
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Low-temperature chemical vapor deposition of cobalt oxide thin films from a dicobaltatetrahedrane precursor†
Marcel Melzer a,
Charan K. Nichenametlaa,
Colin Georgib,
Heinrich Langc and
Stefan E. Schulz*ab
a,
Charan K. Nichenametlaa,
Colin Georgib,
Heinrich Langc and
Stefan E. Schulz*ab
aChemnitz University of Technology, Center for Microtechnologies, 09126 Chemnitz, Germany. E-mail: stefan.schulz@zfm.tu-chemnitz.de
bFraunhofer Institute for Electronic Nanosystems, Department Back-End of Line, Technologie-Campus 3, 09126 Chemnitz, Germany
cTechnische Universität Chemnitz, Institute of Chemistry, Inorganic Chemistry, 09107 Chemnitz, Germany
First published on 27th October 2017
Abstract
Cobalt oxides are a promising anode material for lightweight rechargeable lithium-ion batteries. Thus, the low temperature deposition of cobalt oxide is a key-technology for the production of flexible energy storage systems enabling novel application opportunities such as wearables. To satisfy the emerging process requirements the dicobaltatetrahedrane precursor [Co2(CO)6(η2-H–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–nC5H11)] was investigated for the low-temperature chemical vapor deposition of cobalt oxides. Oxygen, water vapor and a combination of both were examined as possible co-reactants. In particular, wet oxygen proves to be an appropriate oxidizing agent providing dense and high purity cobalt oxide films within the examined temperature range from 130 °C to 250 °C. Film growth occurred at temperatures as low as 100 °C making this process suitable for the coating of temperature-sensitive and flexible substrates.
C–nC5H11)] was investigated for the low-temperature chemical vapor deposition of cobalt oxides. Oxygen, water vapor and a combination of both were examined as possible co-reactants. In particular, wet oxygen proves to be an appropriate oxidizing agent providing dense and high purity cobalt oxide films within the examined temperature range from 130 °C to 250 °C. Film growth occurred at temperatures as low as 100 °C making this process suitable for the coating of temperature-sensitive and flexible substrates.
Introduction
Cobalt forms the two stable oxides CoO and Co3O4. These oxides have gained great interest due to their possible application as catalyst, e.g. for the combustion of hydrocarbons1,2 or as sensitive materials in gas sensors.3,4 Both amorphous and crystalline cobalt oxide layers are a promising anode material for thin film lithium-ion batteries enabling lighter batteries compared to standard graphite electrodes.5–8 Furthermore Co3O4, is an ideal material for absorber layers in solar thermal collectors due to its high absorptance across the complete solar spectrum.9,10 Various processes are available for the deposition of cobalt oxides such as molecular beam epitaxy,4 sputtering,11 spray pyrolysis,12 electrochemical deposition,13,14 thermal oxidation14 and chemical vapor deposition.15 Thermal oxidation of pre-deposited metallic cobalt is not feasible in terms of temperature and rate requirements, since temperatures above 375 °C are required to accelerate diffusion for a sufficiently fast oxidation.16 For the conformal coating of non-planar substrates the application of CVD is necessary with respect to the high volume production of flexible devices. All other processes mentioned are either not applicable for cost-effective roll-to-roll processes or they would lead to non-conformal deposition on structured substrates due to their directional deposition mode.Various cobalt precursors have been developed and investigated for the deposition of CoxOy (x = 1, y = 1; x = 3, y = 4) in the past decades. This development was driven by the need for conformal, low cost CVD processes with a high deposition rate. These precursors include [Co(acac)2] (acac = acetylacetonate),15,17–25 [Co(acac)3],26 [Co(hfac)2] (hfac = hexafluoroacetylacetonate),8,27,28 [Co(thd)2] (thd = tetramethylheptanedionate)22,29,30 and [CpCo(CO)2] (Cp = cyclopentadienyl),31,32 see Table 1. All these precursors require process temperatures above 200 °C for the thermal CVD of cobalt oxide films or the utilization of plasma-enhancement. However, plasma-enhanced processes lead to limitations with respect to the uniform coating of structures with high aspect ratios, but such structures are needed for advanced energy storage systems with high energy density. Only recently Amin-Chalhoub et al. addressed the low temperature thermal CVD of cobalt oxide.33 It was shown that pure CoO films can be grown with high rates (>100 nm min−1) from Co2(CO)8 at temperatures between 120 °C and 190 °C without addition of any co-reactant. However, the deposition rate is extremely sensitive to temperature changes, which makes a stable and homogeneous process problematic.
| Precursor | Temp. [°C] | Co-reactant | Ref. |
|---|---|---|---|
| a Plasma-enhanced process (PECVD).b (TMEDA = N,N,N′,N′-tetramethylethylenediamine). | |||
| Cu(acac)2 | 625 | O2 | 25 |
| Co(acac)2 | 400–550 | O2 | 24 |
| Co(acac)2 | 400–550 | O2 | 20 |
| Co(hfac)2·TMEDAb | 400–500 | O2 | 8 |
| Cu(hfac)2·H2O·tryglyme | 400 | O2 | 28 |
| Co(acac)2 | 375–550 | O2, N2O | 21 |
| Co(acac)2 | 350–600 | O2 | 22 |
| Co(acac)2·TMEDAb | 350–600 | O2 | 22 |
| Co(thd)2 | 350–600 | O2 | 22 |
| Co(thd)2·TMEDAb | 350–600 | O2 | 22 |
| Co(thd)2·monoglyme | 350–540 | O2 | 30 |
| Cu(acac)3 | 350–540 | O2 | 26 |
| Co(thd)2 | 350–500 | O2 | 29 |
| Co(hfac)2·H2O | 350–500 | O2 | 27 |
| Cu(hfac)2·H2O·dyglyme | 350 | O2 | 28 |
| Co(acac)2 | 300–500 | H2O | 15 |
| Co(acac)2 | 250–400 | Air | 19 |
| CpCo(CO)2 | 200–650 | O2 | 31 |
| Co(acac)2 | 200–500 | O2, — | 23 |
| Co(acac)2 | 150–400 | Oa | 17 |
| Co(acac)2 | 150–400 | Oa | 18 |
| Co2(CO)8 | 120–190 | — | 33 |
| CpCo(CO)2 | 25 | Oa | 32 |
In the present paper the dicobaltatetrahedrane [Co2(CO)6(η2-HCCnC5H11)] was applied as cobalt precursor for the CVD of cobalt oxide. The precursor is shown in Fig. 1. The used precursor is characterized by a high vapor pressure of 26.3 hPa at 40 °C.34 Furthermore, the precursor is liquid at ambient temperature. These two properties lead to the fact that the precursor can be vaporized at a high rate with constant flow by simple technical means in contrast to the solid Co2(CO)8. In this regard, the here investigated [Co2(CO)6(η2-HCCnC5H11)] proves to be promising for industrial use. So far, this precursor has not been studied with respect to its suitability for the CVD of cobalt oxide films. This work is committed to the development of a novel low-temperature CVD process for substrate temperatures below 150 °C. These low temperatures are required to enable the coating of flexible polymer substrates.35 Oxygen, water vapor as well as a combination of both were investigated as possible oxidation agents since the pyrolysis of [Co2(CO)6(η2-HCCnC5H11)] alone results in cobalt films with considerable carbon impurities.36
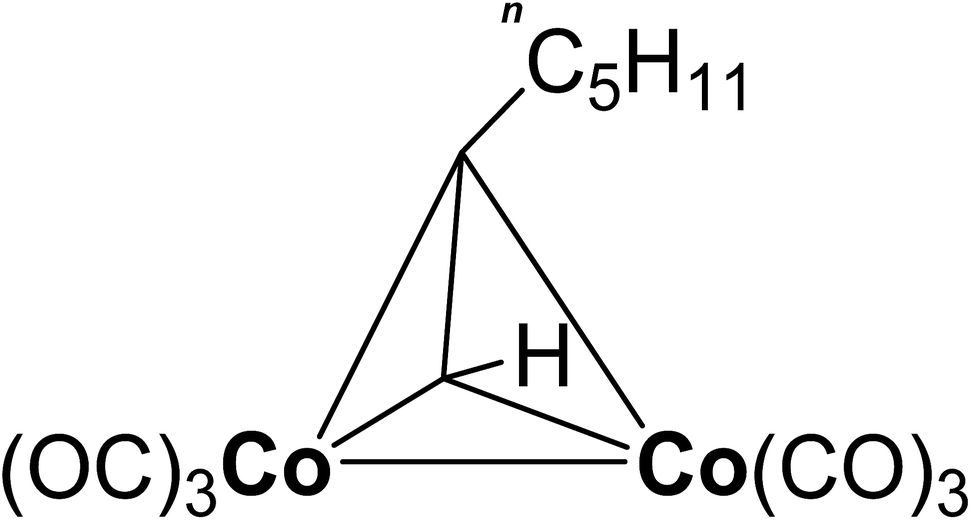 | ||
