
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterostructural MnO2@NiS2/Ni(OH)2 materials for high-performance pseudocapacitor electrodes†
Ying Jia,
Wei Liua,
Ziqing Zhanga,
Ying Wanga,
Xudong Zhaoa,
Benxian Lia,
Xiaofeng Wang*a,
Xiaoyang Liu *a,
Bingbing Liub and
Shouhua Fenga
*a,
Bingbing Liub and
Shouhua Fenga
aState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, 2699 Qianjin Street, Changchun 130012, P. R. China. E-mail: liuxy@jlu.edu.cn; wangxf103@jlu.edu.cn; Fax: +86-431-85168316; Fax: +86-431-85168601; Tel: +86-431-85168316 Tel: +86-431-85168601
bState Key Laboratory of Superhard Materials, College of Chemistry, Jilin University, 2699 Qianjin Street, Changchun 130012, P. R. China
First published on 14th September 2017
Abstract
A new MnO2@NiS2/Ni(OH)2 heterostructure material was synthesized using a two-step hydrothermal process. The presence of 1D MnO2 nanosticks provided a substrate for the junction and facilitated the charge transmission. The grafting of NiS2/Ni(OH)2 sheets onto the surface of MnO2 enlarged the specific surface area (SSA) of the material. As a result, the SSA of the electrode material was improved and the capacitor performance was optimized. The material also showed a high rate capacity and long-term cycling capability.
1. Introduction
The demand for high-performance energy storage systems that are sustainable and renewable is high. There is an urgent need to develop the enabling technology, partly because of the fast depletion of fossil fuels and the severe environment pollution.1–4 The electrochemical energy storage technologies attract strong interest for their direct conversion of chemical energy to electrical energy, with little environmental pressure.5–8 Electrochemical capacitors are considered as promising candidates for effective energy storage, because of their high power density, fast charging–discharging processes, long life cycle, and low maintenance cost.9–12 In the past decade, much effort has been devoted to the advancement of electrical capacitors. Among them, redox-type supercapacitors are widely investigated and accepted for their high specific capacitances. A series of metal oxides, conductive polymers, and relevant carbon-based composites are put forward as potential alternative materials.13–16The transition metal chalcogenides with variable valence, such as MnO2, CoSx, and NiSx, are considered as the most promising pseudocapacitive materials.17–21 However, the poor electrical conductivity of metal oxides and the relatively low energy density of chalcogenides limit their applications in high-performance supercapacitors. The low energy density is due to its charge storage mechanism, which is based on ions adsorption or redox reactions on the surface layer.22,23 In a typical charge–discharge process, only a limited number of molecules can be utilized for the electronic and ionic transportation, thus limiting kinetically the rapid faradic reduction and oxidation reactions within the bulk material.24,25 One important approach to improve the performance of supercapacitors at high charge/discharge rates is to reduce their charge-transfer resistance and increase the surface area of the active materials.
Over the past few years, nanoparticle materials in various morphologies have been synthesized to enlarge the specific surface area (SSA) of pseudocapacitive materials.26–30 Tremendous efforts have been made to combine carbon-based materials to lower the charge-transfer resistance.31–33 Herein, we constructed a bran-new core–shell heterostructure material of MnO2@NiS2/Ni(OH)2 basing on the MnO2 nanosticks and NiS2/Ni(OH)2 nanosheets as a pseudocapacitive material. The synthesized electrode materials showed excellent electrochemical properties for use in supercapacitor. The 1D MnO2 nanosticks acted as the substrate insuring the high cycling performance, and facilitated electron transportation during the electrochemical process. The 2D NiS2/Ni(OH)2 nanosheets contributed to the remarkable capacitive property. The existence of heterostructure made of MnO2 nanosticks and NiS2/Ni(OH)2 sheets effectively increased the SSA and reduced the charge-transfer resistance among the different capacitive materials.
2. Experimental
2.1 Preparation of α-MnO2 nanosticks
All reagents used in the experiments were of analytical grade and used as received without further purification. The potassium permanganate, hydrochloric acid, nickel nitrate, thiourea, and potassium hydroxide were obtained from Shanghai Chemical Reagent Co. Nickel foam was purchased from Kunshan Dessco Electron Co., Ltd. Acetylene black was purchased from Alfa Aesar. 0.274 g of KMnO4 and 0.3 ml of HCl (37%) were mixed and dissolved in 10 ml of deionized water under magnetic stirring. The precursor solution was then transferred into a 15 ml of Teflon-lined stainless steel autoclave. The autoclave was sealed and kept in an oven at 120 °C for 4 hours (h). After cooling to room temperature, the products were purified by washing with deionized water and ethanol for several times. The resultant α-MnO2 nanosticks were dried at 60 °C for the following experiments.2.2 Preparation of MnO2@NiS2/Ni(OH)2 heterostructured materials
40 mg of prepared α-MnO2 nanosticks were added into a 20 ml aqueous solution, which contained 60 mg Ni(NO3)2·6H2O and 60 mg thiourea. After ultrasonic treatment for 1 h, the solution was added into a Teflon-lined stainless steel autoclave and heated to 120 °C. The dwell time for the reaction varied from 1 h to 24 h to optimize the structure and performance of the products for electrochemical applications. Later in this paper, we expressed 4 h, 12 h, and 24 h as samples with corresponding reaction times.2.3 Characterization of materials
The phase purity of the synthesized products was examined by X-ray powder diffraction (XRD) on a Rigaku D/max 2550VB diffractometer at a scanning rate of 10° min−1 in the 2θ range from 10° to 80°. The morphology and size of the synthesized products were determined with a scanning electron microscope (SEM, JSM-6700F, JEOL, Japan) and a transmission electron microscope (TEM, Hitachi H-800 electron microscope) equipped with a CCD camera at an accelerating voltage of 200 kV. High-resolution TEM (HRTEM) images were collected on a JEM-2100F electron microscope. The elementary analysis was performed on a VG ESCALAB MKII X-ray photoelectron spectrometer (XPS) equipped with a Mg K excitation (1253.6 eV) source. The binding energy was calibrated with C 1s at 284.6 eV. The Brunauer–Emmett–Teller (BET) surface area of the synthesized products was measured with N2 adsorption at 77 K on an ASAP 2420 Micromeritic system (Micromeritic, Japan). The electrochemical property was characterized by a CHI 660C electrochemical analyzer, CH Instruments.For the electrochemistry tests, a three-electrode system was employed. Hg/HgO electrode acted as a reference electrode. A Pt plate electrode was used as the counter electrode. The working electrode was fabricated by a traditional method. Firstly, the synthesized products of MnO2@NiS2 (70 wt%), acetylene black (25 wt%) and poly(tetrafluorene ethylene) binder (5 wt%) were mixed together. And then, the mixture was pressed onto a nickel foam, which acted as a current-collector, to produce the working electrode. The electric capacity properties of synthesized products were evaluated by using cyclic voltammogram and galvanostatic charge–discharge methods.
