
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, cytotoxic activity and drug combination study of tertiary amine derivatives of 2′,4′-dihydroxyl-6′-methoxyl-3′,5′-dimethylchalcone†
Chen Wangac,
Ping Wua,
Xiao-Ling Shend,
Xiao-Yi Wei*a and
Zi-Hua Jiang *b
*b
aKey Laboratory of Plant Resources Conservation and Sustainable Utilization, South China Botanical Garden, Chinese Academy of Sciences, Xingke Road 723, Tianhe District, Guangzhou 510650, People's Republic of China. E-mail: wxy@scbg.ac.cn; Tel: +86-20-37252538
bDepartment of Chemistry, Lakehead University, 955 Oliver Road, Thunder Bay, Ontario P7B 5E1, Canada. E-mail: zjiang@lakeheadu.ca; Tel: +1-807-766-7171
cUniversity of Chinese Academy of Sciences, Yuquanlu 19A, Beijing 100049, People's Republic of China
dLaboratory of Herbal Drug Discovery, Tropical Medicine Institute, Guangzhou University of Chinese Medicine, Guangzhou 510405, People's Republic of China
First published on 12th October 2017
Abstract
In an effort to develop more water soluble anticancer drugs based on 2′,4′-dihydroxyl-6′-methoxyl-3′,5′-dimethylchalcone (DMC), a group of DMC derivatives bearing polar and ionizable tertiary amine functionalities were synthesized. Cell based MTT assay resulted in the discovery of one compound (2b) which shows a broad spectrum of cytotoxic activity (IC50 < 5 μM against all tested sensitive cancer cells) and moderate selectivity between normal and tumor cells. Further drug combination study uncovered that all of the semi-synthetic DMC derivatives (2a–2f) acted synergistically with anticancer drug Taxol® to achieve improved anti-proliferative efficacy against drug resistant HeLa/Tax cells. The combinations of Taxol® with each of the two most effective compounds (2b and 2d) displayed weighted average Combination Index (CI) values of 0.14 and 0.15, respectively, and significantly reduced the dosage of Taxol® while maintaining the same level of efficacy. Our findings suggested that the introduction of a polar and ionizable tertiary amine functionality into the 4′-OH of DMC is a feasible way to improve its aqueous solubility, anticancer activity, and cancer selectivity.
1. Introduction
Chalcones constitute an important class of plant-derived natural products with a wide range of biological and pharmacological properties. Several review articles comprehensively summarized their anticancer, anti-infection, anti-inflammatory, antioxidant, and anti-hyperglycemic activities as well as their identified molecular targets.1–9 A number of chalcones have been approved for clinical use, including the choleretic drug metochalcone and the gastrointestinal medication sofalcone (Fig. 1).8 Structurally, chalcones carry an α,β-unsaturated carbonyl system, which is further conjugated with two aromatic rings at both end (Fig. 1). As a member of this huge family, 2′,4′-dihydroxyl-6′-methoxyl-3′,5′-dimethylchalcone (DMC, Fig. 1), featuring a 3′,5′-dimethyl substituted ring A, was originally isolated from the leaves of Myrica gale in 1977 (ref. 10) and later found in several other species including Syzygium samarangense,11 Psorothamnus polydenius,12 and Cleistocalyx operculatus.13 It is abundantly accumulated in the buds of Cleistocalyx operculatus which has been used for preparing tonic drinks in southern China for centuries.