
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ternary Eu(III) and Tb(III) β-diketonate complexes containing chalcones: photophysical studies and biological outlook†
Zafar Abbas,
Srikanth Dasari and
Ashis K. Patra *
*
Department of Chemistry, Indian Institute of Technology Kanpur, Kanpur 208016, Uttar Pradesh, India. E-mail: akpatra@iitk.ac.in
First published on 13th September 2017
Abstract
Four ternary lanthanide(III) complexes, namely [Eu(Anpp)(TTA)3] (1), [Tb(Anpp)(TTA)3] (2), [Eu(Pypp)(TTA)3] (3) and [Tb(Pypp)(TTA)3] (4), [TTA = 1,1,1-trifluoro-3-(2-theonyl)acetone, Anpp = 3-(anthracen-9-yl)-1-(pyridine-2-yl)prop-2-en-1-one, Pypp = 3-(pyren-1-yl)-1-(pyridine-2-yl)prop-2-en-1-one] were designed, synthesized and characterized by various spectroscopic techniques like FT-IR, 1H-NMR, ESI-MS, UV-Vis and spectral emission studies. [Eu(Pypp)(TTA)3] (3) was structurally characterized, showing an eight coordinated {EuN1O7} square antiprismatic geometry with one N,O-donor chalcone and three O,O-donor β-diketonate ligands. Detailed photophysical properties like excited state lifetime (τ) and hydration number (q) were determined. The presence of three TTA ligands inhibits non-radiative deactivation with lifetimes in H2O in the range of 0.306–0.445 ms. The calculated hydration number (q) values (0.37–0.63) from the excited state lifetime measurements show the absence of any bound H2O molecules in the coordinatively saturated complexes (q < 1). Complexes 1–4 were studied for their biological interactions with DNA and protein and their DNA photocleavage activity was investigated. The binding constants with CT-DNA (Kb = 1.15–8.8 × 105 M−1) and HSA (KHSA = 6.5–8.1 × 105 M−1) of complexes 1–4 show their significant affinity towards these biological targets. Incorporation of chalcone ligands containing anthracene or pyrene as a photosensitizing moiety in these complexes were effective in generating reactive oxygen species (ROS). Complexes 1–4 display moderate photocleavage of supercoiled (SC)-DNA to its nicked circular (NC) form on exposure to UV-A light of 365 nm through the generation of singlet oxygen (1O2) and hydroxyl radicals (˙OH) under physiological conditions.
Introduction
Lanthanide complexes have been widely explored as sensors, MRI contrast agents, responsive optical probes, luminescent MOFs, display materials, chemo and radio therapeutic agents, and cellular imaging agents owing to their intricate optical properties and high redox stability of the trivalent Ln3+ state.1–11 Lanthanides are used abundantly in various industries like optical, lighting, display, laser, ceramics, energy or non-invasive imaging technologies. The very fascinating optical features of the lanthanide ions originate from their [Xe] 4fn (n = 0–14) electronic configuration because of shielded f-orbitals by the filled 5s and 5p orbitals, giving rise to various electronic levels of defined energy which are hardly sensitive to the surrounding chemical environment. The f–f transitions are symmetry forbidden and have very weak absorption (ε ∼ 0.1–1 M−1 cm−1), therefore resulting in very faint luminescence, which can be enhanced by attachment of a suitably chosen organic chromophore to excite the lanthanide by the so called ‘antenna effect’, which provides a large Stokes shift and avoids photobleaching.4,5 Such indirect sensitization in fact relaxes the strict selection rules that otherwise limit the f → f transitions in Ln3+ and results in significant luminescence enhancement. Currently polyaminocarboxylate gadolinium complexes, namely gadopentatic acid (Magnevist®) and gadoteric acid (Artirem®) are in clinical use as magnetic resonance imaging (MRI) contrast agents, showing promising role of the lanthanides in biomedical imaging.12 Photodynamic therapy is now being widely appreciated as a promising noninvasive methodology for curb and cure of cancer which relies on the generation of cytotoxic reactive oxygen species (mainly 1O2) by irradiating some nontoxic photosensitizers at a specific wavelength, that causes cell death in the region of irradiation.13–15 Photofrin®, the first clinically accepted PDT agent, is an oligomeric mixture of natural hematoporphyrin, generates reactive oxygen species (1O2) in a type II energy transfer pathway at 633 nm.14–16 Weak absorbance in phototherapeutic window, prolonged skin sensitivity and hepatotoxicity are the major drawbacks with porphyrin-based PDT agents.17,18 Although there are several transition metal complexes in the literature showing remarkable photocytotoxicity and DNA photocleavage activity, very few are reported with lanthanides.19–24 Lanthanide(III) complexes with a range of coordination numbers ranging from 7–12 and unusual optical properties will be niche in the development of efficient photocytotoxic agents and the excellent redox stability of Ln(III) is also beneficial for their use in biological media against various reducing agents in biology like thiols and ascorbates. Some of the Ln(III) ions have the advantage of bright visible light luminescence over other lanthanides and transition metals and they have been widely explored as sensors in physiological conditions or in bioassays.4–7 Currently we are exploring luminescent Ln(III) complexes for their interaction with biological targets, photoinduced DNA cleavage activity and cellular imaging agents.24We describe here, four lanthanide complexes of Eu(III) and Tb(III) viz. [Eu(Anpp)(TTA)3] (1), [Tb(Anpp)(TTA)3] (2), [Eu(Pypp)(TTA)3] (3) and [Tb(Pypp)(TTA)3] (4) [TTA = 1,1,1-trifluoro-3-(2-theonyl)acetone, Anpp = 3-(anthracen-9-yl)-1-(pyridine-2-yl)prop-2-en-1-one, Pypp = 3-(pyren-1-yl)-1-(pyridine-2-yl)prop-2-en-1-one], were synthesized, characterized and studied for their photophysical properties and interaction with DNA and serum proteins along with photoinduced DNA cleavage activity. The complexes containing three anionic β-diketonate ligand and one bidentate chalcone conjugated to a flurophore have been designed as coordinatively saturated systems to avoid non-radiative transitions from the Ln(III) excited states through vibrational energy transfer (VET) to O–H, N–H and C–H oscillators (Scheme 1).2,4,7,25 The β-diketonate ligands having various conjugated aromatic planar groups are established as efficient sensitizers in developing visible light emitting lanthanide (Eu3+, Sm3+ and Tb3+) complexes. Use of polyfluorinated β-diketonates have the advantage over non-fluorinated ones because of the vibrational energy of the C–H oscillator (ν = 2800–3000 cm−1) is higher than that of C–F oscillator (ν = 1120–1350 cm−1). Fluorination induced inductive effect of highly electron withdrawing –CF3 group decreases the non-radiative loss of Ln(III) excited state energy compared to the high energy C–H oscillators, resulting in enhanced luminescence intensity and rigidity of the complexes due to tight bond between ligand and Ln(III).26,27 The choice of two different chalcones as secondary ligands having α,β-unsaturated ketone as central core conjugated to anthracene or pyrene as light harvesting photosensitizing moiety. Chalcones show various biological effects like anti-inflammatory, anticancer, antimalarial, antifungal, anti-infective activity and some other features like sensing to non-linear optics.28–33 Moreover these moieties can also act as DNA intercalators and photosensitizers. We have evaluated biological interaction of complexes 1–4 with nucleic acid and protein. They show good binding affinity towards ds-DNA through the intercalation of planar aromatic pyrene and anthracene moieties between the DNA base pairs. HSA binding affinity was determined by quenching in emission of Trp-214 residue. They exhibit photoinduced DNA photocleavage activity through the generation of singlet oxygen (1O2) in Type-II pathways and ˙OH involving photoredox pathway at physiological conditions.