3. Results and discussion
Alpha-phase MnO2 was prepared and its X-ray diffraction (XRD) pattern is presented in Fig. 1a. The different diffraction peaks corresponded to the facets of alpha-phase MnO2 (JCPDS card: no. 44-0141).34 No redundant peak was observed, which confirmed that the products were highly pure and crystalline. Moreover, the widths at half maximum of the XRD peaks were rather broad, suggesting that the synthesized MnO2 was nanosticks. After compositing the α-MnO2 with Ni nitrate, nickel cations were absorbed onto some active sites of MnO2, and reacted with thiourea, along with the existence of H2O in the system, to form the NiS2/Ni(OH)2 as a shell on the surface of MnO2 nanosticks. The whole process by forming this heterostructure is shown in Scheme 1. And the corresponding XRD pattern of 4 h is shown in Fig. 1b. In addition to the peaks of MnO2, diffraction peaks of NiS2 and Ni(OH)2 were observed, which are classified and listed in Table 1. XRD pattern indicated that Ni(OH)2 was produced along with NiS2, yielding nanoparticles of MnO2@NiS2/Ni(OH)2. The introduction of Ni(OH)2 into the heterostructured nanoparticles may have advantageous effect on the electric capacity properties.35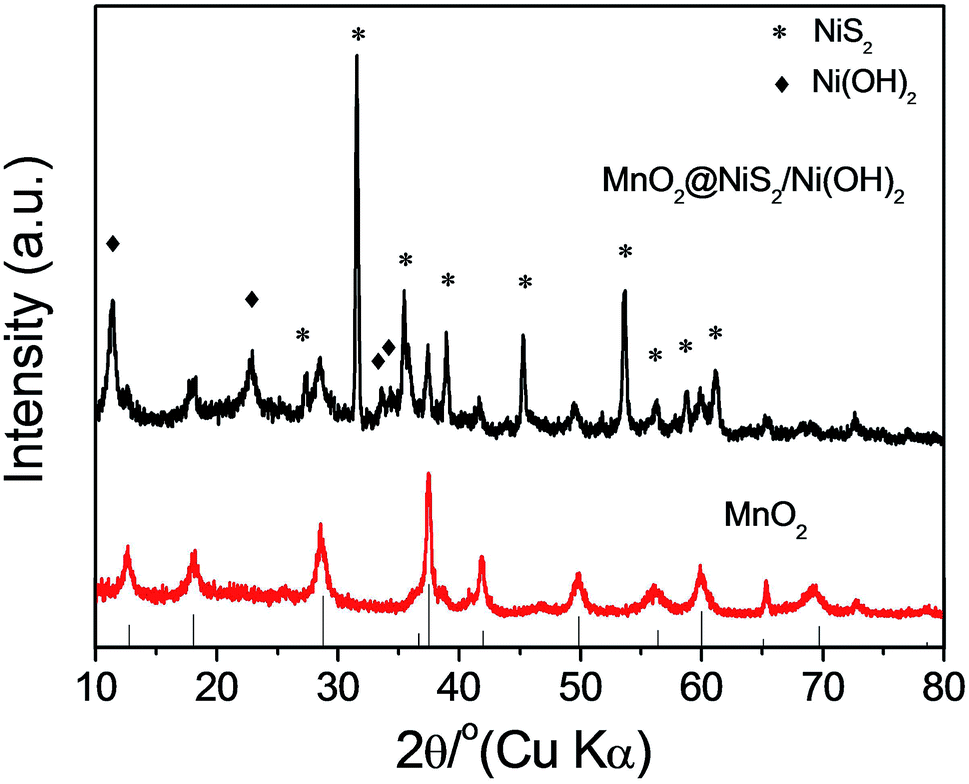 | ||
| Fig. 1 XRD pattern of as synthesized (a) MnO2 and (b) MnO2@NiS2/Ni(OH)2. | ||
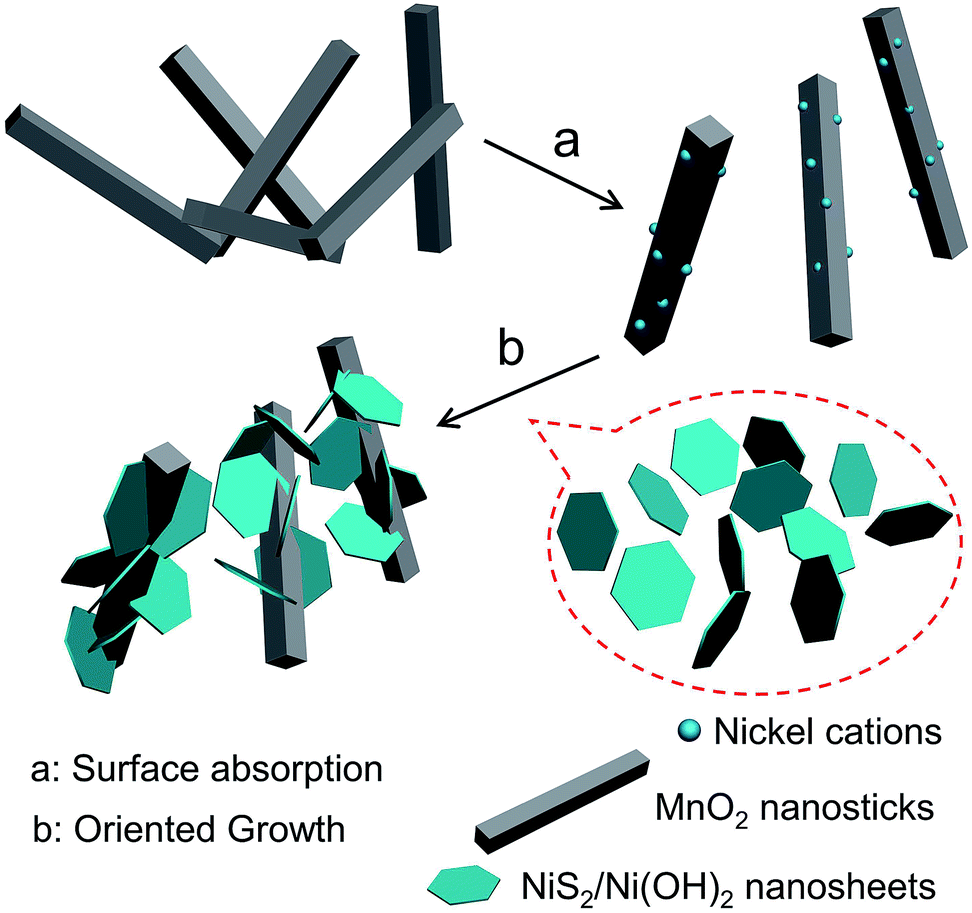 | ||
| Scheme 1 The forming process of this heterostructured MnO2@NiS2/Ni(OH)2. | ||
| MnO2 | NiS2 | Ni(OH)2 | |||
|---|---|---|---|---|---|
| Peaks/degree | Lattice plane | Peaks/degree | Lattice plane | Peaks/degree | Lattice plane |
| 12.6 | (111) | 22.7 | (111) | 11.4 | (003) |
| 18.1 | (200) | 31.5 | (200) | 22.7 | (006) |
| 28.6 | (310) | 35.4 | (210) | 33.5 | (101) |
| 37.4 | (211) | 38.8 | (211) | 34.4 | (012) |
| 41.8 | (301) | 45.2 | (220) | ||
| 49.6 | (411) | 53.5 | (311) | ||
| 60.0 | (521) | 56.2 | (222) | ||
| 65.4 | (002) | 58.6 | (023) | ||
| 69.3 | (541) | 61.1 | (321) | ||
| 72.8 | (312) | ||||
Fig. 2a shows the scanning electron microscopy (SEM) image of MnO2, which exhibited a stick-like structure with a length of 1 μm and a width of 200 nm. Moreover, smooth surface was observed in the SEM image of MnO2, which are consistent with previous reports.34,36 The one-dimensional (1D) stick-like structure of MnO2 is beneficial to the transmission of electrons and can enhance its electrochemical property.37 Employing the prefabricated MnO2 as the substrate, we deposited nickel cations on MnO2 