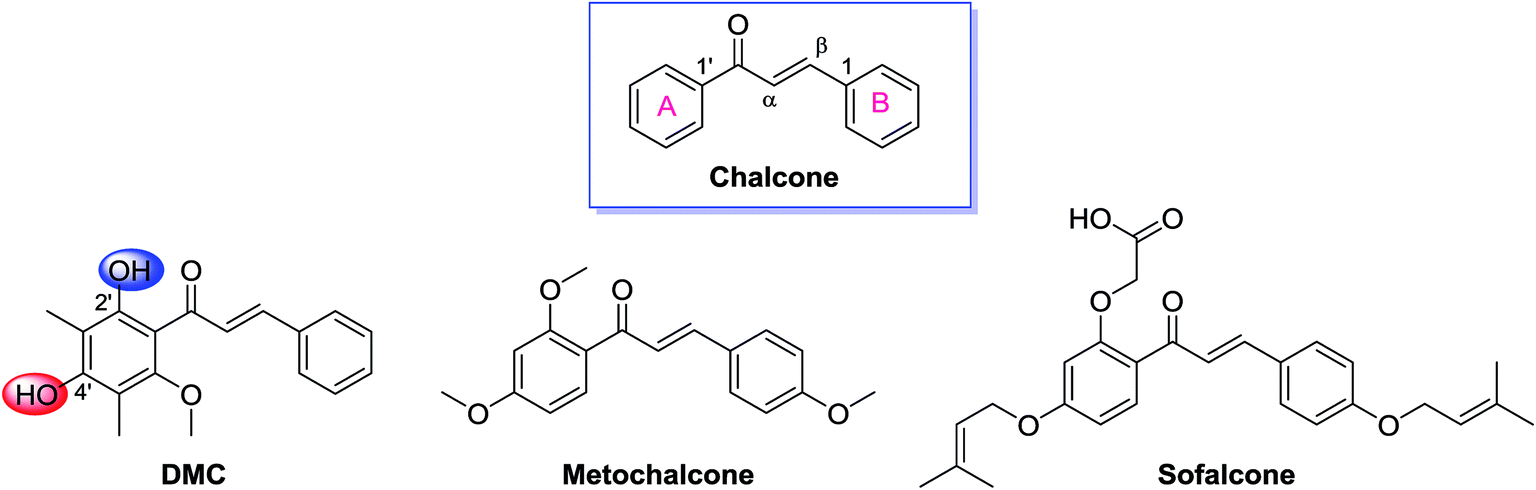 | ||
| Fig. 1 Structures of chalcone, DMC, and two clinically approved drugs, metochalcone and sofalcone. | ||
The pharmacological profile of this C-methylated chalcone has been extensively studied.14–22 Ye et al. disclosed that DMC not only effectively inhibited the growth of six human cancer cell lines at micromolar concentration in cell based assay,14 but also significantly suppressed the tumorous development of solid human carcinoma in xenograft model.15 More interestingly, Qian et al. discovered that DMC was able to potentiate the cytotoxicity of doxorubicin against drug resistant KB-A1 cells both in vitro and in vivo by increasing the intracellular accumulation of doxorubicin and modulating the expression of MDR1 gene which encodes the transporter P-glycoprotein.16 Similar multidrug resistance (MDR) reversal effect of DMC was also observed by Huang et al. in a human hepatocellular carcinoma BEL-7402/5-FU cell line.17,18 This intriguing pharmacological profile of DMC makes it a promising anticancer drug lead in a world crying for alternative drugs to treat patients who are suffering from multidrug resistant cancers.
However, DMC is barely soluble in aqueous solution due to its lipophilic nature. This physicochemical shortcoming severely hampers its development for clinical use. Earlier studies have shown that the incorporation of polar and ionizable functional group(s) into the structure of a lead compound is a feasible approach to solve this troublesome issue.23 In a pool of suitable ionizable groups, tertiary amine functionality appears to be a superior choice by virtue of its excellent ability to ionize, the easiness of introducing an aminoalkly group onto the phenolic hydroxyl of DMC, and the fact that installation of heterologous atom containing side chain usually benefits biological activities of the lead compound. By following these designing logic, we synthesized a group of DMC derivatives bearing various polar and ionizable tertiary amine groups using DMC as the starting material. The cytotoxicity of these semi-synthetics was investigated against five cell lines (A549, HepG2, MCF-7, HeLa, and Vero), and a drug combination study was conducted to evaluate the synergistic effect of these compounds with Taxol® against a multi-drug resistant cancer cell line (HeLa/Tax). To the best of our knowledge, this is the first chemical modification study of DMC aiming to improve its aqueous solubility and anticancer property.