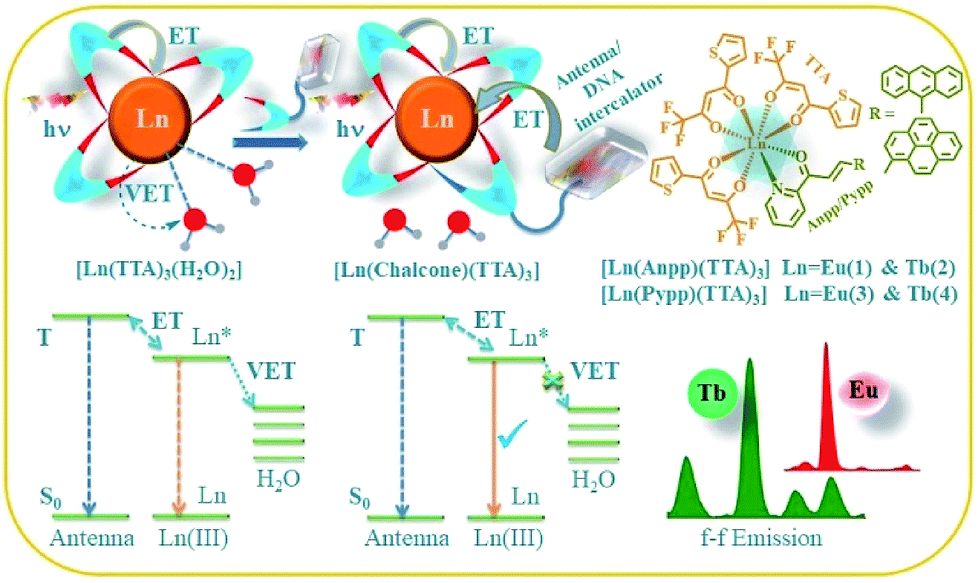 | ||
| Scheme 1 Schematic design principle for complexes 1–4 showing deactivation of Ln* in [Ln(TTA)3(H2O)2] via nonradiative vibrational energy transfer (VET) through lanthanide coordinated H2O (left); incorporation of bidentate chalcones inhibit this quenching with increased emission intensity (middle) and representation of [Ln(chalcone)(TTA)3] (1–4) with luminescence from of Eu3+ and Tb3+ (right). | ||
Results and discussion
Synthesis and general aspects
Herein, we have synthesized two chalcone derivatives namely Anpp [(E)-3-(anthracen-9-yl)-1-(pyridin-2-yl)prop-2-en-1-one] and Pypp [(E)-3-(pyren-1-yl)-1-(pyridin-2-yl)prop-2-en-1-one] by Claisen–Schmidt condensation reaction and characterized by 1H-NMR, UV-Vis and ESI-MS studies.34,35 Four TTA-based lanthanide complexes containing these chalcones were then synthesized by reacting a solution of Ln(TTA)3·2H2O in THF with chalcones (Scheme 2). Complexes were isolated in good yields and characterized by FT-IR, ESI-MS, UV-Vis and emission spectroscopy and through structural determination of complex 3 by single crystal X-ray crystallography. FT-IR spectra shows a lowering in frequency in the stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CC bonds from 1676 and 1632 cm−1 in the free HTTA to around 1600 and 1535 cm−1 in the complexes 1–4 while the bands at 490 cm−1 correspond to νLn–O stretching vibrations and at 519 cm−1 to Ln–N stretching vibrations.26b,26c,36,37 ESI-MS spectra of the complexes 1, 2 and 4 assignable to [M-TTA]+ and for complex 3 to [M − Na]+ in DMF with matching isotopic distribution profiles indicating the formation of complexes 1–4 (Fig. S1, ESI†).
O and CC bonds from 1676 and 1632 cm−1 in the free HTTA to around 1600 and 1535 cm−1 in the complexes 1–4 while the bands at 490 cm−1 correspond to νLn–O stretching vibrations and at 519 cm−1 to Ln–N stretching vibrations.26b,26c,36,37 ESI-MS spectra of the complexes 1, 2 and 4 assignable to [M-TTA]+ and for complex 3 to [M − Na]+ in DMF with matching isotopic distribution profiles indicating the formation of complexes 1–4 (Fig. S1, ESI†).
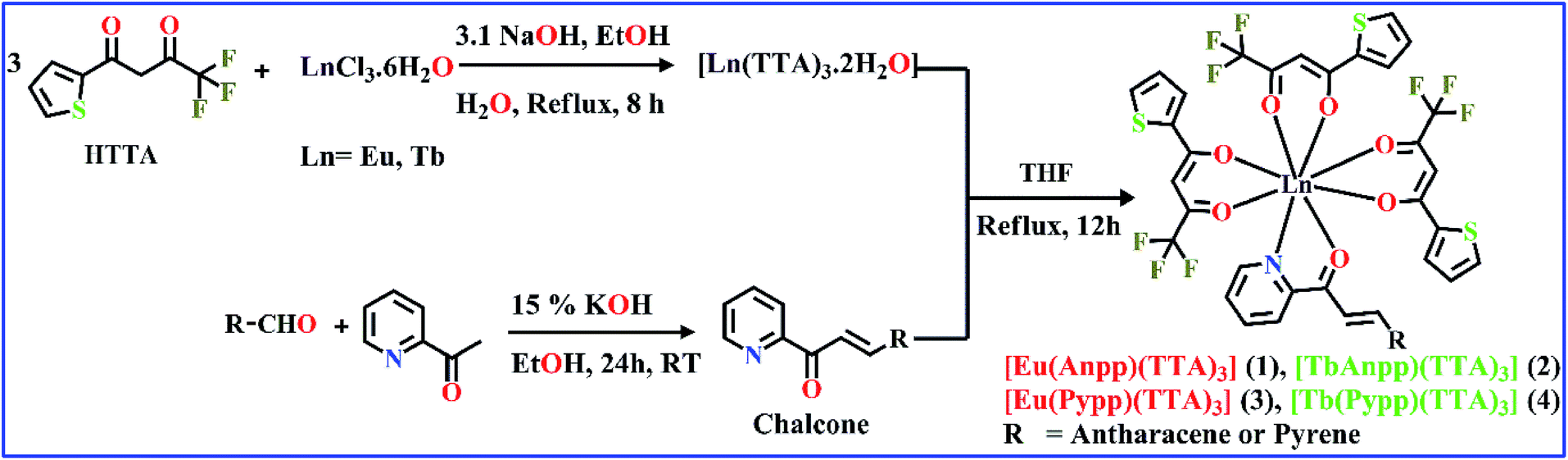 | ||
| Scheme 2 Synthetic route for the chalcone ligands and [Ln(chalcone)(TTA)3] (1–4) complexes. | ||
The UV-Vis absorption spectra of the complexes show the ligand-centered absorption band corresponding to π–π* transitions in the range of 270 to 480 nm. Complexes 1 and 2 containing Anpp ligand displayed close resemblance in their absorption spectra with λmax at 270 nm and 346 nm originated from the Anpp and TTA ligands. On the contrary, complexes 3 and 4 with Pypp ligands shows overlapping spectra with λmax at 273 nm, 346 nm and 440 nm originated from the Pypp and TTA ligands (Fig. 1(a), S2 and S3 in ESI†). Close resemblance in the UV-Vis spectra of 1–4 with identical ligands is independent of Ln(III), also suggests minimal or no perturbation of the electronic levels of Ln(III). Absorption spectra of the complexes 1–4 in DMF recorded for a period of 4 h at 298 K shows no appreciable change in the band intensity suggesting their stability in solution state (Fig. S4, ESI†).
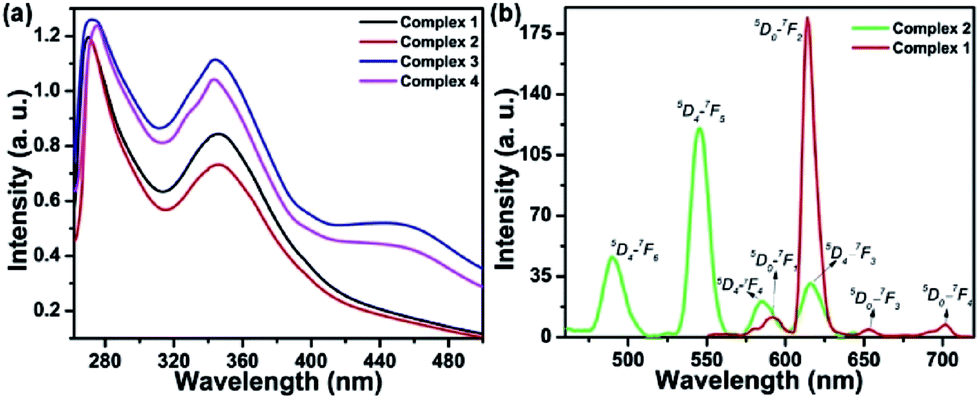 | ||
| Fig. 1 (a) UV-visible absorption spectral traces of complexes 1–4 ([1] and [2] = 20 μM, [3] and [4] = 15 μM) in DMF at 298 K. (b) Time-delayed luminescence spectra for complexes 1 and 2 at 298 K. [delay time = 0.1 ms, gate time = 0.1 ms, λex = 340 nm, slit width = 10 nm, [1] and [2] = 20 μM]. | ||
Photophysical properties
Time-delayed luminescence spectra of the complexes shows emission bands corresponds to 5D0 → 7FJ (J = 0–4) and 5D4 → 7FJ (J = 6–3) transitions for Eu3+ and Tb3+ respectively (Fig. 1(b)). Excited state lifetime of the complexes in H2O were found in the range of 0.306 ms to 0.445 ms are depicted in Table 1 (Fig. S5, ESI†).| Complexes | IRa (cm−1) νCO |
τDMFb (μs) | τH2Oc (μs) | τD2Od (μs) | qe |
|---|---|---|---|---|---|
| a IR vibrational stretching frequency in KBr phase.b Excited state lifetime (τ ± 15%) measured in DMF.c Excited state lifetime (τ ± 15%) measured in H2O.d Excited state lifetime (τ ± 15%) measured in D2O.e Hydration number (q ± 10%) determined using modified Horrock's equation. | |||||
| Complex 1 | 1601 (s) | 464 | 445 | 633 | 0.50 |
| Complex 2 | 1603 (s) | 420 | 395 | 426 | 0.62 |
| Complex 3 | 1602 (s) | 457 | 390 | 560 | 0.63 |
| Complex 4 | 1602 (s) | 391 | 306 | 319 | 0.37 |
The lower τ value for complex 4 possibly due to the smaller energy gap between the 5D4 state of Tb3+ and the triplet excited state of TTA moiety, that may result in thermal back energy transfer processes and consequently an ineffective radiative transition from Tb3+ 5D4 excited state.38 Solution state speciation of complexes in aqueous media was established by recording their excited state lifetimes in H2O and D2O. Their hydration number (q) was calculated using modified Horrock's equation that gave values less than one, confirming the absence of H2O molecule in the complexes summarised in Table 1 (Fig. S6, ESI†).39 Absence of H2O molecule at Ln(III) center arrest the nonradiative vibrational energy transfer (VET) from Ln(III) to O–H oscillators of Ln(III) bound H2O, thus results in longer lifetimes.