using a hydrothermal process, to form MnO2@NiS2/Ni(OH)2 nanoparticles. And a heterostructure of 4 h is clearly observed in Fig. 2b, in which several sheets extend from the original smooth surface of MnO2 nanosticks. The NiS2/Ni(OH)2 sheets can be easily identified in the transmission electron microscopy (TEM) image (Fig. 2c). The products were consisted of two types of domains with different contrast, indicating the formation of heterostructure.
 | ||
| Fig. 2 (a) SEM image of MnO2 nanosticks. (b) SEM, (c) TEM and (d) HRTEM images of MnO2@NiS2/Ni(OH)2 heterostructure. | ||
The high-resolution TEM (HRTEM) image in Fig. 2d clearly shows an interplanar distance of 2.4 Å, which fits well with the (211) facet of alpha-phase MnO2 crystal. Another dominant interplanar spacing of 3.1 Å was detected, representing the lattice plane of (310). Moreover, for the sheet structure with light contrast, an interplanar distance of 2.3 Å was observed, representing the (211) crystallographic facets of NiS2 crystal. The similar interplanar distances of MnO2 and NiS2 allow the formation of a heterostructure. The formed NiS2 nanocrystals were observed to be sphere-like, embedded inside the extended sheet and formed the structure with light contrast in Fig. 2c and d. The other structure with invisible space lattice is predicted to be Ni(OH)2, constituting the framework for NiS2.
To determine the valence states of Mn and Ni, X-ray photoelectron spectroscopy (XPS) of 4 h was obtained (Fig. 3). The two characteristic peaks of Mn were clearly detected at 642.1 eV and 654.0 eV, with a standard splitting of 11.9 eV (Fig. 3a). They are assigned to the 2p of Mn, indicating the existence of Mn4+.38 The Ni 2p spectrum exhibits two narrow and symmetric peaks at 855.9 eV and 873.6 eV with satellite peaks, corresponding to the binding energy for lattice Ni2+.39 In addition, scanning transmission electron microscopy (STEM) elemental mapping analysis unambiguously shows the presence of a heterostructure for MnO2@NiS2/Ni(OH)2. The structure and composition of this heterostructured MnO2@NiS2/Ni(OH)2 of 4 h are further characterized by high-angle annular dark-field scanning TEM-energy dispersive X-ray spectroscopy (HAADF-STEM-EDX) in Fig. 4. The element mappings clearly indicate that manganese element is only distributed in the middle part as a core. While nickel and sulfur locate homogeneously outside as a shell, constructing a heterostructure. No traditional permeation between Mn and Ni is observed.
