS can denature BSA completely, indicating that Fe3O4@TMOS has a substantial ability to denature protein.
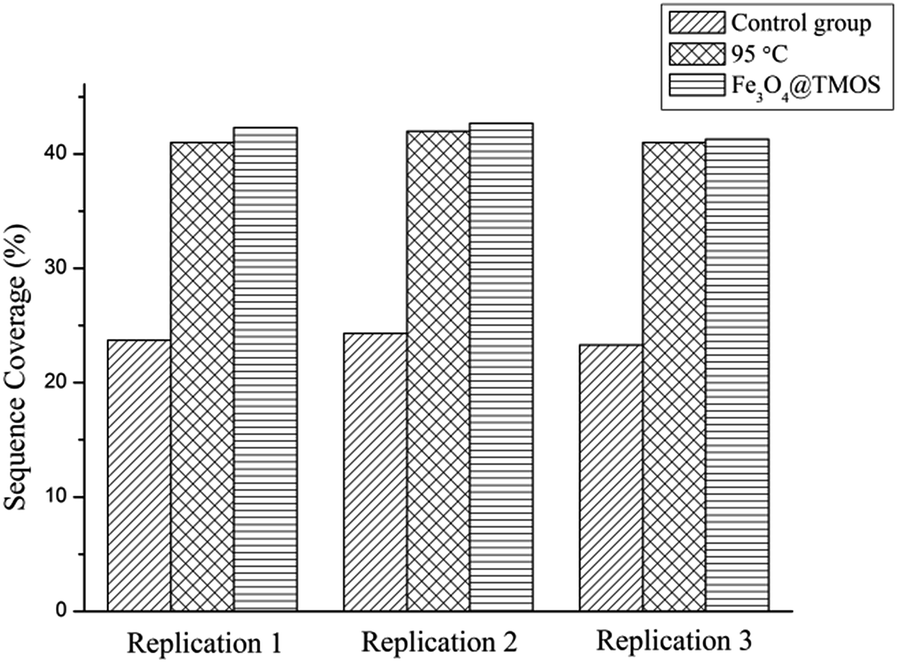 | ||
| Fig. 6 Sequence coverages of BSA identified by mass spectrometry for three times (control group: undenatured BSA digested for 1 h; 95 °C: BSA digested for 1 h after denatured with 95 °C; Fe3O4@TMOS: BSA digested for 1 h after denatured with Fe3O4@TMOS). | ||
3.3 Examination of protein denaturation and solubilization abilities of the immobilized surfactant using a complex sample
To further examine the protein extraction, denaturation and solubilization performance of the immobilized surfactant, a complex sample, HeLa cells, was processed with the immobilized surfactant and compared with extraction by H2O, 0.1% SDS and 8 M UA (Table 2).| Lysis reagents | Peptide number | Protein number |
|---|---|---|
| Fe3O4@TMOS | 27388 |
3668 |
| H2O | 12688 |
2716 |
| 0.1% SDS | 24136 |
3547 |
| 4% SDS | 17415 |
2934 |
| 8 M UA | 24151 |
3671 |
The ability of Fe3O4@TMOS to extract proteins from a complex sample was demonstrated by comparing the results to those of using H2O alone, which is a pollution and interference free method. The number of identified proteins increased by 35.05% using Fe3O4@TMOS, compared to the number of proteins identified when using H2O alone as a lysis reagent.
A frequently used lysis buffer, 0.1% SDS,38 has a robust ability to solubilize and denature membrane proteins. Compared with this reagent, Fe3O4@TMOS extracted slightly more proteins. However, SDS is hard to remove39 and interferes with subsequent protein analysis by MS.40 When the concentration of SDS was increased to 4% followed by FASP purification, a large number of proteins were lost, and the number of identified proteins by MS decreased to 2934. It is interesting to note that Fe3O4@TMOS can effectively avoid this problem because of its easy removal by a magnet (Fig. 2); thus, there is no impact on MS analysis.
Another widely used lysis buffer is 8 M UA13,26 which is highly efficient at denaturing and solubilizing proteins by decreasing the hydrophobic effect and directly binding to proteins via hydrogen bonds.41–43 The number of proteins extracted from HeLa cells with Fe3O4@TMOS and identified by MS was nearly the same as the number extracted using 8 M UA, which is regarded as a good cell lysate reagent. UA has shortcomings, however, including poor thermal stability and a high salt concentration, thus, it cannot be heated, or else protein amidation can occur, and it must be diluted to less than 2 M before enzymatic hydrolysis. In contrast, the samples can be heated to shorten the reaction time when Fe3O4@TMOS is used, and it can be easily removed to avoid enzyme deactivation in the next processing step.
Three replicated experiments were performed (Fig. S5–S7†) and a total of 5092 proteins extracted by Fe3O4@TMOS were identified. In all the above results indicated that Fe3O4@TMOS is comparable to SDS and UA when used for protein lysis, but unlike these commonly used reagents, Fe3O4@TMOS can be heated and then be removed by a magnet to shorten the reaction time, simplify the experimental steps and reduce sample loss, no negative impact on mass spectrometry analysis. These suggest that Fe3O4@TMOS has a strong ability to denature and solubilize proteins and can be used in future proteomics studies.