| Fig. 1 Structure of the used cobalt precursor [Co2(CO)6(η2- HCCnC5H11)]. | ||
Experimental section
Thin film deposition
The CVD experiments were carried out in a Varian 100 mm vertical flow cold-wall reactor. The system consists of a stainless steel reactor and a load-lock system to keep air and moisture from the deposition chamber enabling reproducible process conditions. Depositions were carried out on 100 nm silica 4 inch Si wafers. The oxide was thermally grown in a Centrotherm tube furnace prior to the experiments. The substrates were exposed to air before the CVD processes without any further pretreatment. Table 2 provides an overview of the relevant process parameters and the gases used.| Substrate | 100 nm SiO2 on 4 in Si wafers |
| Substrate temperature | 100 °C–250 °C |
| Gas line temperature | 60 °C |
| Reactor wall temperature | 60 °C |
| Precursor solvent (n-hexane, >99%, Merck) | 80.0% by mass |
| Precursor solution flow rate | 45 mg min−1 |
| Precursor carrier gas flow rate (Ar 6.0) | 700 sccm |
| Precursor evaporation temperature | 55 °C |
| Oxygen flow rate (O2 5.0) | 100 sccm |
| Water vapor flow rate | 20 mg min−1 |
| Water carrier gas flow rate (Ar 6.0) | 200 sccm |
| Total pressure | 13.0 hPa |
| Deposition duration | 15 min |
The dicobaltatetrahedrane [Co2(CO)6(η2-HCCnC5H11)] was synthesized according to a published reaction procedure.36 The precursor was stored at ambient temperature in a stainless steel cylinder. Since the precursor is both harmful to health and sensitive to moisture, contact with the air must be prevented by appropriate handling. Suitable measures include the use of Schlenk line during synthesis and storage in a tightly closed container under dry inert gas. A 20.0% by mass solution of the precursor in n-hexane was utilized to facilitate the dosing. Previous experiments at 250 °C have shown that the used solvent does not affect the surface chemistry of the applied precursor.37 Argon (6.0) was applied as pressure gas to push the precursor from the steel cylinder into a controlled evaporation and mixing system (CEM) from Bronkhorst. A schematic drawing of the experimental setup for evaporation of the dissolved precursor can be found in the electronic ESI.† The measurement of the precursor solution flow rate in units of mg min−1 is based on the specific heat capacity Cp of the liquid. Since this value is unavailable for [Co2(CO)6(η2-HCCnC5H11)], the flow rate was estimated by using the Cp value of the applied solvent n-hexane (2.3 J gK−1).38 The flow rate of the precursor solution was set to 45 mg min−1 corresponding to a cobalt precursor flow rate of 9 mg min−1. A CEM temperature of 55 °C enabled in combination with 700 sccm argon carrier gas (6.0) the complete evaporation of the precursor solution without any aerosol formation. The gas lines towards the reactor as well as the reactor wall were heated to 60 °C to prevent precursor condensation.
During the CVD process the chamber pressure was regulated by a butterfly valve to 13.0 hPa. The film growth experiments were carried out on silicon wafers with 100 nm thermally grown silicon dioxide on top. Based on previous CVD experiments without co-reactant, a process temperature in the range from 130 °C to 250 °C appeared appropriate for the deposition studies.36 To establish a suitable cobalt oxide CVD process, growth experiments were carried out with oxygen, water vapor and a combination of both called wet oxygen as co-reactant. Except for the initial screening experiments a flow rate of 100 sccm was applied for oxygen (5.0). The water vapor flow was set to 20 mg min−1 H2O in combination with 200 sccm Ar (6.0) as carrier gas for the water bubbling at 50 °C. The same flow rates were applied for the processes using wet oxygen. Thus, the ratio between oxygen and water vapor was fixed at a molar ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for these processes. This ratio is based on the results of previous experiments.39 If not otherwise stated, the process time was 15 min in order to obtain sufficiently thick films for the subsequent ex situ X-ray photoelectron spectroscopy (XPS) analysis.
:1 for these processes. This ratio is based on the results of previous experiments.39 If not otherwise stated, the process time was 15 min in order to obtain sufficiently thick films for the subsequent ex situ X-ray photoelectron spectroscopy (XPS) analysis.
Sample characterization
The film morphology as well as the film thickness were studied using a Zeiss Supra 60 scanning electron microscope (SEM) for top-view and cross-sectional images. Spectroscopic ellipsometry was applied to determine the thickness for samples with a CoxOy thickness below 50 nm. For this purpose a SENTECH SE850 ellipsometer was utilized. The measurements were carried out in the spectral range from 190–830 nm and the incident angle was set to 70°. The ellipsometric data were analyzed by SpectraRay/3. In order to determine accurate values for the film thickness of the CoxOy layer, the substrate layer stack consisting of Si and SiO2 was studied by ellipsometry prior to the CVD experiments. The silicon substrate was modelled using tabulated n and k values from G. E. Jellison Jr.40 A Cauchy model was applied to parameterize the dielectric function of the SiO2. After the deposition the samples were characterized again by ellipsometry adding a Lorentz oscillator to describe the deposited cobalt oxide film.The film composition was analyzed within 24 hours after the deposition by XPS using a R3000 electron energy analyzer with a pass energy of 200 eV and monochromatic Al-Kα radiation from a MX-650 X-ray source both from VG Scienta. Prior to the XPS measurements, the samples were cleaned by 15 min Ar+ sputtering in order to remove contaminations which have formed due to air contact during the sample transport. The ion energy was set to 4 keV. A sputter time of 15 min was selected for all experiments based on primary XPS depth profiles which have shown a steady sample composition after approximately 10 minutes sputtering. An electron flood gun was applied during the XPS measurement in order to compensate the charging of the semiconducting cobalt oxide. The parameters of the flood gun were chosen in such a way that the cobalt 2p3/2 peak was shifted to 780.0 eV binding energy corresponding to CoO as well as Co3O4.41 For the analysis of the XPS spectra CasaXPS Version 2.3.16 Pre-rel 1.4 was applied using Scofield relative sensitivity factors in combination with a Shirley background. A value of −0.75 was set for the depth correction.
The structural properties of the deposited films were analyzed by powder X-ray diffraction (PXRD) in (θ–2θ) geometry, using a XRD 3000 PTS diffractometer from Seifert-FPM with Cu-Kα radiation. The PXRD spectra were measured exclusively in the angular ranges up to a maximum of 66°, in which peaks for CoO and Co3O4 were expected.42 For a qualitative phase analysis the peak positions were used and are shown in the corresponding diagrams.