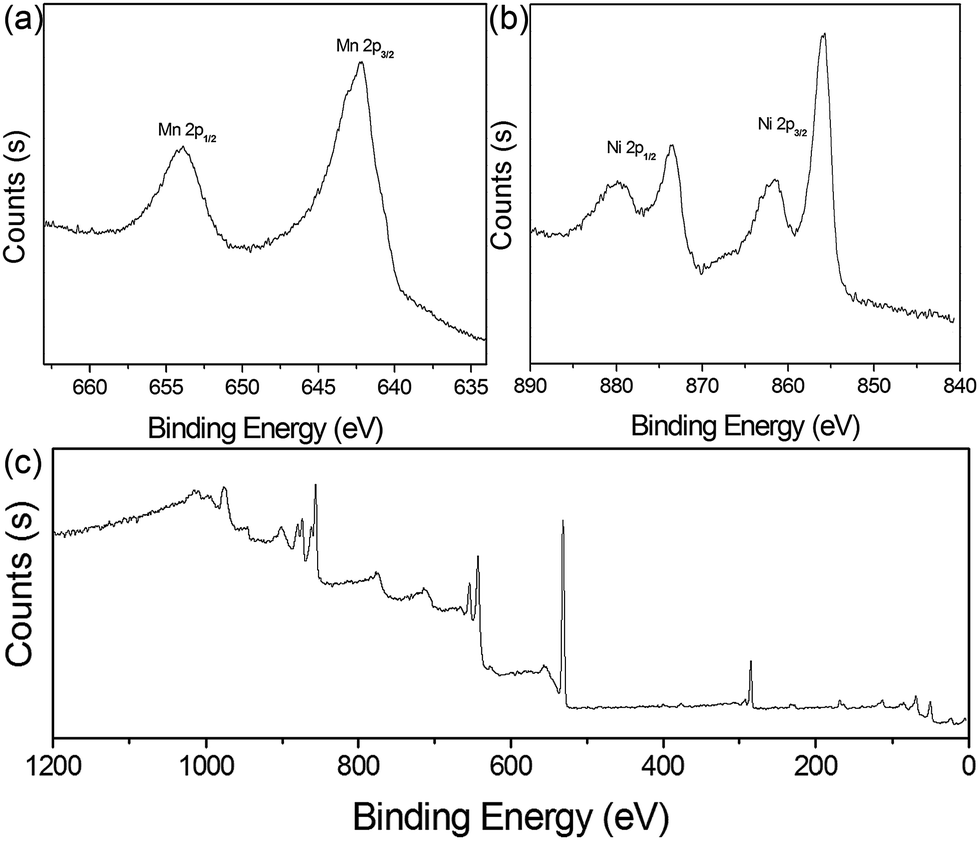 | ||
| Fig. 3 (a) XPS Mn 2p, (b) Ni 2p, and (c) the survey spectra of MnO2@NiS2/Ni(OH)2 heterostructure. | ||
 | ||
| Fig. 4 HAADF-STEM-EDX images of heterostructured MnO2@NiS2/Ni(OH)2 and the relevant mapping images of Mn, Ni and S elements. | ||
By simply adjusting the reaction time, morphologies of the products can be easily changed. When reacted for 4 h, NiS2/Ni(OH)2 sheets were detected on the surface of MnO2 nanosticks (Fig. 5a and d), forming a heterostructure. When reacted for less than 4 h, the NiS2/Ni(OH)2 sheets were discontinuous with irregular distribution. The lower content of NiS2/Ni(OH)2 and incomplete heterostructure might limit the full enhancement of the electrochemical property. When reacted for 12 h, the NiS2/Ni(OH)2 sheets covered almost all the surface of MnO2 nanosticks (Fig. 5b and e) and thus maximized the SSA of the heterostructured material. When reacted for 24 h, NiS2/Ni(OH)2 sheets grew further and the newly formed NiS2/Ni(OH)2 sheets attached onto the surface of MnO2 nanosticks in an disordered fashion (Fig. 5c and f), with much more NiS2/Ni(OH)2 attached. The detailed SEM images of materials prepared at different reaction times are given in Fig. S1.†
 | ||
| Fig. 5 SEM and TEM images of MnO2@NiS2/Ni(OH)2 heterostructure materials synthesized with (a and d) 4 h, (b and e) 12 h, and (c and f) 24 h. | ||
Brunauer–Emmett–Teller (BET) surface areas of the synthesized materials were measured. The N2 adsorption–desorption isotherms are shown in Fig. S2.† The SSA of the heterostructured materials prepared with reaction times of 4 h, 12 h, and 24 h were 22.8 m2 g−1, 30.4 m2 g−1, and 24.9 m2 g−1, respectively. The materials prepared with the treatment time of 12 h possessed the largest SSA. In comparison, materials with shorter or longer reaction time showed smaller SSA. When treated for a short reaction time, the incomplete reaction resulted in smaller SSA. However, too long reaction time produced too much NiS2/Ni(OH)2 deposit, which reduced the SSA.
A quantitative analysis of the heterostructured materials was performed by energy-dispersive X-ray spectroscopy (EDX) (Fig. S3†). The contents of Mn, Ni, along with S, are given in Table 2, which were normalized with the content of manganese. Throughout the growth process from 4 h to 24 h, Ni(OH)2 was grown in a constant state, while NiS2 decrease at first and increase later. Through the analysis of sulfur element content, the change of sulfur content reveals a unique process for the formation of heterostructure. At the initial stage, both NiS2 and Ni(OH)2 generated at the same time judging from the XRD pattern of 4 h product. With the reaction time continuing, H2O reacted with NiS2 to form Ni(OH)2, decreasing the sulfur content. It is worth to mention that, the whole reaction system is inclosed. With prolonging the reaction to 24 h, the abundant H2S underwent a kinetically-favored reaction with nickel, thus forming NiS2 again, resulting an increase of sulfur content. In a word, the generation of Ni(OH)2 are throughout the whole reaction process, and giving an increase nanosheets in SEM and TEM imagines. Along with this, a transformation from NiS2 to Ni(OH)2, and back to NiS2 occurred, showing a variation of content for sulfur. The sulfur content reflected the changing contents of NiS2 and Ni(OH)2 in the composite of MnO2@NiS2/Ni(OH)2. The corresponding XRD patterns are given in Fig. S4.† Clearly, the intensities of NiS2 diffraction peaks marked here varied significantly, especially at 31.5 degree. This indicates the changing amount of NiS2 in this heterostructure, and in agreement with the results of EDX measurements in Table 2.