2. Results and discussion
2.1. Synthesis and structural characterization
DMC possesses two free aromatic hydroxyl groups in the ring A (Fig. 1). The one attached to C-2′ is unfavorable for chemical modification because it is involved in an intra-molecular hydrogen bond with the adjacent carbonyl group and its reactivity is reduced. The reactivity of 4′-OH is much higher and could act as an effective nucleophile in SN2 reactions in the presence of a relatively weak base. Herein, we linked the nitrogen atom of different secondary amines, which represent non-cyclic, cyclic, and aromatic subgroups, with the 4′-OH of DMC via a four carbon bridge (Scheme 1). The starting material DMC was isolated from Cleistocalyx operculatus as previously described (see the Experimental section, 4.1 General, for more details) and treated with 1,4-dibromobutane in the presence of K2CO3 in acetone under reflux to give bromobutyl-substituted 1 in 59% yield. The following coupling of 1 with different secondary amines was conducted in chloroform using K2CO3 as base, providing corresponding tertiary amine derivatives 2a–2f in good yield. All the synthesized compounds were purified by silica gel CC and/or preparative HPLC and their structures were confirmed by comprehensive analysis of their HR-ESIMS data, 1H-NMR and 13C-NMR spectral data.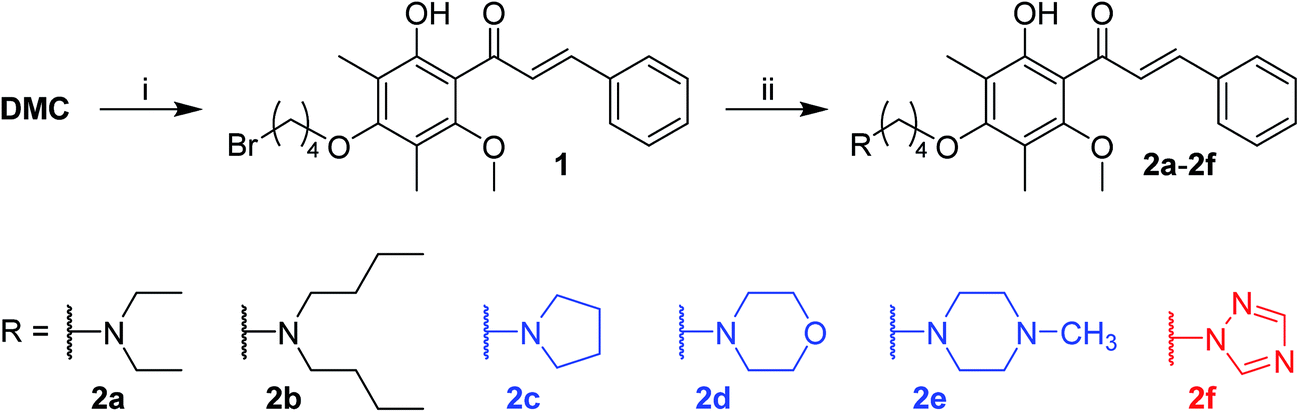 | ||
| Scheme 1 Reagents and conditions: (i) 1,4-dibromobutane, acetone, K2CO3, reflux, 10 h, 59%; (ii) secondary amines, K2CO3, CHCl3, reflux, 40–88%. | ||
2.2. Biological evaluation
| Compound | Cell lines | |||||
|---|---|---|---|---|---|---|
| A549 | HepG2 | MCF-7 | HeLa | HeLa/Tax | Vero | |
| a Values represent means ± SD based on three individual experiments.b n.t. not tested.c Values expressed in nM. | ||||||
| 1 | 7.6 ± 0.76 | 8.5 ± 0.32 | 5.2 ± 0.12 | 10.9 ± 0.10 | n.t.b | 4.1 ± 0.81 |
| 2a | 10.3 ± 0.21 | 14.3 ± 0.07 | 9.6 ± 0.68 | 7.4 ± 0.27 | 13.5 ± 0.004 | 11.7 ± 1.19 |
| 2b | 3.8 ± 0.55 | 4.3 ± 0.57 | 1.3 ± 0.16 | 2.3 ± 0.08 | 7.9 ± 0.63 | 14.9 ± 0.20 |
| 2c | 14.3 ± 0.64 | 17.9 ± 0.21 | 6.5 ± 0.96 | 17.6 ± 0.88 | 19.0 ± 0.57 | 25.6 ± 4.15 |
| 2d | 24.7 ± 3.94 | 19.1 ± 0.44 | 15.4 ± 1.99 | 20.2 ± 1.45 | 22.0 ± 1.18 | 11.2 ± 0.75 |
| 2e | 13.0 ± 0.89 | 10.8 ± 1.09 | 11.3 ± 1.39 | 4.9 ± 0.23 | 12.8 ± 0.71 | 5.6 ± 0.88 |
| 2f | 9.4 ± 1.34 | 11.8 ± 0.43 | 15.1 ± 0.14 | 6.1 ± 0.61 | 7.8 ± 0.16 | 12.3 ± 0.12 |
| DMC | 2.3 ± 0.44 | 8.3 ± 0.22 | 15.1 ± 0.72 | 11.8 ± 0.42 | 15.3 ± 0.19 | 20.3 ± 4.08 |
| Doxorubicin | 0.7 ± 0.06 | 1.2 ± 0.02 | 1.0 ± 0.17 | 0.5 ± 0.05 | n.t.b | 0.4 ± 0.01 |
| Taxol® | n.t.b | n.t.b | n.t.b | 0.2 ± 0.01c | 0.5 ± 0.002 | n.t.b |
In general, all tertiary amine derivatives (2a–2f) exhibited good activity against all tested cancer cell lines with IC50 values ranging from 1.3 to 24.7 μM. Compounds 2a, 2e, and 2f displayed higher activity against HeLa cell line (IC50 < 8 μM) while 2b–2d were more potent against MCF-7 cells. Among these DMC derivatives, 2b is the most active compound, having IC50 values all below 5 μM against tested sensitive cancer cell lines and specifically an IC50 of 1.3 μM against MCF-7 cells. 