4. Conclusions
An immobilized surfactant was prepared by modifying the long carbon chain on the surface of magnetic nanoparticles. The sample solution and the immobilized surfactant can be easily separated by applying an external magnetic field before digestion or LC-MS/MS analysis procedures. This avoids interference from surfactants, which are often difficult to be removed in conventional experiments in downstream MS analysis of proteins. The protein denaturation and solubilization abilities of synthesized Fe3O4@TMOS were verified with BSA and protein extraction of HeLa cells. The experimental results showed that the average amino acid sequence coverage was 42.1% for BSA denatured by immobilized surfactant, which increased by 76.89% compared with that of the undenatured BSA digest. Additionally, Fe3O4@TMOS achieved the same denaturation level as that of the calefaction method. When using immobilized surfactant to extract and denature HeLa proteins, the number of proteins subsequently identified by MS was comparable with that of 8 M UA and 0.1% SDS. These results demonstrate that immobilized surfactant has a good ability to denature and solubilize proteins, avoids the shortcomings of both UA and SDS, and can be utilized in the pretreatment of biological samples for analysis by MS.Conflicts of Interest
The authors declared no conflict of interest.Acknowledgements
This work was supported by National Key R&D Program of China (2017YFA0505002), the National Key Program for Basic Research of China (2013CB911204 and 2016YFA0501403), the National Key Program for Scientific Instrument and Equipment Development (2012YQ12004407 and 2013YQ14040506).Notes and references
- A. Pandey and M. Mann, Nature, 2000, 405, 837 CrossRef CAS PubMed.
- S. Chen and A. C. Harmon, Proteomics, 2006, 6, 5504–5516 CrossRef CAS PubMed.
- S. P. Gygi, Y. Rochon, B. R. Franza and R. Aebersold, Mol. Cell. Biol., 1999, 19, 1720–1730 CrossRef CAS PubMed.
- M. R. Wilkins, J.-C. Sanchez, A. A. Gooley, R. D. Appel, I. Humphery-Smith, D. F. Hochstrasser and K. L. Williams, Biotechnol. Genet. Eng. Rev., 1996, 13, 19–50 CrossRef CAS PubMed.
- M. R. Wilkins, R. D. Appel, J. E. Van Eyk, M. Chung, A. Görg, M. Hecker, L. A. Huber, H. Langen, A. J. Link and Y. K. Paik, Proteomics, 2006, 6, 4–8 CrossRef CAS PubMed.
- V. C. Wasinger, S. J. Cordwell, A. Cerpa-Poljak, J. X. Yan, A. A. Gooley, M. R. Wilkins, M. W. Duncan, R. Harris, K. L. Williams and I. Humphery-Smith, Electrophoresis, 1995, 16, 1090–1094 CrossRef CAS PubMed.
- C. May, F. Brosseron, P. Chartowski, C. Schumbrutzki, B. Schoenebeck and K. Marcus, Data Mining in Proteomics: From Standards to Applications, 2011, pp. 3–26 Search PubMed.
- M. Wilhelm, J. Schlegl, H. Hahne, A. M. Gholami, M. Lieberenz, M. M. Savitski, E. Ziegler, L. Butzmann, S. Gessulat and H. Marx, Nature, 2014, 509, 582–587 CrossRef CAS PubMed.
- K. Nakata, T. Ichibangase, R. Saitoh, M. Ishigai and K. Imai, Analyst, 2015, 140, 71–73 RSC.
- Y. Zhang, B. R. Fonslow, B. Shan, M.-C. Baek and J. R. Yates III, Chem. Rev., 2013, 113, 2343–2394 CrossRef CAS PubMed.
- A. Armirotti, Curr. Anal. Chem., 2009, 5, 116–130 CrossRef CAS.
- J. M. Chick, D. Kolippakkam, D. P. Nusinow, B. Zhai, R. Rad, E. L. Huttlin and S. P. Gygi, Nat. Biotechnol., 2015, 33, 743–749 CrossRef CAS PubMed.
- A. Schmudlach, J. Felton, C. Cipolla, L. Sun, R. T. Kennedy and N. J. Dovichi, Analyst, 2016, 141, 1700–1706 RSC.
- J. Mayne, Z. Ning, X. Zhang, A. E. Starr, R. Chen, S. Deeke, C.-K. Chiang, B. Xu, M. Wen and K. Cheng, Anal. Chem., 2015, 88, 95–121 CrossRef PubMed.
- C. Evans, J. Noirel, S. Y. Ow, M. Salim, A. G. Pereira-Medrano, N. Couto, J. Pandhal, D. Smith, T. K. Pham and E. Karunakaran, Anal. Bioanal. Chem., 2012, 404, 1011–1027 CrossRef CAS PubMed.