Crystal structure determination
[Eu(Pypp)(TTA)3] (3) have been structurally characterized by single crystal X-ray diffraction studies. It was crystallized in triclinic crystal system with space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) as mononuclear discrete molecule having two molecules in the unit cell. The ORTEP view of complex 3 along with coordination polyhedron of lanthanide core have been shown in Fig. 2. It has an eight coordinated {EuN1O7} coordination geometry around the europium centre, two of which originates from a bidentate N,O-donor neutral chalcone ligand and the other six from three monoanionic bidentate O,O donor TTA ligands, that can be best described as distorted square antiprismatic geometry. Here each square-plane is constituted from N1, O1, O6, O7 and O2, O3, O4, O5 atoms respectively with a dihedral angle of 4.75° between them (Fig. 2(b)). At the center of the square antiprism lies Eu(III) with a distance of 1.251 Å to the oxygen containing plane and 1.440 Å to the opposite plane for complex 3. Eu–N(1) and Eu–O(1) bond lengths of the neutral chalcone were 2.608(6) and 2.453(5) Å respectively. On contrary, Eu–O bond distances from the anionic TTA was found in the range from 2.349(5) to 2.375(6) Å. The ∠N–Eu–O bite angle for the chalcone ligand was 62.43(18)° while ∠O–Eu–O bite angle for TTA ligands were ranging from 71.02(19)–72.15(19)°. All the selected major bond lengths and bond angles have been listed in the Table S1, ESI.† Unit cell packing diagram have been given in Fig. S7 in ESI.†
as mononuclear discrete molecule having two molecules in the unit cell. The ORTEP view of complex 3 along with coordination polyhedron of lanthanide core have been shown in Fig. 2. It has an eight coordinated {EuN1O7} coordination geometry around the europium centre, two of which originates from a bidentate N,O-donor neutral chalcone ligand and the other six from three monoanionic bidentate O,O donor TTA ligands, that can be best described as distorted square antiprismatic geometry. Here each square-plane is constituted from N1, O1, O6, O7 and O2, O3, O4, O5 atoms respectively with a dihedral angle of 4.75° between them (Fig. 2(b)). At the center of the square antiprism lies Eu(III) with a distance of 1.251 Å to the oxygen containing plane and 1.440 Å to the opposite plane for complex 3. Eu–N(1) and Eu–O(1) bond lengths of the neutral chalcone were 2.608(6) and 2.453(5) Å respectively. On contrary, Eu–O bond distances from the anionic TTA was found in the range from 2.349(5) to 2.375(6) Å. The ∠N–Eu–O bite angle for the chalcone ligand was 62.43(18)° while ∠O–Eu–O bite angle for TTA ligands were ranging from 71.02(19)–72.15(19)°. All the selected major bond lengths and bond angles have been listed in the Table S1, ESI.† Unit cell packing diagram have been given in Fig. S7 in ESI.†
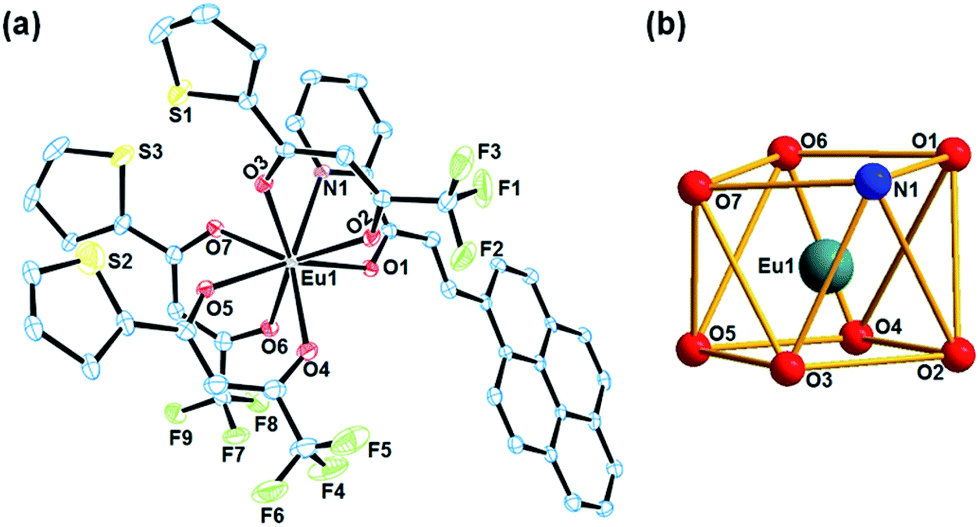 | ||
| Fig. 2 (a) ORTEP view of [Eu(Pypp)(TTA)3] (3) with thermal ellipsoids drawn at the 50% probability level. (b) The {EuN1O7} coordination polyhedra of the Eu(III) core showing distorted square antiprism geometry. | ||
DNA binding studies
For the development of potential chemotherapeutic agents, DNA has been the primary biological target of many pharmacologically active drugs. These drug molecules mainly bind with the target either through covalent or noncovalent interactions. There have been two major classes of noncovalent DNA binding agents: intercalators and groove binders. Intercalation involves the insertion of planar moiety between the DNA base pairs, resulting a decrease in DNA helical twist and its elongation whereas groove binding does not affect large conformational changes which fit into the major or minor grooves of DNA.40,41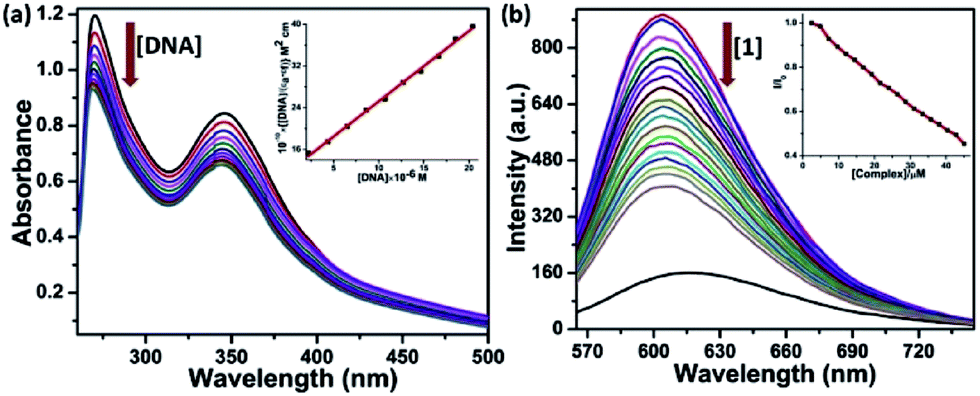 | ||
| Fig. 3 (a) UV-visible traces for [Eu(Anpp)(TTA)3] (20 μM) in 5 mM Tris buffer (pH 7.2) with increasing [CT-DNA] at 298 K; inset: [DNA]/Δεaf versus [DNA] plot for complex 1. (b) Emission spectral traces for EB-bound CT-DNA with increasing concentration for complex 1 in 5 mM Tris buffer (pH 7.2) 298 K; λex = 546 nm, λem = 603 nm, [DNA] = 212 μM, [EB] = 12 μM; inset: a plot of I/I0 vs. [complex 1]. | ||
| Complexes | Kb/M−1a (×105) | Kapp/M−1b (×107) | KHSA/M−1c (×105) | nd, K/M−1e (×105) |
|---|---|---|---|---|
| a Kb: intrinsic DNA binding constant.b Kapp: apparent DNA binding constant.c KHSA: Stern–Volmer quenching constant of HSA emission.d n: number of binding sites in protein.e K: HSA binding constant. | ||||
| Complex 1 | 1.15 ± 0.03 | 3.56 | 7.3 ± 0.04 | 0.69, 2.5 |
| Complex 2 | 8.8 ± 0.07 | 3.77 | 6.5 ± 0.02 | 0.72, 3.6 |
| Complex 3 | 1.64 ± 0.11 | 3.59 | 8.1 ± 0.03 | 0.79, 9.3 |
| Complex 4 | 3.68 ± 0.10 | 3.65 | 7.1 ± 0.03 | 0.71, 3.5 |
Human serum albumin (HSA) binding studies