| Elements | Mn | Ni | S |
|---|---|---|---|
| Contents/4h | 1.00 | 0.15 | 0.35 |
| 12h | 1.00 | 0.22 | 0.02 |
| 24h | 1.00 | 1.34 | 0.16 |
The electrochemical behavior of heterostructured materials prepared with different reaction times were studied with cyclic voltammetry (CV) and galvanostatic charging–discharging techniques. A three-electrode system was employed, with a working electrode, a Hg/HgO electrode as the reference electrode, and a Pt electrode as the counter electrode. The CV curves with a potential range from 0 V to 0.5 V at a scan rate of 100 mV s−1 are presented in Fig. 6a, and the relevant galvanostatic charge–discharge curves are presented in Fig. 6b. All the electrodes exhibited redox peaks, indicating that, in addition to the electric double-layer capacitance, the manganese oxides also exhibit faradaic capacitance resulting from the intercalation and de-intercalation of protons.40,41 The distinguishable redox peaks in Fig. 6a indicate the characteristics of faradaic pseudocapacitance.42,43 Namely, the capacitance depends not only on the charge storage on the electric double layer, but also the redox reactions between electrode and electrolyte solution.44 This feature ensures a much higher electric capacity than the traditional double-layer capacitor.
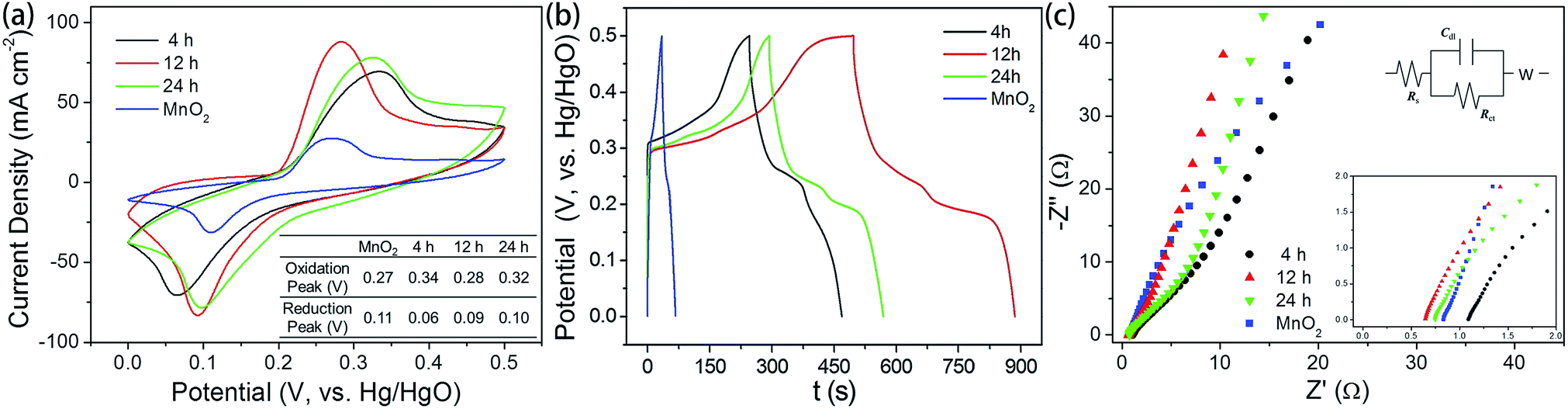 | ||
| Fig. 6 (a) Cyclic voltammetry curves for MnO2 and MnO2@NiS2/Ni(OH)2 heterostructure materials synthesized with different time. (b) Galvanostatic charge–discharge curves of the corresponding electrode materials. (c) Nyquist plots at the range of 0.1 Hz to 100 kHz and the equivalent circuit. | ||
Apparently, a pair of redox peaks is observed for the MnO2@NiS2/Ni(OH)2 heterostructure material electrode, which originates from faradaic reactions related to Ni–O/Ni–O–OH associated with anions OH−1. And the peaks are symmetric, revealing a reversible electrochemical reaction between electrode and the alkaline solution. Remarkably, a similar CV shape is still found for the MnO2 nanosticks, and the 1D MnO2 nanosticks are also efficient for the transfer of electrons. It is obvious that MnO2@NiS2/Ni(OH)2 heterostructure electrode possess a larger area of CV curve, indicating a higher level of charge storage and performing a better electrochemical behaviour than native MnO2 nanosticks. Our measurement indicated the heterostructured MnO2@NiS2/Ni(OH)2 composites presented much higher redox currents, comparing with the native MnO2 nanosticks. This enhancement may be caused by the interface between MnO2 and NiS2/Ni(OH)2, which facilitates the separation and transportation of electrons. And the 1D MnO2 nanosticks offer a stable and efficient backbone for charge transport.45 Meanwhile, introduction of NiS2/Ni(OH)2 nanosheets onto the surface of MnO2 further increased the SSA. The symmetrically shape and larger area of the CV curve shows that the 12 h MnO2@NiS2/Ni(OH)2 heterostructure electrode has the best pseudo-capacitive behaviour, which is shown in Fig. 6a.