2b is 11.6 times and 5.1 times more potent than DMC against MCF-7 cells and HeLa cells, respectively, and its efficacy on MCF-7 cells is comparable to the clinically approved anticancer drug doxorubicin. 2a, 2c, 2e, and 2f also showed potency enhancement against certain cancer cell line when compared to DMC (2c: 6.5 μM vs. DMC: 15.1 μM against MCF-7 cells; 2a: 7.4 μM, 2e: 4.9 μM and 2f: 6.1 μM vs. DMC: 11.8 μM against HeLa cells). Additionally, the bromobutyl-substituted synthetic intermediate (1) exhibited a broad range of cytotoxic activity as well (Table 1).
Several studies have shown that DMC is able to reverse cancer cell multidrug resistance by suppressing the expression of MDR1 gene.16–18 In this study we included a Taxol®-resistant HeLa cell subline, HeLa/Tax, to show whether our DMC derivatives are active against multidrug-resistant cancer cells. The HeLa/Tax cells were established by continuous exposing HeLa cells to Taxol® at a gradient concentration, which became resistant to Taxol® and other chemodrugs (see Experimental for more details). Most of the synthesized compounds that are cytotoxic to HeLa cells also display cytotoxicity to the Taxol®-resistant HeLa/Tax cells, albeit at somewhat reduced potency (Table 1). Compounds 2b and 2f have IC50 below 10 μM, indicating that they are quite potent against Taxol®-resistant HeLa/Tax cells. Whether these DMC derivatives are able to reverse multidrug resistance requires further studies (see also the Drug combination study in section 2.2.2 below).
In order to find out whether these DMC derivatives are toxic to normal cells, their anti-proliferative activity against monkey kidney epithelial cells (Vero) were evaluated, and the results were shown in Table 1. Although the tertiary amine derivatives showed considerable growth inhibitory activity to Vero cells, some of them, such as 2b and 2c, were found to be less toxic to normal cells than to certain type of cancer cells (2b: 1.3 μM to MCF-7 cells vs. 14.9 μM to Vero cells; 2c: 6.5 μM to MCF-7 cells vs. 25.6 μM to Vero cells), and hence these compounds have moderate selectivity towards cancer cells over normal cells.
A preliminary structure–activity relationship (SAR) analysis of these DMC derivatives indicates that, for DMC derivatives carrying non-cyclic tertiary amine groups, longer aliphatic chains on the nitrogen atom is superior to shorter ones in improving cytotoxicity (2b is 7 times more potent than 2a against MCF-7 cells and 3 times more potent than 2a against A549, HepG2, and HeLa cells). In the case of cyclic tertiary amine substituents, compound 2d with morpholine moiety is less active than those with N-methyl piperazine and pyrrolidine (2e and 2c), which might be related to the reduced basicity of the nitrogen atom in morpholine. Compound 2f with a 1,2,4-triazole moiety displays higher potency than 2c (with a pyrrolidine moiety) against most cell lines except MCF-7. In fact, 2f is the most potent compound against the Taxol®-resistant HeLa/Tax cells. These observations indicate that multiple nitrogen atoms introduced in DMC are tolerated and may benefit the anticancer effect of such derivatives.