Serum albumin is the most abundant protein in human blood plasma (around 40 mg mL−1), has been among the most widely studied proteins. It has very crucial role as a transporter of a variety of fatty acids and capable of binding effectively with metabolites and drug molecules, therefore governing the distribution of such entities in plasma.44 The binding of complexes 1–4 was investigated using native tryptophan emission quenching. The emission of HSA is mainly because of the tryptophan residue (Trp-214) located in a hydrophobic environment. Fig. 4 shows the quenching of emission intensity along with hypsochromic shift upon gradual increasing concentration of the complexes at λex = 295 nm. The observed spectral changes reveals that the binding of complexes leads to a change in the secondary structure of the protein through various hydrophobic interaction, electrostatic interaction, collisional quenching and ground state complex formation.45 The dynamic quenching constant (KHSA) of the complexes were calculated from the slope of I0/I vs. [complex] linear plot using Stern–Volmer equation (eqn (3)).46 The values of binding constant (K) and the number of binding sites (n) was obtained from the linear log(I0/I − 1) vs. log[complex] plot using Scatchard equation (eqn (4)).47 The corresponding plots have been shown in Fig. 4 and S14–S16 in ESI† and values are listed in Table 2. Obtained values of binding constant are of the order ∼105 M−1 suggesting strong binding affinity of the complexes towards target protein.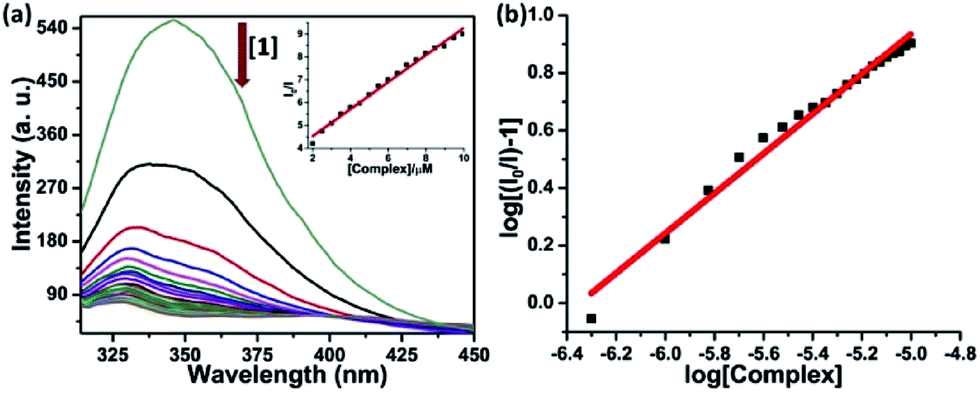 | ||
| Fig. 4 (a) The emission quenching of HSA on addition of complex 1 in 5 mM Tris buffer (pH 7.2) at 298 K; inset: a plot of I0/I versus [complex 1]; λex = 295 nm, λem = 345 nm, [HSA] = 5 μM. (b) Scatchard plot: log[(I0 – I)/I] vs. log[complex] for HSA in the presence of complex 1. | ||
Synchronous fluorescence studies
Synchronous fluorescence spectroscopy study provides crucial information in determining the molecular microenvironment of chromophoric groups in protein cavity. As stated by Miller, change of Δλ, which is the difference of emission and excitation wavelength (Δλ = λem − λex) gives the spectra of different nature. If Δλ = 60 nm, emission spectra will reflect the characteristics of tryptophan (Trp) residue and Δλ = 15 nm will suggest the characteristics of tyrosine (Tyr) residue in HSA protein.48 The synchronous spectra was recorded with increasing concentration of complexes 1–4 both at Δλ = 60 nm and 15 nm. The fluorescence spectra at Δλ = 60 nm in Fig. 5(a) and S17(a)–S19(a) in ESI† showing dramatic quenching of emission intensity by 82–87% without any significant changes in λem maxima. Similarly, the synchronous spectra at Δλ = 15 nm (Fig. 5(b) and S17(b)–S19(b) in ESI†) showing quenching of emission intensity by 35–40% with no apparent shift in emission maxima. It is quite obvious from these results that the complexes are affecting the microenvironments of both Tyr and Trp residues, but the effect was much pronounced in microenvironment around Trp.49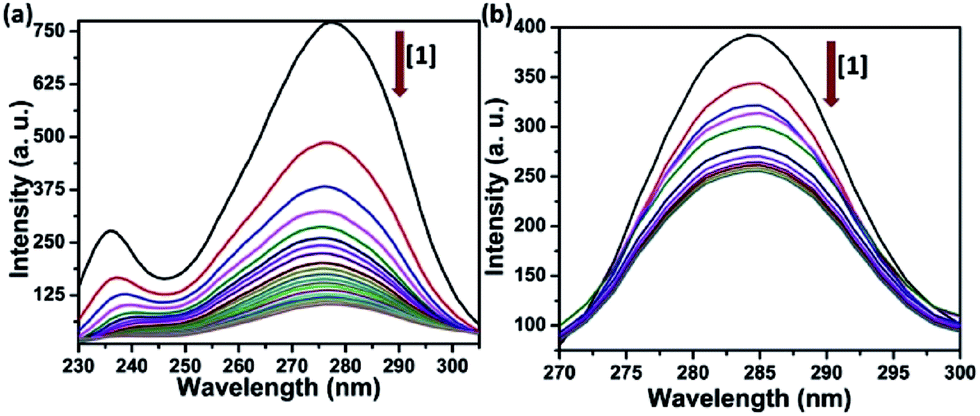 | ||
| Fig. 5 Synchronous emission spectra of HSA (5 μM) showing effect of increasing concentration of complex 1 (a) with Δλ = 60 nm and (b) with Δλ = 15 nm at 298 K in Tris–HCl buffer (pH = 7.2). | ||
Photoactivated DNA cleavage activity
The DNA cleavage activity by photoactivation for the complexes 1–4 was carried out in 5 mM Tris HCl/NaCl buffer (pH = 7.2) using supercoiled (SC) pUC19 DNA (30 μM, 0.2 μg) by exposing the sample to a radiation of 365 nm UV-A light by a low power monochromatic lamp (6 W). The control experiment with only SC-DNA did not displayed any noticeable photocleavage while the ligands HTTA, Anpp and Pypp were showing 6, 9 and ∼15% cleavage of SC-DNA (Fig. 6, lanes 1–4). However EuCl3·6H2O and TbCl3·6H2O were not showing any photocleavage (Fig. 6, lanes 5, 6, 11–14). Complexes 2 and 4 displayed better photocleavage capability compared to 1 and 3 at 40 μM, mainly due to the possible involvement of the pyrene ring. Treatment of SC-DNA with the complexes 1–4 in dark for 1.5 h did not show any apparent cleavage, rules out the possibility of any hydrolytic cleavage by Ln(III) complexes. | ||
| Fig. 6 Photocleavage of SC pUC19DNA (30 μM, 0.2 μg) with complexes 1–4 and controls (40 μM) in 50 mM Tris–HCl buffer (pH 7.2) at 37 °C for after 1.5 h exposure with UV-A light (λ = 365 nm, 6 W). L1: DNA control; L2: DNA + HTTA; L3: DNA + Anpp; L4: DNA + Pypp; L5: DNA + EuCl3·6H2O; L6: DNA + TbCl3·6H2O; L7: DNA + 1 (dark); L8: DNA + 2 (dark); L9: DNA + 3 (dark); L10: DNA + 4 (dark); L11: DNA + 1; L12: DNA + 2; L13: DNA + 3; L14: DNA + 4; L15: DNA + MG (methyl green); L16: DNA + MG + 1; L17: DNA + MG + 2; L18: DNA + MG + 3; L19: DNA + MG + 4. | ||
The groove binding nature of the complexes were also studied utilizing methyl green (MG), which is a major groove binder of SC-DNA and showing ∼10% nicking of SC-DNA. All the complexes bound with SC-DNA showed an inhibition of their DNA photocleavage activity to its nicked circular form in the presence of methyl green reveals the complexes 1–4 are the major groove binders (Fig. 6, lanes 15–19).23b
The mechanism of DNA photocleavage activity of the complexes 1–4 was carried out in order to gain some insight into the process of photocleavage using various additives like 1O2 quenchers (L-Histidine, NaN3), ˙OH scavengers (DMSO, KI, catalase).50–52 Photoinduced DNA cleavage reactions mainly involve two ROS-mediated pathways: either via Type-II pathway through 1O2 generation or via photoredox pathway that involves ˙OH generation. Addition of L-His and NaN3 to SC-DNA as 1O2 quenchers, displayed partial inhibition of the photocleavage activity. Photocleavage of DNA by the complexes through 1O2 was further confirmed by using D2O as solvent showing an enhancement of the cleavage because of the longer lifetime of 1O2 in D2O than in H2O.53 Addition of DMSO, KI and catalase as hydroxyl radical scavengers showed partial inhibition of photocleavage, suggesting the generation of ˙OH during the process. All the data shown in Fig. 7 and S22–S26 in ESI† confirms that photoinduced DNA cleavage here involves both Type-II and photoredox pathways and is consistent with earlier reported lanthanide complexes.23,24