The charge–discharge behaviors of the heterostructured materials were measured with a potential ranging from 0 V and 0.5 V at a current density of 1 A g−1 (Fig. 6b). The nonlinear variation between potential and time indicated a typical pseudocapacitance behavior, resulting from the electrochemical redox reaction. This result is in good agreement with the results from the CV curves. A negligible asymmetry was observed in the charge–discharge curves, caused by the incompletely reversible redox reaction.46 As shown, the specific capacitances for heterostructure MnO2@NiS2/Ni(OH)2 were significantly increased comparing with the native MnO2. The products of 12 h reaction possessed the highest specific capacitance of 1010 F g−1, and that for 4 h and 24 h reaction products were 502 F g−1 and 612 F g−1, respectively. The specific capacitance was calculated using the following equation of C = IΔt/(mΔV), where C (F g−1) is the specific capacitance; I (mA) is the discharge current; Δt (s) is the discharge time; ΔV (V) is the potential change during discharge; and m (mg) is the mass of active material.47 The highest specific capacitance for the composite prepared with 12 h reaction time is in agreement with the results of SSA. Moreover, the product of 12 h contained the least amount of sulfur and thus the increased content of Ni(OH)2. Based on the mechanism of NiS2 reacting with alkaline solution, Ni(OH)2 could facilitate this reaction and enhance the capacitance. Therefore, both the optimal structure and composition determined the highest capacitance for the 12 h composite, as high as 1010 F g−1.
The Nyquist plots for the MnO2 and MnO2@NiS2/Ni(OH)2 are tested at the range of 0.1 Hz to 100 kHz and given in Fig. 6c, as well as the equivalent circuit. And for this experiment, Ni(OH)2 and NiS2 are generated simultaneously. Thus, no MnO2@NiS2 can be produced without Ni(OH)2. So all samples produced here are MnO2@NiS2/Ni(OH)2 with a heterostructure. The only difference for various reaction times is the content of sulfur. The Nyquist plot consisted of a linear slope in the low frequency region, while no semicircle in the high frequency region. The semicircle suggested a charge-transfer resistance Rct, resulted from the electrical conductivity of material. The negligible semicircle in high frequency region shows higher electron conductivity, and a lower charge-transfer resistance. And the linear slope represents a ion diffusion impedance during the electrode reaction. According to the electrochemical impedance spectroscopy (EIS) results (Fig. 6c), MnO2@NiS2/Ni(OH)2 almost have the same diffusion resistance W (nearly parallel to each other at the linear part corresponding to the low frequency range), but better diffusion properties than MnO2 nanosticks. The fact, 24 h sample has the lowest diffusion resistance, demonstrates that NiS2/Ni(OH)2 nanosheets are beneficial for electron transfer.
The electrochemical properties of composites prepared with 12 h of reaction were further studied at different current densities (Fig. S5a and b†). Calculated from the galvanostatic charge–discharge curves, the specific capacitances were much higher than 1010 F g−1 at a current density lower than 1 A g−1 (Fig. S5a†). When further increase the current density, up to 20 A g−1, the specific capacitances reduced gradually (Fig. S5b†). This may be caused by the insufficient redox reactions. When at a lower current density, there is enough time for electrolyte ions to diffuse into the probe to allow a sufficient interaction between electrode and electrolyte. But for a higher current density, there could be no sufficient time to allow this interaction. As a result, the capacitance decreased with the increase of current density.
Furthermore, the charge–discharge stability of the composite materials at 1 A g−1 is presented in Fig. 7a. The capacitance was still about 785 F g−1 after 3000 cycles of the charging and discharging process, retaining nearly 78% of the value. The capacitance stabilized after the first 300 cycles, and a negligible decrease was observed, indicating a high capacitance stability. And there was no ascending tendency at the initial stage, demonstrating that there was no activation occurred throughout the process. And the decrease may be improved with the two aspects. One is the SSA of the electrode material, which has been changed during the charge and discharge process because of the immersion of the electrolyte. This is caused by the change of inner microstructure. And it can be improved by changing the electrode making method or by using a milder electrolyte. On the other hand, there is a great relationship between the cyclic performance and the degree of crystallinity of materials. Good crystallinity of the material ensures a good cycle performance. According to the previous literature data, if using a carbon material such as graphene coated, the cycle performance of electrode material will be a qualitative promoted. However, this paper focuses on the synthesis of the heterogeneous structure of the electrode material with a novel structure, and which has good electrochemical properties. The EIS of MnO2@NiS2/Ni(OH)2 heterostructure before and after cycling is shown in Fig. 7b. After cycling, the slope of straight line in the low frequency region decreases, which shows that the diffusion resistance becomes larger. This may be due to the redox reaction occurred during the cycling, resulting in the dissolution of some compounds, as well as the change of micro morphology or pore structure on the electrode. And the SEM of MnO2@NiS2/Ni(OH)2 core–shell nanosheets after 3000 cycling are test, which is placed in Fig. 7d. We can observe that the morphology of the core–shell nanosheets arrays retained almost intact, which further demonstrated its relatively good stability.