- T. H. Steinberg, B. J. Agnew, K. R. Gee, W. Y. Leung, T. Goodman, B. Schulenberg, J. Hendrickson, J. M. Beechem, R. P. Haugland and W. F. Patton, Proteomics, 2003, 3, 1128–1144 CrossRef CAS PubMed.
- A. Politis, F. Stengel, Z. Hall, H. Hernández, A. Leitner, T. Walzthoeni, C. V. Robinson and R. Aebersold, Nat. Methods, 2014, 11, 403–406 CrossRef CAS PubMed.
- A. H. Smits and M. Vermeulen, Trends Biotechnol., 2016, 34, 825–834 CrossRef CAS PubMed.
- Q. Qi, G. Yan, C. Deng and X. Zhang, Anal. Bioanal. Chem., 2016, 408, 8437–8445 CrossRef CAS PubMed.
- M. des Plantes, C. ProtØines, F. Mauguio and D. Pathologique, Electrophoresis, 2000, 21, 3329–3344 CrossRef.
- L. Zhang, J. Xie, X. e. Wang, X. Liu, X. Tang, R. Cao, W. Hu, S. Nie, C. Fan and S. Liang, Proteomics, 2005, 5, 4510–4524 CrossRef CAS PubMed.
- F. Jiao, R. Zhai, J. Huang, Y. Zhang, Y. Zhang and X. Qian, RSC Adv., 2016, 6, 84113–84118 RSC.
- A. Shevchenko, H. Tomas, J. Havlis, J. V. Olsen and M. Mann, Nat. Protoc., 2007, 1, 2856–2860 CrossRef PubMed.
- G. Menschaert, W. Van Criekinge, T. Notelaers, A. Koch, J. Crappé, K. Gevaert and P. Van Damme, Mol. Cell. Proteomics, 2013, 12, 1780–1790 CAS.
- D. Botelho, M. J. Wall, D. B. Vieira, S. Fitzsimmons, F. Liu and A. Doucette, J. Proteome Res., 2010, 9, 2863–2870 CrossRef CAS PubMed.
- J. R. Wisniewski, A. Zougman, N. Nagaraj and M. Mann, Nat. Methods, 2009, 6, 359 CrossRef CAS PubMed.
- A. Serra, H. Zhu, X. Gallart-Palau, J. E. Park, H. H. Ho, J. P. Tam and S. K. Sze, Anal. Bioanal. Chem., 2016, 408, 1963–1973 CrossRef CAS PubMed.
- Z. Sui, Y. Weng, Q. Zhao, N. Deng, F. Fang, X. Zhu, Y. Shan, L. Zhang and Y. Zhang, Talanta, 2016, 161, 541–546 CrossRef CAS PubMed.
- A. J. Nel, S. Garnett, J. M. Blackburn and N. C. Soares, J. Proteome Res., 2015, 14, 1637–1642 CrossRef CAS PubMed.
- A. Zienkiewicz, J. D. Rejón, J. de Dios Alché, M. I. Rodríguez-García and A. J. Castro, Plant Proteomics: Methods and Protocols, 2014, pp. 85–91 Search PubMed.
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167 CrossRef CAS.
- A. H. Lu, E. e. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed.
- H. Teymourian, A. Salimi and S. Khezrian, Biosens. Bioelectron., 2013, 49, 1–8 CrossRef CAS PubMed.
- Y. Zhang, G.-M. Zeng, L. Tang, D.-L. Huang, X.-Y. Jiang and Y.-N. Chen, Biosens. Bioelectron., 2007, 22, 2121–2126 CrossRef CAS PubMed.
- Q. Pankhurst, N. Thanh, S. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2009, 42, 224001 CrossRef.
- R. Zhai, F. Jiao, D. Feng, F. Hao, J. Li, N. Li, H. Yan, H. Wang, Z. Jin and Y. Zhang, Electrophoresis, 2014, 35, 3470–3478 CrossRef CAS PubMed.
- Z. Hu, L. Zhao, H. Zhang, Y. Zhang, R. a. Wu and H. Zou, J. Chromatogr. A, 2014, 1334, 55–63 CrossRef CAS PubMed.
- M. A. K. Markwell, S. M. Haas, L. Bieber and N. Tolbert, Anal. Biochem., 1978, 87, 206–210 CrossRef CAS PubMed.
- L. Henderson, S. Oroszlan and W. Konigsberg, Anal. Biochem., 1979, 93, 153–157 CrossRef CAS PubMed.
- R. Pitt-Rivers and F. A. Impiombato, Biochem. J., 1968, 109, 825–830 CrossRef CAS PubMed.
- E. Liepinsh and G. Otting, J. Am. Chem. Soc., 1994, 116, 9670–9674 CrossRef CAS.
- Q. Zou, S. M. Habermann-Rottinghaus and K. P. Murphy, Proteins: Struct., Funct., Bioinf., 1998, 31, 107–115 CrossRef CAS.
- B. J. Bennion and V. Daggett, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 6433–6438 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08874d |
| This journal is © The Royal Society of Chemistry 2017 |