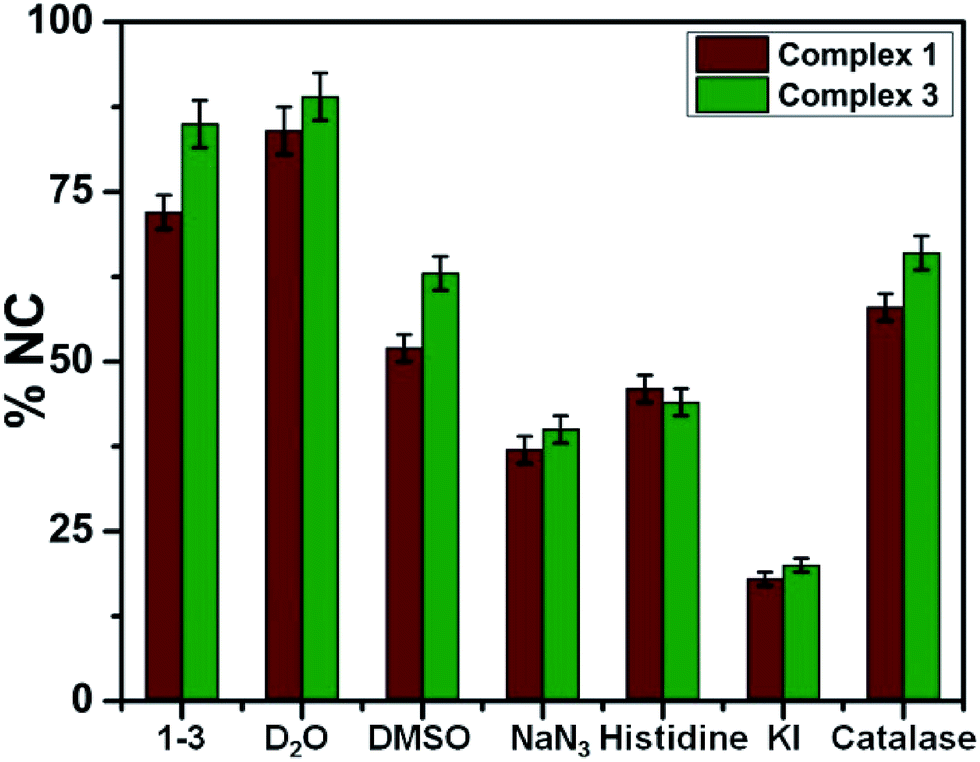 | ||
| Fig. 7 Cleavage of SC pUC19 DNA (30 μM, 0.2 μg) by complexes 1 and 3 (40 μM) (wine) and (olive) on photo-exposure at 365 nm (6 W) for 1.5 h in the presence of various additives in Tris–HCl/NaCl buffer. NaN3, 0.4 mM; KI, 0.4 mM; D2O, 16 μL; L-histidine, 0.4 mM; DMSO, 4 μL; catalase, 4 U. | ||
Conclusions
Four new Eu(III) and Tb(III) complexes containing chalcones as biologically diverse molecules and TTA as ancillary sensitizing ligands have been designed, synthesized and characterized. The structurally characterized complex 3 showing an eight coordinated square antiprismatic {EuN1O7} geometry. Strong luminescence from all the complexes suggest effective energy transfer from sensitizing ligands to Ln(III). Horrock's experiment showed the absence of any inner sphere H2O molecule (q < 1) attached to Ln(III) in solution. Complexes showed good binding affinity with DNA and HSA as evidenced form spectroscopic studies. Both the chalcone ligands containing pyrene or anthracene acts as efficient photosensitizers to generate ROS for DNA photocleavage. These complexes showed effective photoinduced DNA cleavage under UV-A light of 365 nm at 40 μM concentration through the generation of singlet oxygen (1O2) in a type-II pathway and ˙OH in a photoredox pathway. Further chalcone modifications with a series of antenna moieties for the development of highly emissive lanthanide chalcones complexes are in progress to study their various biological potential utilizing lanthanide luminescence.Experimental section
Materials
Chemicals like 9-anthraldehyde, pyrene-1-carbaldehyde and 2-acetyl pyridine were purchased from Alfa-Aesar. Theonyl trifluoro acetylacetone, europium chloride hexahydrate and terbium chloride hexahydrate were form Sigma Aldrich and used without any further purification. Solvents purchased from commercial sources were either of spectroscopic grade or purified by standard literature procedures.54 Ln(TTA)3·2H2O were prepared according to the literature reported procedure.55 Calf thymus (CT)-DNA, albumin from human serum (HSA), agarose (molecular biology grade), methyl green, catalase, gel loading solution and ethidium bromide were procured from Sigma (U.S.A.). Milli-Q water (18.2 MΩ) was used to prepare Tris-(hydroxymethyl)-aminomethane–HCl (Tris–HCl) buffer solution. Supercoiled (SC) pUC19 DNA was from Merck Millipore (India).General measurements
Perkin-Elmer Model 1320 FT-IR spectrometer was used for the infrared spectra with KBr disc in the operating range of 4000–400 cm−1. WATERS Q-TOF Premier mass spectrometer was used for recording electrospray ionization mass spectral (ESI-MS) measurements. 1H NMR spectra were recorded using JEOL-ECX 500 FT (400 MHz) for the characterization of chalcones at 298 K with reference to TMS. UV-Vis spectra were recorded using Varian V670 spectrophotometer at 298 K.Agilent Cary Eclipse fluorescence spectrophotometer was used for fluorescence and time resolved luminescence spectra measurements. Luminescence decay measurements were done with same instrument using a pulsed Xe-source at λex = 340 nm and emission at 616 nm for Eu(III) and 545 nm for Tb(III) complexes respectively. The decay curves were fitted using nonlinear least square method. Calculation for determination of number of H2O molecules coordinated to metal in the complexes 1–4 was done by the measurement of excited state lifetimes in H2O and D2O and thereby applying modified Horrocks equations for ternary europium and terbium complexes respectively.39
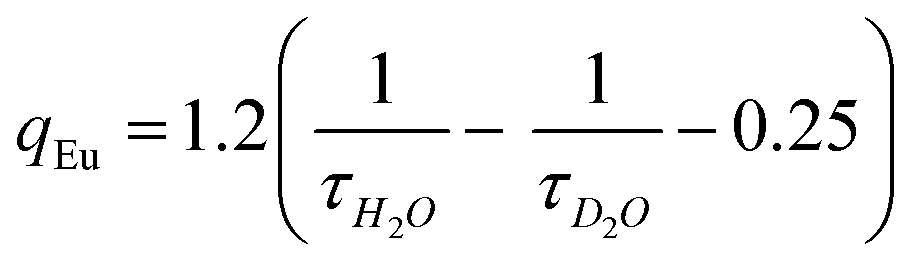
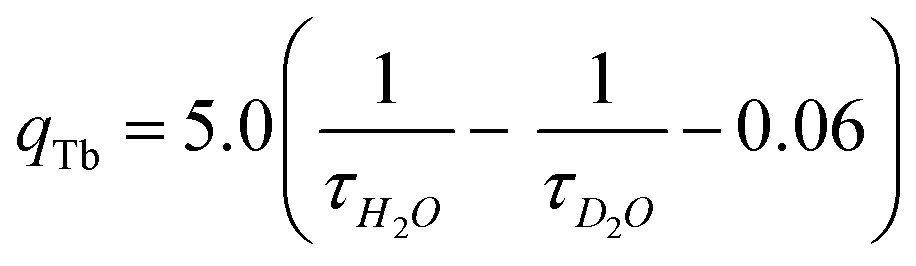
Synthesis and characterization
(E)-3-(Anthracen-9-yl)-1-(pyridin-2-yl)prop-2-en-1-one (Anpp). Yield: 1.24 g (78%), UV-Vis (DMF): λmax (ε/L mol−1 cm−1) = 267 (93
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800), 333 (10800), 351 (19600), 368 (27000), 388 (26900). FT-IR (in KBr/cm−1): 3051 (br, aromatic C–H), 1670 (s, CO), 1599 (s), 1582 (s), 1565 (s). 1H NMR (400 MHz, CDCl3, δ from TMS): 8.9 (d, 1H), 8.68 (1H, d), 8.48 (1H, s), 8.34 (2H, m), 8.29 (1H, d; 1H, s), 8.03 (2H, m), 7.93 (2H, td), 7.5 (4H, m). ESI-MS (m/z): [M + H]+ calcd for C22H16N1O1, 310.123; found, 310.125.
800), 333 (10800), 351 (19600), 368 (27000), 388 (26900). FT-IR (in KBr/cm−1): 3051 (br, aromatic C–H), 1670 (s, CO), 1599 (s), 1582 (s), 1565 (s). 1H NMR (400 MHz, CDCl3, δ from TMS): 8.9 (d, 1H), 8.68 (1H, d), 8.48 (1H, s), 8.34 (2H, m), 8.29 (1H, d; 1H, s), 8.03 (2H, m), 7.93 (2H, td), 7.5 (4H, m). ESI-MS (m/z): [M + H]+ calcd for C22H16N1O1, 310.123; found, 310.125.
(E)-3-(Pyren-1-yl)-1-(pyridin-2-yl)prop-2-en-1-one (Pypp). Yield: 1.14 g (73%), UV-Vis (DMF): λmax (ε/L mol−1 cm−1) = 270 (91
000), 310 (59600), 344 (45300), 390 (70300), 422 (69800). FT-IR (in KBr/cm−1): 3044 (br, aromatic C–H), 1660 (s, CO), 1588 (s), 1574 (s), 1560 (m). 1H NMR: (400 MHz, CDCl3, δ from TMS): 9.10 (1H, d), 8.78 (1H, s), 8.56 (3H, m), 8.18 (5H, m), 8.02 (2H, m), 7.90 (1H, s), 8.69 (1H, d), 7.51 (1H, s). ESI-MS (m/z): [M + H]+ calcd for C24H16N1O1, 334.123; found, 334.124.