Results and discussion
Influence of the co-reactant flowrate
The goal of the initial experiments was to identify a suitable co-reactant flowrate for a fixed precursor flow rate of 9 mg min−1. These investigations form the basis for the further cobalt oxide CVD experiments which are presented in this work. For these screening experiments the process temperature was set to 130 °C and wet oxygen was applied as co-reactant. The ratio between oxygen and water vapor was fixed at a molar ratio of 4:1 based on earlier experiments.39 The composition of the deposited cobalt oxide layers was measured by ex situ XPS.
These measurements exhibited a minimum impurity content for a co-reactant flow rate of 100 sccm O2 in combination with 20 mg min−1 H2O, see Fig. 2. This specific co-reactant flow rate resulted reproducibly in a carbon content of 1.5 at% in the deposited cobalt oxide films. This value is significantly lower than the carbon portion of 8.9 to 19.9 at%, which was achieved with the other tested flowrates ranging from 50–200 sccm O2 in combination with 10–40 mg min−1 H2O. Lower flowrates were not examined since it is known from previous work that the pure pyrolysis of the used precursor results in severe carbon contaminations of 35.2 at% at 250 °C.36 Therefore, the combination of 100 sccm O2 with 20 mg min−1 H2O was applied as co-reactant for the CVD experiments with wet oxygen as co-reactant and 100 sccm O2 or 20 mg min−1 H2O were applied for all other experiments.
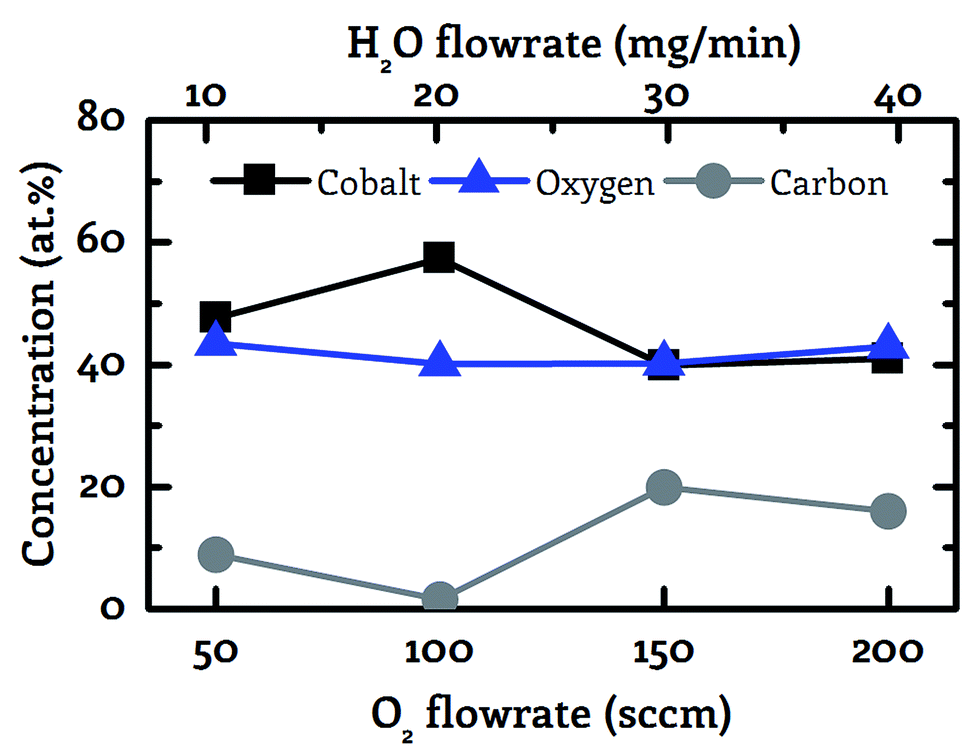 | ||
| Fig. 2 Correlation between the co-reactant flowrate and the sample composition (cobalt – black squares, oxygen – blue triangles, carbon – grey circles). Wet oxygen was applied as co-reactant with a fixed molar ratio of 4:1 between oxygen and water vapor showing a minimum carbon impurity content for 100 sccm oxygen in combination with 20 mg min−1 water vapor as co-reactant at 130 °C. | ||
Composition of the deposited CoxOy films
A key objective of this work was to provide a CVD process, which facilitates the deposition of CoxOy layers with high purity at deposition temperatures below 150 °C. For this purpose, the composition of the deposited layers was examined by XPS. Fig. 3 to 5 show the composition of the deposited layers for the three investigated co-reactants H2O, O2 and H2O + O2 in the temperature range from 130 °C to 250 °C.From these investigations it becomes clear that pure water vapor is not a suitable oxidizing agent for the deposition of cobalt oxide using [Co2(CO)6(η2-HCCnC5H11)] since a carbon content of more than 30.0 at% was detected for all investigated process temperatures (Fig. 3). Oxygen on the other hand facilitated the deposition of impurity-free layers for temperatures greater than or equal to 200 °C, see Fig. 4. No carbon peaks were detectable by XPS for layers deposited at 200 °C and 250 °C using oxygen as co-reactant. This limits the carbon content to the detection limit of XPS of approximately 1.0 at%.
 | ||
| Fig. 3 Correlation between the sample composition (cobalt – black squares, oxygen – blue triangles, carbon – grey circles). Determined by XPS and the process temperature for water as co-reactant. | ||
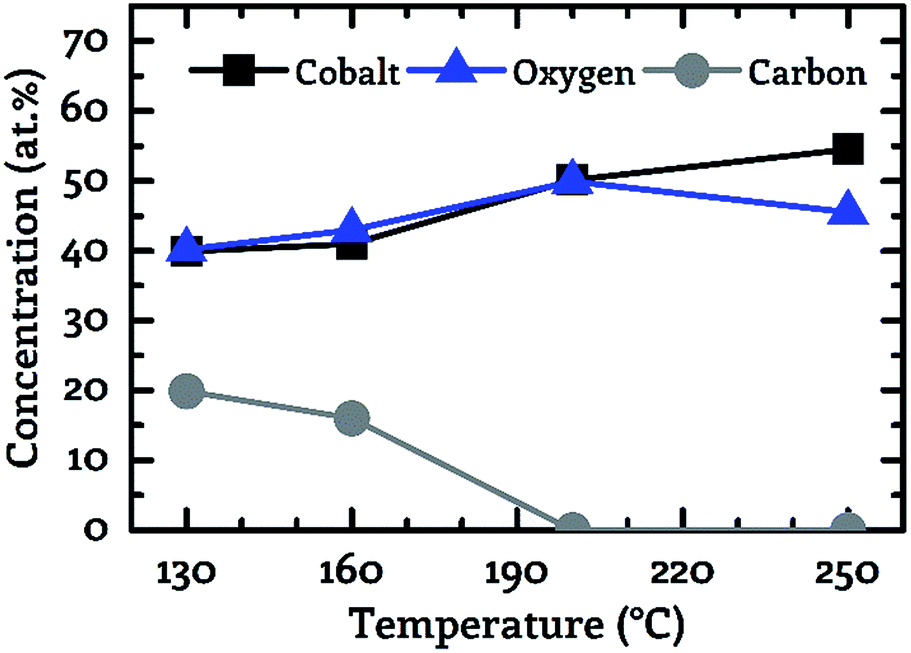 | ||
| Fig. 4 Correlation between the sample composition (cobalt – black squares, oxygen – blue triangles, carbon – grey circles). Determined by XPS and the process temperature for oxygen as co-reactant. | ||
The best results were obtained by the use of wet oxygen as co-reactant enabling the deposition of pure cobalt oxide layers even at temperatures as low as 130 °C, see Fig. 5. A more detailed investigation of the binding conditions based on the XPS spectra is not part of this work since despite the use of an electron flood gun strong non-linear charging effects were observed. These charging effects prevented the accurate determination of the chemical peak shift. However, the precise determination of this peak shift forms the basis for the analysis of the binding conditions of the examined layer.