DMC is known to promote apoptosis in human breast adenocarcinoma MCF-7 cells by activating poly(adenosine diphosphate-ribose) polymerase (PARP),25 which is one of the best biomarker of apoptosis. Ye et al. also reported that DMC was able to induce apoptosis in human hepatocarcinoma SMMC-7721 cells, as indicated by the fragmentation and condensation of chromatin in the cells and the increase on the percentage of hypodiploid cells showed by flowcytometric analysis.14 Furthermore, Zhu et al. demonstrated that DMC inhibited tumor growth of human hepatocarcinoma Bel7402 and lung cancer GLC-82 xenografts in nude mice, and their study showed that DMC inhibited vascular endothelial growth factor receptor (kinase-inserting domain-containing receptor, KDR) tyrosine kinase and shut down KDR-mediated signal transduction.22 The detailed anticancer mechanism of our DMC derivatives needs further investigation.
The term Combination Index (CI) was introduced by Chou and Talaly in 1980s to quantitatively describe drug combination effect as synergism (CI < 1), additive effect (CI = 1), or antagonism (CI > 1).35,36 A corresponding computer software for dose-effect analysis, CI calculation, and Fa–CI plot simulation was developed by Joseph Chou in 1989, and its second (CalcuSyn, 1997)37 and third (CompuSyn, 2005)38 generations were written by Mike Hayball and Nick Martin, respectively. In this study, a drug combination assay using Chou–Talalay method was carried out to investigate whether the synthesized DMC derivatives could work synergistically with Taxol®. For specific drug combination assay, the IC50 values of Taxol® and DMC derivatives against HeLa/Tax cells (Table 1) were used as a guide to choose individual drug concentration.
Each drug combo was prepared at a constant ratio of (IC50)1/(IC50)2 so that each drug contributes equally to the cytotoxic effect.32,36 This was followed by serial duplicate dilutions to afford six gradient concentrations. The Taxol®-resistant HeLa/Tax cells were treated with Taxol® alone, each DMC derivative alone, and the corresponding combination for 72 h. CI was calculated by the CompuSyn software using the general equation: 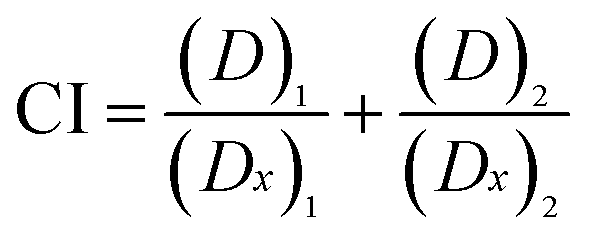 , in which the denominator (Dx)1 and (Dx)2 are the concentration of Drug 1 and Drug 2 alone that inhibit the growth of cells by x%. Similarly, the numerator (D)1 and (D)2 represent the respective dosage of Drug 1 and Drug 2 at which their drug combo inhibits the cell growth by the same x%. The CI values of each drug combo at 50%, 75% and 90% growth inhibition (GI50, GI75, and GI90) were listed in Table 2.
, in which the denominator (Dx)1 and (Dx)2 are the concentration of Drug 1 and Drug 2 alone that inhibit the growth of cells by x%. Similarly, the numerator (D)1 and (D)2 represent the respective dosage of Drug 1 and Drug 2 at which their drug combo inhibits the cell growth by the same x%. The CI values of each drug combo at 50%, 75% and 90% growth inhibition (GI50, GI75, and GI90) were listed in Table 2.