[Eu(Anpp)(TTA)3] (1). Yield: 73.06 mg (65%). UV-Vis (DMF): λmax (ε/L mol−1 cm−1) = 270 (69
400), 346 (49500). FT-IR (in KBr/cm−1): ν = 3432 (w, br, νO–H), 3053 (w), 2925 (w), 1601 (s, νCO), 1537 (s), 1504 (m), 1464 (m), 1411 (s), 1356 (s), 1305 (s), 1247 (m), 1229 (s), 1186 (s), 1139 (s), 1059 (m), 933 (s), 892 (m), 858 (m), 786 (s, νCF3), 767 (s), 735 (s), 720 (s), 680 (m), 619 (s), 580 (s), 519 (w, νEu–N), 491 (w, νEu–O). ESI-MS (m/z): [M-TTA]+ calcd for C38H23N1O5F6S2Eu1, 904.013; found, 904.011.
[Tb(Anpp)(TTA)3] (2). Yield: 75.80 mg (67%). UV-Vis (DMF): λmax (ε/L mol−1 cm−1) = 271 (69
000), 346 (42700). FT-IR (in KBr/cm−1): ν = 3425 (w, br, νO–H), 3084 (w), 2963 (s), 1603 (s, νCO), 1537 (s), 1502 (m), 1460 (m), 1412 (s), 1354 (s), 1305 (s), 1261 (s), 1229 (s), 1183 (s), 1098 (s), 1059 (s), 1020 (s), 932 (s), 877 (m), 858 (m), 799 (s, νCF3), 735 (s), 717 (s), 679 (s), 640 (s), 579 (s), 519 (w, νTb–N), 486 (w, νTb–O). ESI-MS (m/z): [M-TTA]+ calcd for C38H23N1O5F6S2Tb1, 910.0175; found, 910.0154.
[Eu(Pypp)(TTA)3] (3). Yield: 68.90 mg (60%). UV-Vis (DMF): λmax (ε/L mol−1 cm−1) = 273 (88
200), 344 (77700), 440 (36600). FT-IR (in KBr/cm−1): ν = 3432 (w, br, νO–H), 3091 (w), 2962 (s), 2925 (w), 1602 (vs., νCO), 1536 (s), 1505 (m), 1459 (m), 1411 (s), 1353 (s), 1305 (s), 1261 (s), 1229 (s), 1185 (s), 1133 (s), 1092 (s) 1060 (m), 1025 (s), 933 (s), 845 (m), 799 (s, νCF3), 767 (s), 749 (s), 714 (s), 680 (m), 640 (s), 580 (s), 519 (w, νEu–N), 491 cm−1 (w, νEu–O). ESI-MS (m/z): [M + Na]+ calcd for C48H27N1O7F9S3Eu1Na1, 1171.991; found, 1171.970.
[Tb(Pypp)(TTA)3] (4). Yield: 72.80 mg (63%). UV-Vis (DMF): λmax (ε/L mol−1 cm−1) = 273 (86
800), 344 (72800), 440 (31200). FT-IR (in KBr/cm−1): ν = 3433 (w, br, νO–H), 3046 (w), 2963 (s), 2925 (w), 1602 (s, νCO), 1535 (s), 1504 (m), 1464 (m), 1411 (s), 1353 (s), 1304 (s), 1261 (s), 1229 (s), 1184 (s), 1099 (s), 1061 (s), 1023 (s), 932 (s), 844 (m), 799 (s, νCF3), 767 (s), 735 (s), 713 (s), 679 (m), 640 (s), 614 (s), 579 (s), 519 (w, νTb–N), 491 (w, νTb–O). ESI-MS (m/z): [M-TTA]+ calcd for C38H23N1O5F6S2Tb1, 934.0175; found, 934.0212.
Single crystal X-ray crystallography
[Eu(Pypp)(TTA)3] (3) was structurally characterized by single crystal X-ray diffraction technique. A crystal of proper size was mounted on a glass fiber and used for collecting the data. X-ray diffraction data were collected using ω-scan technique (width of 0.5° per frame) at a scan rate of 10 s each frame on a Bruker D8 Quest microfocus X-ray CCD diffractometer with graphite monochromated Mo Kα radiation (λ = 0.71073 Å) at 100(2) K controlled by an APEX2 v2012.4-3 software.56 Data were corrected for Lorentz polarization correction thereafter processed with Bruker's SAINT software which was collected using ω–2θ scan mode.57 Absorption corrections were applied on a SADABS program to the collected reflections and space group was determined from XPREP.58 The structure solved on a SHELXL-97 by direct methods, were refined using SHELXTL-6.14 program on a F2 full-matrix least square method. Subsequently obtained structure was refined on a SHEXTL-97 associated to WinGX-1.70 crystallographic package.59–61 C38 atom in one TTA ligand having site occupancy factor (SOF) of 0.5 in the asymmetric unit. A perspective view of the complex was obtained using ORTEP (Fig. 2) after including all the hydrogen atoms in geometrically calculated positions refined with riding hydrogen model and all other non-hydrogen atoms were anisotropically refined.62 Selected bond length and bond angles are shown in Table S1.† CCDC deposition number: CCDC 1566126. Crystallographic data for 3: C47.5H26.5Eu1F9N1O7S3, M = 1142.34, triclinic, space group P, a = 10.5489(9), b = 10.8247(9), c = 21.7672(19) Å, α = 99.053(2), β = 101.429(2), γ = 105.634(2)°, V = 2286.9(3) Å3, Z = 2, Dx (g cm−3) = 1.659, T = 100(2) K, θ range = 2.00–25.07°, μ = 1.598 mm−1, F(000) = 1133, reflections collected = 45431, unique reflections = 8100 (Rint = 0.0588) Tmax/Tmin, = 0.7619/0.7406, data/restraints/parameters = 8100/2/604, GOF on F2 = 1.109, R1 and wR2 [I > 2σ(I)] = 0.0663, 0.1726; 45431 collected reflections, 8100 unique reflections, Rint = 0.0588, final R1 = 0.0806 and wR2 = 0.1867 (all data).
DNA binding experiments
| [DNA]/(εa − εf) = [DNA]/(εb − εf) + 1/Kb(εb − εf) | (1) |
| Kapp × C50 = KEB × [EB] | (2) |
000 dm3 mol−1 cm−1) and the recorded absorbance (A278) at 278 nm.67,68 After taking a 1 μM solution of HSA, a quenching in emission intensity at 345 nm (λexc = 295 nm) of the tryptophan residue present in protein was recorded upon increasing concentration of the complexes. The quenching constants were calculated using Stern–Volmer equation:46| I0/I = 1 + KHSA[Q] | (3) |
The values of binding constant (K) and number of binding sites (n) was obtained from the linear log(I0/I − 1) vs. log[Q] plot using Scatchard equation:47
|
log[(I0 – I)/I] = logK + nlog[Q]
| (4) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank IIT Kanpur for infrastructure and the Science and Engineering Research Board (SERB) for financial support (EMR/2016/000521). ZA thanks to University Grants Commission (UGC) and SD to Ministry of Human Resources and Development (MHRD) for the research fellowships.Notes and references
- S. Cotton, Lanthanides and actinide chemistry, John Wiley & Sons Ltd., England, 2006 Search PubMed.
- (a) M. C. Hefferin, L. M. Matosziuk and T. J. Meade, Chem. Rev., 2014, 114, 4496–4539 CrossRef PubMed; (b) J. L. Major and T. J. Meade, Acc. Chem. Res., 2009, 42, 893–903 CrossRef CAS PubMed; (c) S. Shuvaev, M. Starck and D. Parker, Chem.–Eur. J., 2017, 23, 9974–9989 CrossRef CAS PubMed.
- (a) P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed; (b) P. Caravan, Chem. Soc. Rev., 2006, 35, 512–523 RSC; (c) M. Mikawa, H. Kato, M. Okumura, M. Narazaki, Y. Kanazawa, N. Miwa and H. Shinohara, Bioconjugate Chem., 2001, 12, 510–514 CrossRef CAS.
- (a) J.-C. G. Bünzli, Chem. Rev., 2010, 110, 2729–2755 CrossRef PubMed; (b) S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
- (a) E. G. Moore, A. P. S. Samuel and K. N. Raymond, Acc. Chem. Res., 2009, 42, 542–552 CrossRef CAS PubMed; (b) A. J. Amoroso and S. J. A. Pope, Chem. Soc. Rev., 2015, 44, 4723–4742 RSC.
- (a) S. Gai, C. Li, P. Yang and J. Lin, Chem. Rev., 2014, 114, 2343–2389 CrossRef CAS PubMed; (b) H. Terraschke and C. Wickleder, Chem. Rev., 2015, 115, 11352–11378 CrossRef CAS PubMed.
- (a) J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC; (b) H. Tsukube and S. Shinoda, Chem. Rev., 2002, 102, 2389–2403 CrossRef CAS PubMed; (c) H. Tsukube and S. Shinoda, Analyst, 2011, 136, 431–435 RSC.