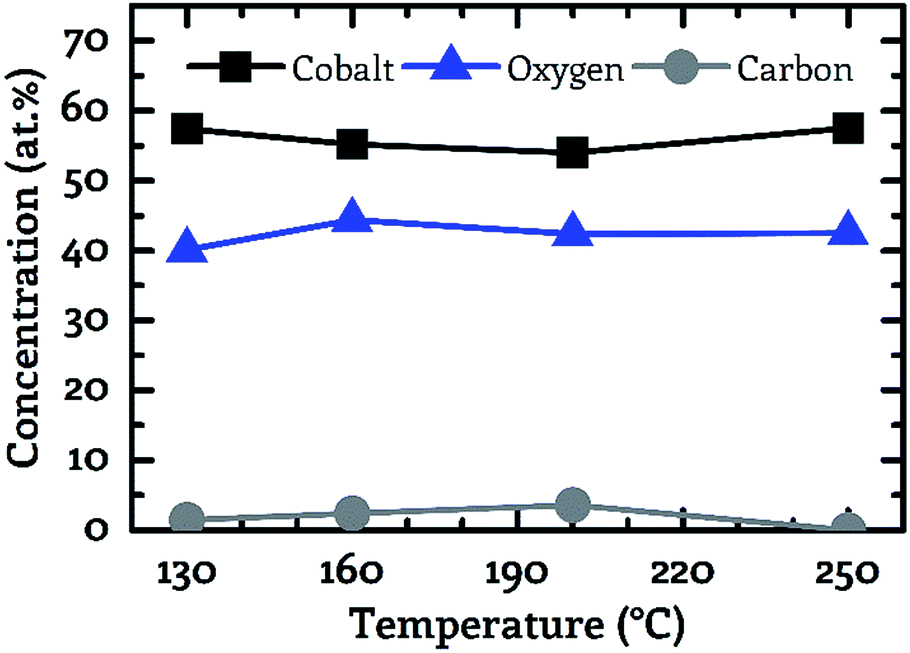 | ||
| Fig. 5 Correlation between the sample composition (cobalt – black squares, oxygen – blue triangles, carbon – grey circles). Determined by XPS and the process temperature for wet oxygen as co-reactant. | ||
Furthermore, a meaningful analysis of the cobalt oxide XPS data was prevented by the fact that the ion bombardment during sputtering leads to a reduction of the oxide from Co3O4 to CoO.30 Thus, a distinction of these two oxides by XPS is inhibited. For these reasons, a detailed analysis and interpretation of the cobalt oxide XPS peaks with their complex satellite structure including up to 5 peaks does not seem to be reliable.43
However, based on existing literature33,44,45 and the obtained composition of the deposited films conclusions are drawn about the surface chemistry of the presented CVD process and thus possible sources of the carbon impurities are discussed. Kwon et al. have investigated the surface chemistry of the similar dicobaltatetrahedrane precursor [Co2(CO)6(η2-HCCtC4H9)].44 This precursor differs from the precursor investigated in the present work in the alkyl ligand used. Based on the findings of Kwon et al., it is suggested that the observed carbon impurities are caused by the alkyl ligand, especially at low process temperatures, and not by the CO ligands. Kwon et al. proved that the tBuCCH ligand absorbs on the surface at 140 °C but desorbs during an annealing to 300 °C. This correlates well with our results for oxygen as co-reactant, where it was detected that the carbon impurities can be completely removed by a temperature increase from 160 °C to 200 °C. The CO ligands appear unlikely as sources of the carbon contamination since they can be separated from the cobalt atom at temperatures below 130 °C during various CVD processes using Co2(CO)8 enabling the deposition of layers without carbon impurities.33,45 Furthermore, the Co–CO bonds are weakened by the oxidation of the cobalt during the presented CVD process due to the reduction of the electron density at the metal center, whereby the desorption of the CO ligands is further amplified.44
The observation that wet oxygen is a more reactive co-reactant compared to its sole components oxygen and water vapor, is consistent with previous works on the ALD of transition metals.39,46,47 For example, it was observed by Alnes et al. that the combination of oxygen and water vapor enables the deposition of copper oxide from Cu(acac)2 in contrast to pure oxygen as co-reactant which does not result in any film growth.47 In the context of the CoxOy CVD experiments presented herein it is suggested that the high redox potential EØ of water in presence of oxygen enables the deposition of layers without any impurities even at low temperatures. Pure water exhibits a redox potential EØ of −0.42 V for an assumed pH neutral environment, whereas the redox potential EØ raises to +0.82 V in the presence of oxygen indicating the stronger oxidation behavior of wet oxygen.48 The observed reactivity of the co-reactants H2O + O2 > O2 > H2O is consistent with the standard redox potential EØ of the investigated oxidizing agents as shown in Table 3.
| Co-reactant | Standard reduction potential EØ | C@130 °C [at%] | |
|---|---|---|---|
| Water | 2H2O + 2e− → H2 + 2OH− | −0.83 V | 31.2 |
| Oxygen | O2 + e− → O2− | −0.56 V | 19.9 |
| Wet oxygen | O2 + 2H2O + 4e− → 4OH− | +0.40 V | 1.5 |
Growth kinetics of the CoxOy films
The dependency of the achieved CoxOy film thickness on the applied co-reactant and the process temperature was investigated by preparing cobalt oxide layers in the temperature range from 130 °C to 250 °C using a fixed process time of 15 min. The obtained values are depicted in Fig. 6. The film thickness was in the range from 34 nm to 735 nm. The deposition on 100 mm wafers is homogeneous over the entire wafer surface, as demonstrated by wafer images in the electronic ESI.† A scaling of the process on larger substrates appears feasible. The smallest value was observed for water vapor as co-reactant at 130 °C and the highest value was detected for oxygen at 200 °C. The general trend suggests a surface reaction limited deposition. As the process temperature was increased, the film thickness increased for all three investigated oxidizing agents except for oxygen in the range from 200 °C to 250 °C. In this case the thickness of the deposited CoxOy layer decreased from 735 nm at 200 °C to 500 nm at 250 °C, see Fig. 6.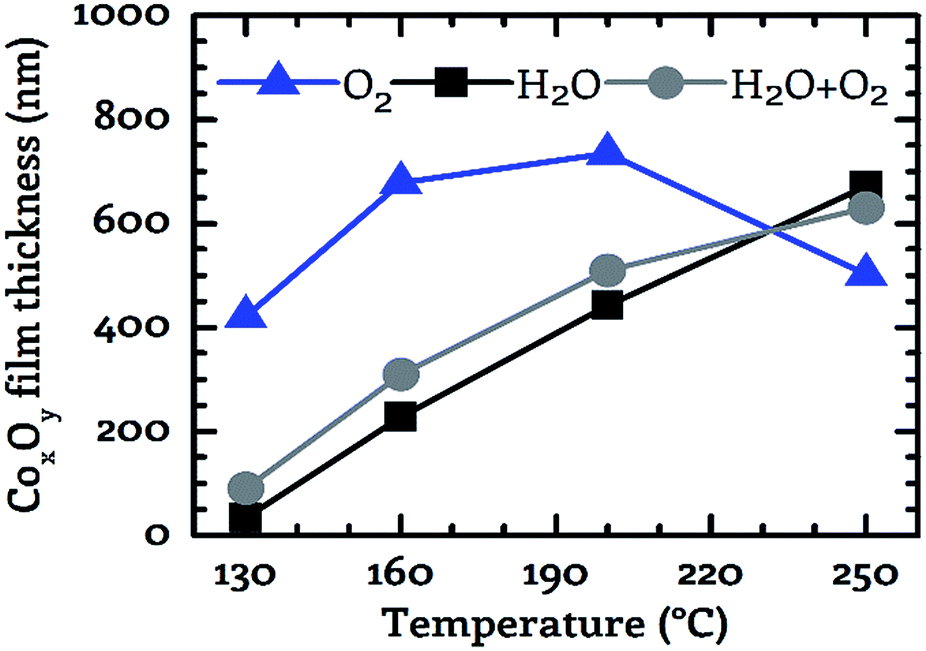 | ||
| Fig. 6 For the three investigated co-reactants (oxygen – blue triangles, water – black squares, wet oxygen –grey circles) the layer thickness of the deposited films is depicted as a function of the substrate temperature. The duration of each process was 15 minutes. Measurement points are connected to guide the eye. | ||
In contrast to this, a monotonically increasing film thickness was measured with increasing process temperature for water vapor and wet oxygen as a co-reactant. The slope of the film thickness was similar for these two reactants in the temperature range from 130 °C to 200 °C (Fig. 6). By using wet oxygen the film thickness was higher compared to pure water vapor for temperatures of up to 200 °C indicating a positive effect of the additional oxygen with respect to the obtained film thickness. This observation is underlined by the fact that in the temperature range from 130 °C to 200 °C the CoxOy films deposited using pure oxygen as co-reactant were significantly thicker than those grown with water vapor or wet oxygen. However, if the process temperature was further increased to 250 °C the largest film thickness was achieved by the water vapor process. Thus, for temperatures above 200 °C the addition of oxygen does not appear to produce a positive effect with respect to the film thickness anymore. This is in contrast to temperatures below 250 °C. These results show that the use or admixture of oxygen leads to an increased deposition rate as long as a process temperature of 200 °C is not exceeded.