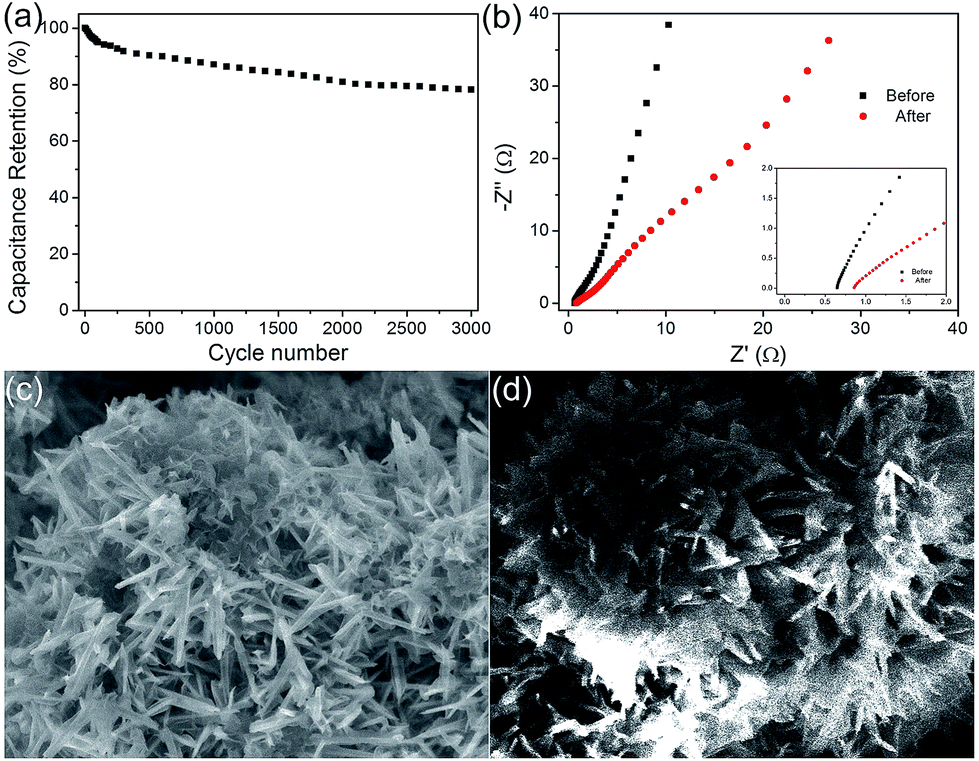 | ||
| Fig. 7 (a) Cycling performance of the 12 h MnO2@NiS2/Ni(OH)2 heterostructure electrode (3000 cycles). (b) Nyquist plots before and after cycling at the range of 0.1 Hz to 100 kHz. (c and d) SEM images of MnO2@NiS2/Ni(OH)2 heterostructure materials before and after cycling. | ||
4. Conclusions
In summary, heterostructured MnO2@NiS2/Ni(OH)2 was synthesized via a hydrothermal method. The reaction time was optimized to control the structure and sulfur content for desired electrochemical properties. The heterostructure exhibited high SSA for the Faraday energy storage. The highest capacitance reached 1010 F g−1, as revealed by galvanostatic charge–discharge measurement. The supercapacitive property was attributed to the specific heterostructure, where the 1D MnO2 nanosticks served as a stable and efficient backbone for charge transport, and the 2D nanosheets provided a high external surface for efficient contact with electrolytes. The MnO2@NiS2/Ni(OH)2 composites with high capacitance and specific structure could be used as supercapacitor materials in performance enhancement and device construction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21271082, 21301066 and 21371068), and the Postdoctoral Science Foundation of China (No. 801141080411).Notes and references
- J. R. Miller and P. Simon, Science, 2008, 321, 651–652 CrossRef CAS PubMed.
- H. Jiang, C. Z. Li, T. Sun and J. Ma, Chem. Commun., 2012, 48, 2606–2608 RSC.
- H. L. Wang, H. S. Casalongue, Y. Y. Liang and H. J. Dai, J. Am. Chem. Soc., 2010, 132, 7472–7477 CrossRef CAS PubMed.
- J. Jiang, Y. Y. Li, J. P. Liu, X. T. Huang, C. Z. Yuan and X. W. Lou, Adv. Mater., 2012, 24, 5166–5180 CrossRef CAS PubMed.
- Rusi and S. R. Majid, Electrochim. Acta, 2014, 138, 1–8 CrossRef CAS.
- H. J. Qiu, J. L. Kang, P. Liu, A. Hirata, T. Fujita and M. W. Chen, J. Power Sources, 2014, 247, 896–905 CrossRef CAS.
- T. C. Zhang, C. J. Kim, Y. W. Cheng, Y. W. Ma, H. B. Zhang and J. Liu, Nanoscale, 2015, 7, 3285–3291 RSC.
- C. F. Zhang, H. B. Wu, C. Z. Yuan, Z. P. Guo and X. W. Lou, Angew. Chem., 2012, 124, 9730–9733 CrossRef.
- Y. W. Cheng, H. B. Zhang, S. T. Lu, C. V. Varanasi and J. Liu, Nanoscale, 2013, 5, 1067–1073 RSC.
- L. Yu, G. Q. Zhang, C. Z. Yuan and X. W. Lou, Chem. Commun., 2013, 49, 137–139 RSC.
- J. Duay, S. A. Sherrill, Z. Gui, E. Gillette and S. B. Lee, ACS Nano, 2013, 7, 1200–1214 CrossRef CAS PubMed.
- L. Kou, Z. Liu, T. Q. Huang, B. N. Zheng, Z. Y. Tian, Z. S. Deng and C. Gao, Nanoscale, 2015, 7, 4080–4087 RSC.
- S. D. Min, C. J. Zhao, G. R. Chen, Z. M. Zhang and X. Z. Qian, Electrochim. Acta, 2014, 135, 336–344 CrossRef CAS.
- H. Chen, S. X. Zhou and L. M. Wu, ACS Appl. Mater. Interfaces, 2014, 6, 8621–8630 CAS.
- J. Y. Ji, L. L. Zhang, H. X. Ji, Y. Li, X. Zhao, X. Bai, X. B. Fan, F. B. Zhang and R. S. Ruoff, ACS Nano, 2013, 7, 6237–6243 CrossRef CAS PubMed.
- S. J. Peng, L. L. Li, H. B. Wu, S. Madhavi and X. W. Lou, Adv. Energy Mater., 2015, 5, 1401172 CrossRef.
- K. Cheng, F. Yang, K. Ye, Y. J. Li, S. N. Yang, J. L. Yin, G. L. Wang and D. X. Cao, J. Mater. Chem. A, 2013, 1, 14246–14252 CAS.
- Y. W. Cheng, H. B. Zhang, C. V. Varanasi and J. Liu, Energy Environ. Sci., 2013, 6, 3314–3321 CAS.