- (a) L. V. Meyer, F. Schönfeld and K. M. Buschbaum, Chem. Commun., 2014, 50, 8093–8108 RSC; (b) J. Heine and K. M. Buschbaum, Chem. Soc. Rev., 2013, 42, 9232–9242 RSC; (c) J. Rocha, L. D. Carlos, F. A. A. Paz and D. Ananias, Chem. Soc. Rev., 2011, 40, 926–940 RSC.
- (a) S. M. Larson, J. A. Carrasquillo, N.-K. V. Cheung and O. W. Press, Nat. Rev. Cancer, 2015, 15, 347–360 CrossRef CAS PubMed; (b) R. D. Teo, J. Termini and H. B. Gray, J. Med. Chem., 2016, 59, 6012–6024 CrossRef CAS PubMed.
- (a) M. D. Gross, G. L. Nelsestuen and R. Kumar, J. Biol. Chem., 1987, 262, 6539–6545 CAS; (b) J. M. Harrowfield, M. I. Ogden, W. R. Richmond and A. H. Whit, J. Chem. Soc., Dalton Trans., 1991, 2153–2160 RSC.
- (a) S. P. Fricker, Chem. Soc. Rev., 2006, 35, 524–533 RSC; (b) K. Wang, R. Li, Y. Cheng and B. Zhu, Coord. Chem. Rev., 1999, 190–192, 297–308 CrossRef CAS.
- (a) M. Bottrill, L. Kwok and N. J. Long, Chem. Soc. Rev., 2006, 35, 557–571 RSC; (b) P. Hermann, J. Kotek, V. KubÍček and I. Lukeš, Dalton Trans., 2008, 23, 3027–3047 RSC.
- R. Bonnett, Chemical Aspects of Photodynamic Therapy, Gordon & Breac, London, UK, 2000 Search PubMed.
- (a) D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 23, 380–387 CrossRef PubMed; (b) J. L. Sessler and R. A. Miller, Biochem. Pharmacol., 2000, 59, 733–739 CrossRef CAS PubMed; (c) G. Thiabaud, R. McCall, G. He, J. F. Arambula, Z. H. Siddik and J. L. Sessler, Angew. Chem., Int. Ed., 2016, 55, 12626–12631 CrossRef CAS PubMed.
- (a) M. Ethirajan, Y. Chen, P. Joshi and R. K. Pandey, Chem. Soc. Rev., 2011, 40, 340–362 RSC; (b) M. R. Detty, S. L. Gibson and S. J. Wagner, J. Med. Chem., 2004, 47, 3897–3915 CrossRef CAS PubMed; (c) M. Triesscheijn, P. Baas, J. H. M. Schellens and F. A. Stewart, Oncologist, 2006, 11, 1034–1044 CrossRef CAS PubMed.
- (a) J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. L. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 110, 2795–2838 CrossRef CAS PubMed; (b) K. Szaciłowski, W. Macyk, A. D. Matuszek, M. Brindell and M. Stochel, Chem. Rev., 2005, 105, 2647–2694 CrossRef PubMed.
- (a) S. I. Moriwaki, J. Misawa, Y. Yoshinari, I. Yamada, M. Takigawa and Y. Tokura, Photodermatol., Photoimmunol. Photomed., 2001, 17, 241–243 CrossRef CAS; (b) M. Ochsner, J. Photochem. Photobiol., B, 1996, 32, 3–9 CrossRef CAS.
- R. R. Allison and C. H. Sibata, Photodiagn. Photodyn. Ther., 2010, 7, 61–75 CrossRef CAS PubMed.
- (a) F. S. Mackay, J. A. Woods, P. Heringová, J. Kaspárková, A. M. Pizarro, S. A. Moggach, S. Parsons, V. Brabec and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20743–20748 CrossRef CAS PubMed; (b) N. J. Farrer, J. A. Woods, L. Salassa, Y. Zhao, K. S. Robinson, G. Clarkson, F. S. Mackay and P. J. Sadler, Angew. Chem., Int. Ed., 2010, 49, 8905–8908 CrossRef CAS PubMed; (c) N. J. Farrer, L. Salassa and P. J. Sadler, Dalton Trans., 2009, 10690–10701 RSC.
- (a) D. A. Lutterman, P. K.-L. Fu and C. Turro, J. Am. Chem. Soc., 2006, 128, 738–739 CrossRef CAS PubMed; (b) A. M. Angeles-Boza, H. T. Chifotides, J. D. Aguirre, A. Chouai, P. K.-L. Fu, K. R. Dunbar and C. Turro, J. Med. Chem., 2006, 49, 6841–6847 CrossRef CAS PubMed.
- (a) K. Mitra, S. Gautam, P. Kondaiah and A. R. Chakravarty, Angew. Chem., Int. Ed., 2015, 54, 13989–13993 CrossRef CAS PubMed; (b) W. Lu, D. A. Vicic and J. K. Barton, Inorg. Chem., 2005, 44, 7970–7980 CrossRef CAS PubMed.
- (a) S. Swavey and K. J. Brewer, Inorg. Chem., 2002, 41, 6196–6198 CrossRef CAS PubMed; (b) A. A. Holder, S. Swavey and K. J. Brewer, Inorg. Chem., 2004, 43, 303–308 CrossRef CAS PubMed.
- (a) A. Hussain, D. Lahiri, M. S. A. Begum, S. Saha, R. Majumdar, R. R. Dighe and A. R. Chakravarty, Inorg. Chem., 2010, 49, 4036–4045 CrossRef CAS PubMed; (b) A. Hussain, S. Gadadhar, T. K. Goswami, A. A. Karande and A. R. Chakravarty, Eur. J. Med. Chem., 2012, 50, 319–331 CrossRef CAS PubMed; (c) A. Hussain, K. Somyajit, B. Banik, S. Banerjee, G. Nagaraju and A. R. Chakravarty, Dalton Trans., 2013, 42, 182–195 RSC.
- (a) S. Dasari and A. K. Patra, Dalton Trans., 2015, 44, 19844–19855 RSC; (b) S. Dasari, S. Singh, S. Sivakumar and A. K. Patra, Chem.–Eur. J., 2016, 22, 17387–17396 CrossRef CAS PubMed; (c) A. Chandra, K. Singh, S. Singh, S. Sivakumar and A. K. Patra, Dalton Trans., 2016, 45, 494–497 RSC; (d) K. Singh, S. Singh, P. Srivastava, S. Sivakumar and A. K. Patra, Chem. Commun., 2017, 53, 6144–6147 RSC.
- M. Sy, A. Nonat, N. Hildebrandt and L. J. Charbonnihre, Chem. Commun., 2016, 52, 5080–5095 RSC.
- (a) V. Divya, M. L. P. Reddy and R. Pavithran, Dalton Trans., 2013, 42, 15249–15262 RSC; (b) D. B. A. Raj, S. Biju and M. L. P. Reddy, Inorg. Chem., 2008, 47, 8091–8100 CrossRef CAS PubMed; (c) P. Martín-Ramos, C. Coya, Á. L. Álvarez, M. R. Silva, C. Zaldo, J. A. Paixao, P. Chamorro-Posada and J. Martín-Gil, J. Phys. Chem. C, 2013, 117, 10020–10030 CrossRef; (d) J. Shi, Y. Hou, W. Chu, X. Shi, H. Gu, B. Wang and Z. Sun, Inorg. Chem., 2013, 52, 5013–5022 CrossRef CAS PubMed.
- (a) J. Yu, L. Zhou, H. Zhang, Y. Zheng, H. Li, R. Deng, Z. Peng and Z. Li, Inorg. Chem., 2005, 44, 1611–1618 CrossRef CAS PubMed; (b) Y. Zheng, J. Lin, Y. Liang, Q. Lin, Y. Yu, Q. Meng, Y. Zhou, S. Wang, H. Wang and H. Zhang, J. Mater. Chem., 2001, 11, 2615–2619 RSC.
- C. Zhuang, W. Zhang, C. Sheng, W. Zhang, C. Xing and Z. Miao, Chem. Rev., 2017, 117, 7762–7810 CrossRef CAS PubMed.
- (a) J. Li, D. Li, Y. Xu, Z. Guo, X. Liu, H. Yang, L. Wu and L. Wang, Bioorg. Med. Chem. Lett., 2017, 27, 602–606 CrossRef CAS PubMed; (b) Z. Liu, L. Tang, P. Zou, Y. Zhang, Z. Wang, Q. Fang, L. Jiang, G. Chen, Z. Xu, H. Zhang and G. Liang, Eur. J. Med. Chem., 2014, 74, 671–682 CrossRef CAS PubMed.
- (a) N. J. Lawrence, R. P. Patterson, L.-L. Ooi, D. Cook and S. Ducki, Bioorg. Med. Chem. Lett., 2006, 16, 5844–5848 CrossRef CAS PubMed; (b) N. Mateeva, S. V. K. Eyunni, K. K. Redda, U. Ononuju, T. D. Hansberry, C. Aikens and A. Nag, Bioorg. Med. Chem. Lett., 2017, 27, 2350–2356 CrossRef CAS PubMed.