A similar diminishing effect of oxygen at elevated temperatures was observed by Fuji et al. at 400 °C for the deposition of CoxOy using a plasma-enhanced CVD process based on Co(acac)2 as precursor and oxygen as co-reactant.18 However, the investigations do not allow any conclusion to the possible cause of this behavior.
Structure and morphology of the deposited CoxOy films
The deposited layers were analyzed using SEM and PXRD in order to study the morphology and the crystal structure of the grown CoxOy layers. Oxygen as co-reactant resulted for all investigated temperatures in a columnar growth, see Fig. 7. In the top-view SEM images, voids are clearly visible between the individual columns. The observed columnar growth mode is typical for CVD films especially for cobalt oxides.18,33,49 | ||
| Fig. 7 Top-view and cross-sectional SEM images of the cobalt oxide films deposited using oxygen as co-reactant: (a)/(b) 130 °C; (c)/(d) 160 °C; (e)/(f) 200 °C and (g)/(h) 250 °C. | ||
For a process temperature of 250 °C the application of water vapor as co-reactant resulted in a similar columnar growth mode (Fig. 8). However, if the process temperature was reduced below 250 °C the morphology of the deposited layers changed (Fig. 8). The deposited layers of this water vapor process appear denser and no longer show the columnar growth observed for oxygen as co-reactant. Furthermore, the top-view SEM images suggest a smoother surface of the films deposited via water vapor as co-reactant. The application of wet oxygen as co-reactant resulted in columnar films which were similar to those deposited using oxygen as oxidizing agent (Fig. 9).
 | ||
| Fig. 8 Top-view and cross-sectional SEM images of the cobalt oxide films deposited using water vapor as co-reactant: (a)/(b) 130 °C; (c)/(d) 160 °C; (e)/(f) 200 °C and (g)/(h) 250 °C. | ||
 | ||
| Fig. 9 Top-view and cross-sectional SEM images of the cobalt oxide films deposited using wet oxygen as co-reactant: (a)/(b) 130 °C; (c)/(d) 160 °C; (e)/(f) 200 °C and (g)/(h) 250 °C. | ||
However, crystallites with a triangular and a quadratic base are recognizable in the top view images of the layers which were deposited at 200 °C via wet oxygen (Fig. 9e). Similar structures also occurred at 160 °C but less pronounced (Fig. 9c). Such quadratic and triangular crystallites occur for the (111) and (200) growth direction of face-centered cubic crystals such as CoO.49 However, an increase of the process temperature to 250 °C led to a disappearance of the crystalline morphology of the cobalt oxide columns indicating an amorphous growth mode again. This is in contrast to the expected behavior that the crystallinity should increase for higher temperatures.7
The PXRD measurements were performed to determine the crystallinity of the deposited layers. For the layers whose crystallinity was accessible by PXRD, it can be further verified based on the characteristic diffraction pattern whether CoO or Co3O4 has been deposited. Both quantities, the crystallinity and the phase of the oxide, are important parameters for some applications such as the use of cobalt oxide as electrode material in intercalation lithium ion batteries. However, in the case of the PXRD measurements it must be noted that the signal strength of the peaks depends also on the layer thickness. The cobalt oxide films investigated in this work have different thicknesses due to the constant process time of 15 min and process dependent growth kinetics, see previous Section 3.3. Furthermore, the crystallites must exceed a minimum size of approximately 2–3 nm to be detectable by PXRD.50 Layers with smaller crystallites exhibit amorphous PXRD patterns without any diffraction peaks.