- D. P. Cai, D. D. Wang, B. Liu, L. L. Wang, Y. Liu, H. Li, Y. R. Wang, Q. H. Li and T. H. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 5050–5055 CAS.
- G. X. Gao, H. B. Wu, S. J. Ding, L. M. Liu and X. W. Lou, Small, 2015, 11, 804–808 CrossRef CAS PubMed.
- J. X. Li, M. Yang, J. P. Wei and Z. Zhou, Nanoscale, 2012, 4, 4498–4503 RSC.
- J. L. Zhang, H. D. Liu, P. Shi, Y. J. Li, L. H. Huang, W. J. Mai, S. Z. Tan and X. Cai, J. Power Sources, 2014, 267, 356–365 CrossRef CAS.
- X. L. Dong, Z. Y. Guo, Y. F. Song, M. Y. Hou, J. Q. Wang, Y. G. Wang and Y. Y. Xia, Adv. Funct. Mater., 2014, 24, 3405–3412 CrossRef CAS.
- J. Q. Sun, W. Y. Li, B. J. Zhang, G. Li, L. Jiang, Z. G. Chen, R. J. Zou and J. Q. Hu, Nano Energy, 2014, 4, 56–64 CrossRef CAS.
- H. Chen, L. F. Hu, Y. Yan, R. C. Che, M. Chen and L. M. Wu, Adv. Energy Mater., 2013, 3, 1636–1646 CrossRef CAS.
- D. Z. Kong, J. S. Luo, Y. L. Wang, W. N. Ren, T. Yu, Y. S. Luo, Y. P. Yang and C. W. Cheng, Adv. Funct. Mater., 2014, 24, 3815–3826 CrossRef CAS.
- X. Y. Liu, S. J. Shi, Q. Q. Xiong, L. Li, Y. J. Zhang, H. Tang, C. D. Gu, X. L. Wang and J. P. Tu, ACS Appl. Mater. Interfaces, 2013, 5, 8790–8795 CAS.
- G. Lee, C. V. Varanasi and J. Liu, Nanoscale, 2015, 7, 3181–3188 RSC.
- X. W. Ma, J. W. Liu, C. Y. Liang, X. W. Gong and R. C. Che, J. Mater. Chem. A, 2014, 2, 12692–12696 CAS.
- M. Yang, J. X. Li, H. H. Li, L. W. Su, J. P. Wei and Z. Zhou, Phys. Chem. Chem. Phys., 2012, 14, 11048–11052 RSC.
- P. Y. Tang, L. J. Han and L. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 10506–10515 CAS.
- L. Gu, Y. W. Wang, R. Lu, W. Wang, X. S. Peng and J. Sha, J. Power Sources, 2015, 273, 479–485 CrossRef CAS.
- G. Lee, Y. W. Cheng, C. V. Varanasi and J. Liu, J. Phys. Chem. C, 2014, 118, 2281–2286 CAS.
- L. H. Du, P. H. Yang, X. Yu, P. Y. Liu, J. H. Song and W. J. Mai, J. Mater. Chem. A, 2014, 2, 17561–17567 CAS.
- S. J. Peng, L. L. Li, H. T. Tan, R. Cai, W. H. Shi, C. C. Li, S. G. Mhaisalkar, M. Srinivasan, S. Ramakrishna and Q. Y. Yan, Adv. Funct. Mater., 2014, 24, 2155–2162 CrossRef CAS.
- J. Liu, R. Younesi, T. Gustafsson, K. Edstrom and J. F. Zhu, Nano Energy, 2014, 10, 19–27 CrossRef CAS.
- D. B. Yu, J. F. Yao, L. Qiu, Y. F. Wang, X. Y. Zhang, Y. Feng and H. T. Wang, J. Mater. Chem. A, 2014, 2, 8465–8471 CAS.
- Z. P. Sun, S. Firdoz, E. Y. X. Yap, L. Li and X. M. Lu, Nanoscale, 2013, 5, 4379–4387 RSC.
- X. H. Xia, J. P. Tu, Y. Q. Zhang, Y. J. Mai, X. L. Wang, C. D. Gu and X. B. Zhao, J. Phys. Chem. C, 2011, 115, 22662–22668 CAS.
- W. Song, G. J. Shao, G. L. Wang, Z. P. Ma, S. Liu, J. J. Song and C. X. Wang, J. Solid State Electrochem., 2014, 18, 3173–3180 CrossRef CAS.
- M. Yang, Y. R. Zhong, X. L. Zhou, J. J. Ren, L. W. Su, J. P. Wei and Z. Zhou, J. Mater. Chem. A, 2014, 2, 12519–12525 CAS.
- W. Tian, X. Wang, C. Y. Zhi, T. Y. Zhai, D. Q. Liu, C. Zhang, D. Golberg and Y. Bando, Nano Energy, 2013, 2, 754–763 CrossRef CAS.
- L. C. Wu, Y. J. Chen, M. L. Mao, Q. H. Li and M. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 5168–5174 CAS.
- J. P. Wang, S. L. Wang, Z. C. Huang and Y. M. Yu, J. Mater. Chem. A, 2014, 2, 17595–17601 CAS.
- H. Y. Wang, F. X. Xiao, L. Yu, B. Liu and X. W. Lou, Small, 2014, 10, 3181–3186 CrossRef CAS PubMed.
- G. X. Zhu, C. Y. Xi, M. Q. Shen, C. L. Bao and J. Zhu, ACS Appl. Mater. Interfaces, 2014, 6, 17208–17214 CAS.
- Y. Ji, X. Y. Liu, W. Liu, Y. Wang, H. D. Zhang, M. Yang, X. F. Wang, X. D. Zhao and S. H. Feng, RSC Adv., 2014, 4, 50220–50225 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed characterization of heterostructural MnO2@NiS2/Ni(OH)2. See DOI: 10.1039/c7ra06569h |
| This journal is © The Royal Society of Chemistry 2017 |