- (a) M. Liu, P. Wilairat and M. L. Go, J. Med. Chem., 2001, 44, 4443–4452 CrossRef CAS PubMed; (b) J. N. Dominguez, C. Leon, J. Rodrigues, N. Gamboa de Dominguez, J. Gut and P. J. Rosenthal, J. Med. Chem., 2005, 48, 3654–3658 CrossRef CAS PubMed.
- P. M. Sivakumar, T. M. Kumar and M. Doble, Chem. Biol. Drug Des., 2009, 74, 68–79 CAS.
- (a) S. Shettigar, K. Chandrasekharan, G. Umesh, B. K. Sarojini and B. Narayana, Polymer, 2006, 47, 3565–3567 CrossRef CAS; (b) Y. J. Jung, K.-I. Son, Y. E. Oh and D.-Y. Noh, Polyhedron, 2008, 27, 861–867 CrossRef CAS.
- (a) P. M. Gallego, H. D. Dulk, J. Brouwer, H. Kooijman, A. L. Spek, O. Roubeau, S. J. Teat and J. Reedijk, Inorg. Chem., 2008, 47, 11171–11179 CrossRef PubMed; (b) B. Song, G. Wang, M. Tan and J. Yuan, J. Am. Chem. Soc., 2006, 128, 13442–13450 CrossRef CAS PubMed.
- (a) M. Chhatwal, A. Kumar, R. D. Gupta and S. K. Awasthi, RSC Adv., 2015, 5, 51678–51681 RSC; (b) K. C. Rout and B. Mondal, Inorg. Chim. Acta, 2015, 437, 54–58 CrossRef CAS.
- (a) J. Yu, L. Zhou, H. Zhang, Y. Zheng, H. Li, R. Deng, Z. Peng and Z. Li, Inorg. Chem., 2005, 44, 1611–1618 CrossRef CAS PubMed; (b) J. Li, H. Li, P. Yan, P. Chen, G. Hou and G. Li, Inorg. Chem., 2012, 51, 5050–5057 CrossRef CAS PubMed; (c) J. Sun, B. Song, Z. Ye and J. Yuan, Inorg. Chem., 2015, 54, 11660–11668 CrossRef CAS PubMed; (d) J. Shi, Y. Hou, W. Chu, X. Shi, H. Gu, B. Wang and Z. Sun, Inorg. Chem., 2013, 52, 5013–5022 CrossRef CAS PubMed.
- P. Martín-Ramos, C. Coya, Á. L. Álvarez, M. R. Silva, C. Zaldo, J. A. Paixão, P. Chamorro-Posada and J. MartÍn-Gil, J. Phys. Chem. C, 2013, 117, 10020–10030 Search PubMed.
- (a) P. Lenaerts, K. Driesen, R. V. Deun and K. Binnemans, Chem. Mater., 2005, 17, 2148–2154 CrossRef CAS; (b) P. Lenaerts, E. Ryckebosch, K. Driesen, R. V. Deun, P. Nockemann, C. G. Walrand and K. Binnemans, J. Lumin., 2005, 114, 77–84 CrossRef CAS; (c) R.-J. Zhang, K.-Z. Yang, A.-C. Yub and X.-S. Zhao, Thin Solid Films, 2000, 363, 275–278 CrossRef CAS.
- (a) W. D. Horrocks Jr and D. R. Sudnick, J. Am. Chem. Soc., 1979, 101, 334–340 CrossRef; (b) A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 1, 1999, 493–504 RSC.
- (a) E. C. Long and J. K. Barton, Acc. Chem. Res., 1990, 23, 271–273 CrossRef CAS; (b) A. C. Komor and J. K. Barton, Chem. Commun., 2013, 49, 3617–3630 RSC.
- (a) T. M. Kelly, A. B. Tossi, D. J. McConnell and T. C. Strekas, Nucleic Acids Res., 1985, 13, 6017–6034 CrossRef PubMed; (b) V. A. Bloomfield, D. M. Crothers and I. Tinocoo Jr, Physical Chemistry of Nucleic Acids, Harper & Row, New York, 1974, p. 432 Search PubMed; (c) J. B. Chaires, Curr. Opin. Struct. Biol., 1998, 8, 314–320 CrossRef CAS PubMed.
- (a) J. Olmsted III and D. R. Kearns, Biochemistry, 1977, 16, 3647–3754 CrossRef; (b) D. L. Boger, B. E. Fink, S. R. Brunnette, W. C. Tse and M. P. Hedrick, J. Am. Chem. Soc., 2001, 123, 5878–5891 CrossRef CAS PubMed.
- (a) M. J. Waring, J. Mol. Biol., 1965, 13, 269–282 CrossRef CAS PubMed; (b) B. C. Baguley and E.-M. Falkenhaug, Nucleic Acids Res., 1978, 5, 161–171 CrossRef CAS PubMed.
- (a) S. Curry, H. Mandelkow, P. Brick and N. Franks, Nat. Struct. Biol., 1998, 5, 827–835 CrossRef CAS PubMed; (b) J. Ghuman, P. A. Zunszain, I. Petitpas, A. A. Bhattacharya, M. Otagiri and S. Curry, J. Mol. Biol., 2005, 353, 38–52 CrossRef CAS PubMed.
- (a) T. Peters, Adv. Protein Chem., 1985, 37, 161–245 CrossRef CAS PubMed; (b) Y.-Q. Wang, H.-M. Zhang, G.-C. Zhang, W.-H. Tao and S.-H. Tang, J. Lumin., 2007, 126, 211–218 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- G. Scatchard, Ann. N. Y. Acad. Sci., 1949, 51, 660–672 CrossRef CAS.
- (a) J. Tang, F. Luan and X. Chen, Bioorg. Med. Chem., 2006, 14, 3210–3217 CrossRef CAS PubMed; (b) J. N. Miller, Proc. Anal. Div. Chem. Soc., 1979, 16, 203–208 CAS.
- N. Selvakumaran, N. S. P. Bhuvanesh, A. Endo and R. Karvembu, Polyhedron, 2014, 14, 95–109 CrossRef.
- M. Y. Li, C. S. Cline, E. B. Koker, H. H. Carmichael, C. F. Chignell and P. Bilski, Photochem. Photobiol., 2001, 74, 760–764 CrossRef CAS PubMed.
- S. M. Klein, G. Cohen and A. I. Cederbaum, Biochemistry, 1981, 20, 6006–6012 CrossRef CAS PubMed.
- J. S. Beckman, T. W. Beckman, J. Chen, P. A. Marshall and B. A. Freeman, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 1620–1624 CrossRef CAS.
- (a) A. U. Khan, J. Phys. Chem., 1976, 80, 2219–2228 CrossRef CAS; (b) P. B. Merkel and D. R. Kearns, J. Am. Chem. Soc., 1972, 94, 1029–1030 CrossRef CAS.
- D. D. Perrin, W. L. F. Armarego and D. R. Perrin, Purification of Laboratory Chemicals, Pergamon Press, Oxford, 1980 Search PubMed.
- L. R. Melby, N. J. Rose, E. Abramson and J. C. Caris, J. Am. Chem. Soc., 1964, 86, 5117–5125 CrossRef CAS.
- APEX2 v2012.4, Bruker AXS, Madison, WI, 1999 Search PubMed.
- N. Walker and D. Stuart, Acta Crystallogr., Sect. A: Found. Crystallogr., 1983, 39, 158–166 CrossRef.
- G. M. Sheldrick, SADABS, Area Detector Absorption Correction, University of Göttingen, Göttingen, Germany, 2001 Search PubMed.
- G. M. Sheldrick, SHELX-97, Program for Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; (b) G. M. Sheldrick, SHELXTL 6.14, Bruker, AXS Inc., Madison, WI, 2000 Search PubMed.
- (a) L. J. Farrugia, WINGX ver 1.70, An Integrated System of Windows Programs for the Solution, Refinement, and Analysis of Single Crystal X-ray Diffraction Data, Department of Chemistry, University of Glasgow, 2005 Search PubMed; (b) L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- M. N. Burnett and C. K. Johnson, ORTEP-III, Report ORNL–6895, Oak Ridge National Laboratory, Oak Ridge, TN, 1996 Search PubMed.
- J. Marmur, J. Mol. Biol., 1961, 3, 208–218 CrossRef CAS.
- M. E. Reichmann, S. A. Rice, C. A. Thomas and P. Doty, J. Am. Chem. Soc., 1954, 76, 3047–3053 CrossRef CAS.
- A. Wolfe, G. H. Shimer and T. Meehan, Biochemistry, 1987, 26, 6392–6396 CrossRef CAS PubMed.
- M. Lee, A. L. Rhodes, M. D. Wyatt, S. Forrow and J. Hartley, Biochemistry, 1993, 32, 4237–4245 CrossRef CAS PubMed.
- T. Peters Jr, All About Albumin: Biochemistry, Genetics and Medical Applications, Academic Press, New York, 1995 Search PubMed.
- E. Alarcon, A. Aspee, M. Gonzalez-Bejar, A. M. Edwards, E. Lissi and J. C. Scaiano, Photochem. Photobiol. Sci., 2010, 9, 455–461 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ESI-MS figures; unit cell packing diagram; selected bond distances and angles; photophysical aspects; lifetime measurements; DNA and protein binding plots; DNA cleavage data. CCDC 1566126. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra08543e |
| This journal is © The Royal Society of Chemistry 2017 |