The application of water vapor as co-reactant resulted in films without any measurable peaks for all tested process temperatures ranging from 130 °C to 250 °C as shown in Fig. 10. The same behavior was observed for oxygen as co-reactant up to a process temperature of 200 °C (Fig. 11). If the process temperature of this oxygen based process was further increased to 250 °C, diffraction patterns were detected indicating the growth of Co3O4 with a (311) preferential growth direction, see Fig. 11. The crystallinity of the deposited layers could be clearly improved by the use of wet oxygen as co-reactant compared to the other tested oxidizing agents (Fig. 12). For wet oxygen as co-reactant peaks with a high intensity were detected for process temperatures above 130 °C indicating a more pronounced crystallinity of the films compared to the processes with oxygen or water vapor. However, at 130 °C process temperature also the layers deposited via wet oxygen appeared amorphous in the PXRD without measurable diffraction peaks similar to the results obtained with the other co-reactants at 130 °C. The diffraction peaks with the highest amplitude were observed in the PXRD patterns for layers deposited at 160 °C and 200 °C, respectively, with wet oxygen as co-reactant. This finding is consistent with the previously described SEM images. Like the SEM images for wet oxygen as co-reactant, the PXRD measurements also indicated an increase of the CoxOy crystallinity if the process temperature was raised from 160 °C to 200 °C. For these two process temperatures the PXRD patterns indicate diffraction patterns which confirm the growth of CoO and show no sign for the deposition of the Co3O4 spinel. In both cases a diffraction reflex at 42.4° verifies a preferential growth direction of (200). For a deposition temperature of 160 °C this is the only crystalline phase, which could be detected by PXRD. In contrast to this, a further CoO diffraction reflexion was measured at 36.5° for the layers deposited at 200 °C indicating a (111) crystal orientation besides the (200) crystallites. If the process temperature was further increased to 250 °C, a phase change occurred and instead of CoO crystallites the PXRD measurement revealed the growth of Co3O4 crystals using wet oxygen as co-reactant (Fig. 12). The preferential growth direction of these crystallites was (111) like the dominant diffraction reflex at 19.0° proves. Such a phase change was expected since CoO can only be deposited under mild oxidation conditions such as low process temperature and low oxidizing agent concentration.8,27 The fact that the choice of the co-reactant has an influence on the crystallinity of the deposited cobalt oxide layer has already been published previously, e.g. by Mane and Shivashankar.21
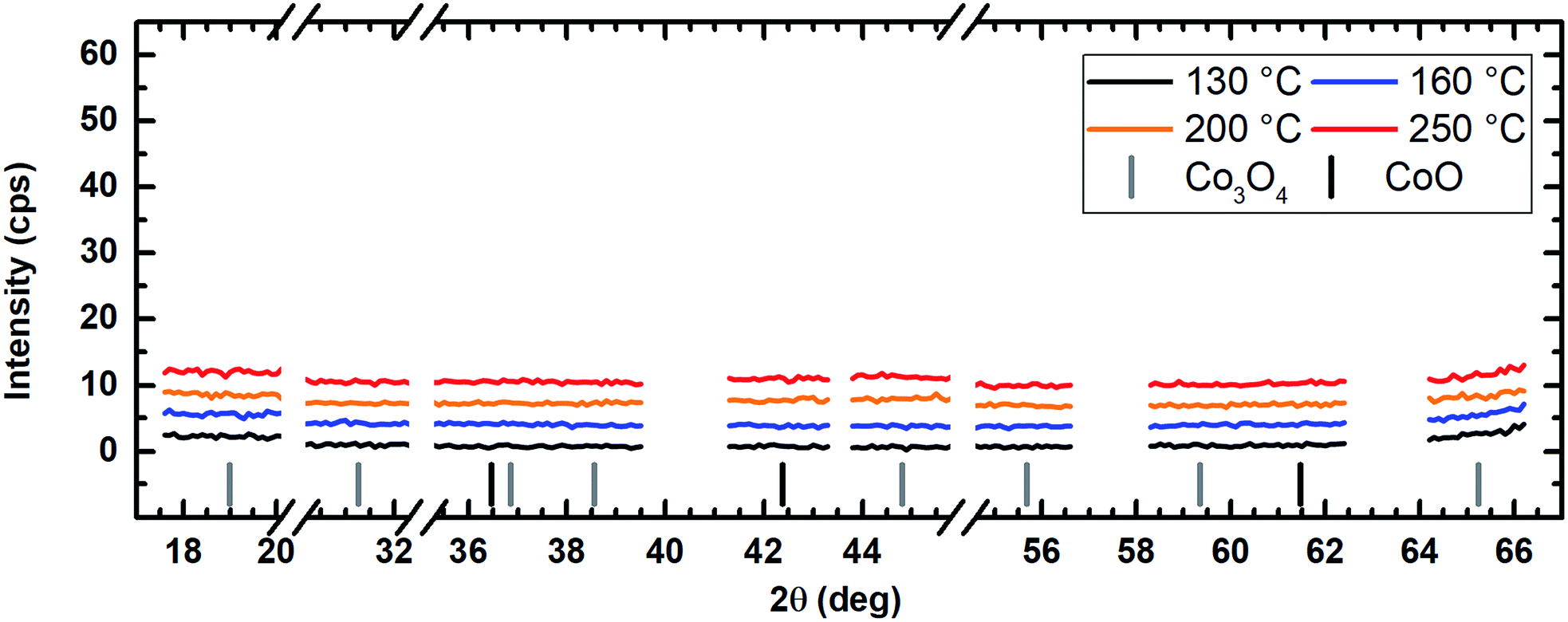 | ||
| Fig. 10 PXRD spectrum of the cobalt oxide films prepared using water vapor as co-reactant showing no diffraction reflexes for all tested deposition temperatures suggesting amorphous film growth. For better comparability, the y-axes are kept the same in Fig. 10–12. An offset of +3 cps has been inserted for better visualization between the measured data for the different temperatures. | ||
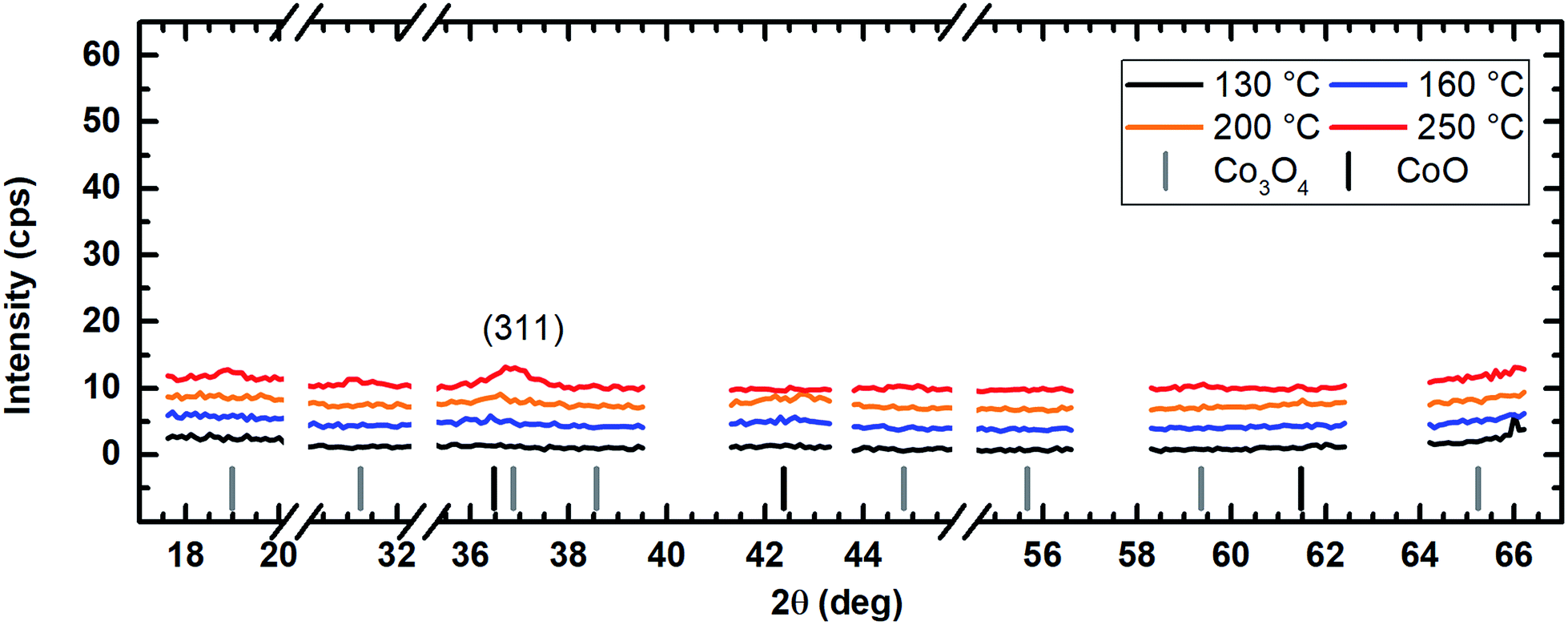 | ||
| Fig. 11 PXRD spectrum of the cobalt oxide films prepared using O2 as co-reactant showing only a weak Co3O4 reflex for the sample for the sample prepared at 250 °C. For better comparability, the y-axes are kept the same in Fig. 10–12. An offset of +3 cps has been inserted for better visualization between the measured data for the different temperatures. | ||
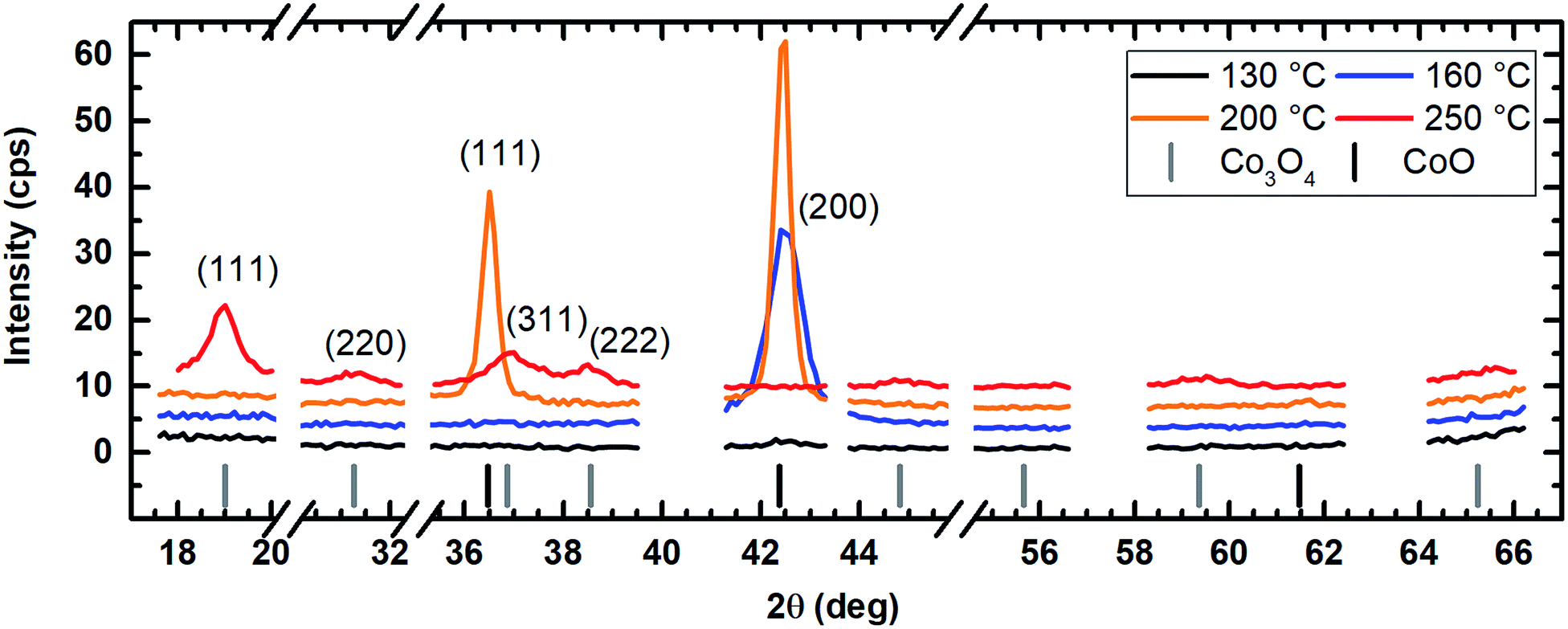 | ||
| Fig. 12 PXRD of the cobalt oxide films prepared using wet oxygen as co-reactant showing the formation of CoO for process temperatures of 160 °C and 200 °C and Co3O4 for a deposition temperature of 250 °C. For better comparability, the y-axes are kept the same in Fig. 10–12. An offset of +3 cps has been inserted for better visualization between the measured data for the different temperatures. | ||
Low temperature CVD of CoxOy films at 100 °C
Based on the promising results obtained with wet oxygen as co-reactant the temperature range was lowered to 100 °C wafer temperature in order to investigate the thermal limits of the presented CVD process. XPS spectra as well as SEM images of layers deposited at 100 °C can be found in the electronic ESI.†Even at 100 °C the impurity content was low enabling the deposition of CoxOy films with 11.7 at% carbon as the only impurity. Furthermore, XPS measurements indicated a cobalt content of 45.1 at% and an oxygen percentage of 43.2 at% suggesting the growth of Co(II) oxide. At 100 °C, the achieved film thickness was 12 nm after a 60 min CVD process indicating a further decrease of the film thickness with respect to a process temperature of 130 °C. This enables the facile and controlled deposition of ultrathin CoxOy layers. The combined results, as shown in Table 4, do not indicate whether the performance of the CVD process is limited by the precursor or the applied co-reactant.
| Co-reactant | T [°C] | Co [at%] | O [at%] | C [at%] | Film thickness [nm] |
|---|---|---|---|---|---|
| a In contrast to the other experiments, the deposition time for this process was 60 minutes in order to obtain sufficiently thick layers for XPS analysis. | |||||
| Water | 130 | 43.2 | 21.4 | 35.3 | 33 |
| 160 | 44.3 | 23.6 | 32.1 | 230 | |
| 200 | 45.1 | 22.7 | 32.2 | 440 | |
| 250 | 41.2 | 17.1 | 41.7 | 670 | |
| Oxygen | 130 | 39.9 | 40.2 | 19.9 | 420 |
| 160 | 41.0 | 43.0 | 16.0 | 680 | |
| 200 | 50.1 | 49.9 | <1.0 | 730 | |
| 250 | 54.5 | 45.5 | <1.0 | 500 | |
| Wet oxygen | 100 | 45.1 | 43.2 | 11.7 | 12a |
| 130 | 57.4 | 40.1 | 1.5 | 90 | |
| 160 | 55.2 | 44.4 | 2.4 | 310 | |
| 200 | 54.0 | 42.4 | 2.5 | 510 | |
| 250 | 57.5 | 42.5 | <1.0 | 630 | |
Conclusions
In summary, the dicobaltatetrahedrane [Co2(CO)6(η2-HCCnC5H11)] precursor dissolved in n-hexane has been proven to be excellently suited for the low-temperature deposition of pure cobalt oxide films by CVD. The use of wet oxygen as a co-reactant enabled the deposition of cobalt oxide layers at temperatures as low as 130 °C. The deposited cobalt oxide films exhibited a purity greater than 98.5 at% with carbon as the only impurity. This is in contrast to experiments in which either water vapor or oxygen were used as oxidizing agent. Water vapor as co-reactant resulted in a carbon content greater than 30.0 at% for all tested process temperatures ranging from 130 °C to 250 °C. Oxygen proved to be more reactive compared to water vapor enabling the deposition of pure cobalt oxide layers for temperatures greater than or equal to 200 °C. The experiments presented here verify that the combination of water vapor and oxygen is more reactive than both reactants on their own. It is suggested that the observed reactivity of the investigated co-reactants H2O + O2 > O2 > H2O is consistent with their standard redox potential EØ.
The deposition of high purity CoxOy at temperatures as low as 130 °C represents a significant extension of the current temperature window for the thermal CVD of cobalt based on liquid precursors. These have traditionally required temperatures above 200 °C. Thus, the examined precursor has been proven to be a promising competitor to the second encouraging precursor Co2(CO)8. The here used liquid precursor seems more advantageous compared to the solid Co2(CO)8 precursor with respect to the process integration, since liquid precursors facilitate reproducible evaporation with high rates in contrast to solid reactants. Thereby, this process appears to be suitable for the coating of flexible, temperature-sensitive substrates as they are required for novel applications such as the manufacturing of flexible power supplies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Cornelia Kowol is acknowledged for the SEM investigations, Dr Patrick Matthes for PXRD measurements, Marcus Kaspar for supporting the initial CVD experiments and Natalia Rüffer for the synthesis of the cobalt precursor. The authors gratefully acknowledge the German Research Foundation (DFG) for funding this work within the International Research Training Group IRTG 1215 ‘‘Materials and Concepts for Advanced Interconnects”.References
- N. Bahlawane, Appl. Catal., B, 2006, 67, 168–176 CrossRef CAS.
- M. M. Natile and A. Glisenti, Chem. Mater., 2002, 14, 3090–3099 CrossRef CAS.
- H. Yamaura, J. Tamaki, K. Moriya, N. Miura and N. Yamazoe, J. Electrochem. Soc., 1997, 144, L158 CrossRef CAS.
- J. Wöllenstein, M. Burgmair, G. Plescher, T. Sulima, J. Hildenbrand, H. Böttner and I. Eisele, Sens. Actuators, B, 2003, 93, 442–448 CrossRef.
- H. J. Qiu, L. Liu, Y. P. Mu, H. J. Zhang and Y. Wang, Nano Res., 2015, 8, 321–339 CrossRef CAS.
- G. X. Wang, Y. Chen, K. Konstantinov, M. Lindsay, H. K. Liu and S. X. Dou, J. Power Sources, 2002, 109, 142–147 CrossRef CAS.
- M. E. Donders, H. C. M. Knoops, W. M. M. Kessels and P. H. L. Notten, J. Power Sources, 2012, 203, 72–77 CrossRef CAS.
- D. Barreca, M. Cruz-Yusta, A. Gasparotto, C. MacCato, J. Morales, A. Pozza, C. Sada, L. Sánchez and E. Tondello, J. Phys. Chem. C, 2010, 114, 10054–10060 CAS.
- G. McDonald, Thin Solid Films, 1980, 72, 83–87 CrossRef CAS.
- K. Chidambaram, L. K. Malhotra and K. L. Chopra, Thin Solid Films, 1982, 87, 365–371 CrossRef CAS.
- L. C. Schumacher, I. B. Holzhueter, I. R. Hill and M. J. Dignam, Electrochim. Acta, 1990, 35, 975–984 CrossRef CAS.
- V. R. Shinde, S. B. Mahadik, T. P. Gujar and C. D. Lokhande, Appl. Surf. Sci., 2006, 252, 7487–7492 CrossRef CAS.
- E. Barrera, I. González and T. Viveros, Sol. Energy Mater. Sol. Cells, 1998, 51, 69–82 CrossRef CAS.
- G. B. Smith, A. Ignatiev and G. Zajac, J. Appl. Phys., 1980, 51, 4186–4196 CrossRef CAS.
- T. Maruyama and T. Nakai, Sol. Energy Mater., 1991, 23, 25–29 CrossRef CAS.
- H. G. Tompkins and J. A. Augis, Oxid. Met., 1981, 16, 355–369 CrossRef CAS.
- E. Fujii, A. Tomozawa, S. Fujii, H. Torii, M. Hattori and R. Takayama, Jpn. J. Appl. Phys., 1993, 32, L1448–L1450 CrossRef CAS.
- E. Fujii, H. Torii, A. Tomozawa, R. Takayama and T. Hirao, J. Mater. Sci., 1995, 30, 6013–6018 CrossRef CAS.
- T. Maruyama and S. Arai, J. Electrochem. Soc., 1996, 143, 1383–1386 CrossRef CAS.
- A. U. Mane, K. Shalini, A. Wohlfart, A. Devi and S. A. Shivashankar, J. Cryst. Growth, 2002, 240, 157–163 CrossRef CAS.
- A. U. Mane and S. A. Shivashankar, J. Cryst. Growth, 2003, 254, 368–377 CrossRef CAS.
- S. Pasko, A. Abrutis, L. G. Hubert-Pfalzgraf and V. Kubilius, J. Cryst. Growth, 2004, 262, 653–657 CrossRef CAS.
- E. F. Rivera, B. Atakan and K. Kohse-Höinghaus, J. Phys., 2001, 11, 629–635 Search PubMed.
- K. Shalini, A. U. Mane, S. a. Shivashankar, M. Rajeswari and S. Choopun, J. Cryst. Growth, 2001, 231, 242–247 CrossRef CAS.
- L. M. Apátiga and V. M. Castaño, Thin Solid Films, 2006, 496, 576–579 CrossRef.
- M. Burriel, G. Garcia, J. Santiso, A. N. Hansson, S. Linderoth and A. Figueras, Thin Solid Films, 2005, 473, 98–103 CrossRef CAS.
- A. Gulino, G. Fiorito and I. Fragalà, J. Mater. Chem., 2003, 13, 861–865 RSC.
- A. Gulino and I. Fragalà, Inorg. Chim. Acta, 2005, 358, 4466–4472 CrossRef CAS.
- D. Barreca, C. Massignan, S. Dalio, M. Fabrizio, C. Piccirillio, L. Armelao and E. Tondello, Chem. Mater., 2001, 13, 588–593 CrossRef CAS.
- M. Burriel, G. Garcia, J. Santiso, A. Abrutis, Z. Saltyte and A. Figueras, Chem. Vap. Deposition, 2005, 11, 106–111 CrossRef CAS.
- S. Schmid, R. Hausbrand and W. Jaegermann, Thin Solid Films, 2014, 567, 8–13 CrossRef CAS.
- J. Tyczkowski, R. Kapica and J. Łojewska, Thin Solid Films, 2007, 515, 6590–6595 CrossRef CAS.
- E. Amin-Chalhoub, T. Duguet, D. Samélor, O. Debieu, E. Ungureanu and C. Vahlas, Appl. Surf. Sci., 2016, 360, 540–546 CrossRef CAS.
- C. Georgi, A. Hildebrandt, A. Tuchscherer, S. Oswald and H. Lang, Z. Anorg. Allg. Chem., 2013, 639, 2532–2535 CrossRef CAS.
- S. Franssila, Introduction to Microfabrication, John Wiley and Sons, 2010 Search PubMed.
- C. Georgi, A. Hildebrandt, T. Waechtler, S. E. Schulz, T. Gessner and H. Lang, J. Mater. Chem. C, 2014, 2, 4676–4682 RSC.
- M. Kaspar, T. Waechtler, R. Ecke, C. Georgi, S. E. Schulz, H. Lang and T. Gessner, in Materials for Advances Metalization (MAM), Grenoble, 2015 Search PubMed.
- B. Kalinowska, J. Jedlinska, W. Wóycicki and J. Stecki, J. Chem. Thermodyn., 1980, 12, 891–896 CrossRef CAS.
- T. Waechtler, S. Oswald, N. Roth, A. Jakob, H. Lang, R. Ecke, S. E. Schulz, T. Gessner, A. Moskvinova, S. Schulze and M. Hietschold, J. Electrochem. Soc., 2009, 156, H453–H459 CrossRef CAS.
- G. E. Jellison Jr, Opt. Mater., 1992, 1, 41–47 CrossRef.
- D. Briggs and M. P. Seah, Practical Surface Analysis by Auger and X-ray Photoelectron Spectroscopy, Wiley & Sons, Chichester, UK, 2nd edn, 1992 Search PubMed.
- S. Kabekkodu and ICDD, PDF-4+ 2016 (Database), International Centre for Diffraction Data, Newtown Square, PA, USA, 2016 Search PubMed.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- J. Kwon, M. Saly, R. K. Kanjolia and Y. J. Chabal, Chem. Mater., 2011, 23, 2068–2074 CrossRef CAS.
- J. H. G. Lee, H. J. Yang, J. H. G. Lee, J. Y. Kim, W. J. Nam, H. J. Shin, Y. K. Ko, J. H. G. Lee, E. G. Lee and C. S. Kim, J. Electrochem. Soc., 2006, 153, G539 CrossRef CAS.
- T. Waechtler, S. Oswald, A. Pohlers, S. Schulze, S. E. Schulz and T. Gessner, in Conference Proceedings AMC XXIII, Advanced Metallization Conference, 2007, p. 23 Search PubMed.
- M. E. Alnes, E. Monakhov, H. Fjellvåg and O. Nilsen, Chem. Vap. Deposition, 2012, 18, 173–178 CrossRef CAS.
- P. W. Atkins and L. Jones, Chemical principles: the quest for insight, Freeman, New York, 1st edn, 1999 Search PubMed.
- J. M. Blocher Jr, J. Vac. Sci. Technol., 1974, 11, 680–686 CrossRef.
- Y. Waseda, E. Matsubara and K. Shinoda, X-Ray Diffraction Crystallography – Introduction, Examples and Solved Problems, Springer-Verlag, 1st edn, 2011 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08810h |
| This journal is © The Royal Society of Chemistry 2017 |