| Comp. | Drug ratio Comp./Taxol®c | CI at | CIavga | DRI of Taxol® at | ||||
|---|---|---|---|---|---|---|---|---|
| GI50 | GI75 | GI90 | GI50 | GI75 | GI90 | |||
| a CI values of each compounds are presented at 50%, 75%, and 90% of growth inhibition (GI50, GI75, and GI90). CIavg = (CI50 + 2CI75 + 3CI90)/6, is the weighted average CI, and CI < 1, CI = 1, and CI > 1 represents synergism, additive effect, and antagonism, respectively.b DRI value of Taxol® in each combination assay is presented at GI50, GI75, and GI90 effect levels.c Each drug combination was prepared at equipotency ratio [(IC50)1/(IC50)2 ratio]. | ||||||||
| 2a | 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
0.33 ± 0.01 | 0.24 ± 0.01 | 0.18 ± 0.001 | 0.22 | 9.36 ± 2.4 | 11.61 ± 0.21 | 14.84 ± 3.30 |
| 2b | 14:1 |
0.26 ± 0.03 | 0.15 ± 0.01 | 0.10 ± 0.005 | 0.14 | 8.60 ± 1.97 | 11.31 ± 0.66 | 15.09 ± 1.72 |
| 2c | 40:1 |
0.36 ± 0.04 | 0.30 ± 0.009 | 0.25 ± 0.02 | 0.29 | 10.49 ± 1.48 | 10.74 ± 0.52 | 11.20 ± 2.65 |
| 2d | 40:1 |
0.28 ± 0.02 | 0.17 ± 0.007 | 0.10 ± 0.02 | 0.15 | 8.26 ± 0.95 | 11.95 ± 1.09 | 17.64 ± 5.20 |
| 2e | 30:1 |
0.45 ± 0.06 | 0.39 ± 0.05 | 0.34 ± 0.03 | 0.38 | 7.41 ± 0.98 | 8.03 ± 0.41 | 8.86 ± 2.07 |
| 2f | 14:1 |
0.60 ± 0.02 | 0.47 ± 0.007 | 0.41 ± 0.007 | 0.46 | 3.39 ± 0.08 | 6.29 ± 0.32 | 11.68 ± 1.44 |
| DMC | 30:1 |
0.67 ± 0.05 | 0.55 ± 0.03 | 0.45 ± 0.02 | 0.52 | 4.40 ± 0.49 | 5.29 ± 0.31 | 6.38 ± 0.03 |
The concept of dose-reduction index (DRI) was originally introduced by Chou JH and Chou TC in 1988 (ref. 39) to describe how many-folds the dose of each drug was reduced at certain effect levels in the drug combo compared with each drug alone.32 The equation for calculating DRI is  and
and 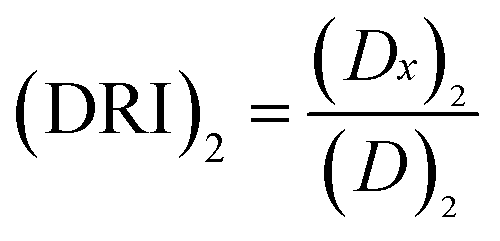 , which is a simple inversion of CI equation, and DRI could be generated by CompuSyn software. The DRI values of Taxol® in each drug combo at 50%, 75%, and 90% cell growth inhibition (GI50, GI75 and GI90) were calculated and listed in Table 2.
, which is a simple inversion of CI equation, and DRI could be generated by CompuSyn software. The DRI values of Taxol® in each drug combo at 50%, 75%, and 90% cell growth inhibition (GI50, GI75 and GI90) were calculated and listed in Table 2.
All tested DMC derivatives display synergistic effects with Taxol® against HeLa/Tax cells, having the weighted average CI (CIavg) values all below 0.5. The combinations of Taxol®/2b and Taxol®/2d exhibit the strongest synergy at different levels of growth inhibition (GI50, GI75, and GI90, Table 2), which result in CIavg of 0.14 and 0.15, respectively. Compounds 2b and 2d reduced the dosage of Taxol® by more than 15-folds at high effect level (with DRI value >15 at GI90, Table 2). Compounds 2a, and 2c are also able to work synergistically with Taxol® (CIavg = 0.22, and 0.29, respectively) and markedly enhance the potency of Taxol® with DRI values around 10 at GI50, GI75 and GI90 effect levels. The other two derivatives 2e and 2f are not as effective, but their drug combos with Taxol® still show stronger synergistic effect than the Taxol®/DMC combination.
The combination index (CI) for each drug combo usually has a different value at a different effect level. A plot of CI values at different effect levels (Fa, fraction affected, i.e., % growth inhibition in this case) can be determined by computer simulation. Such a combination index plot is also called the Fa–CI plot.36 As examples, the Fa–CI plots for drug combos of Taxol® with DMC, 2b or 2d were simulated by CompuSyn and presented in Fig. 2. The plots clearly show the trend that the CI value decreases as the effect level (Fa) increases, indicating that a higher level of synergism is observed at a higher effect level for a drug combo.
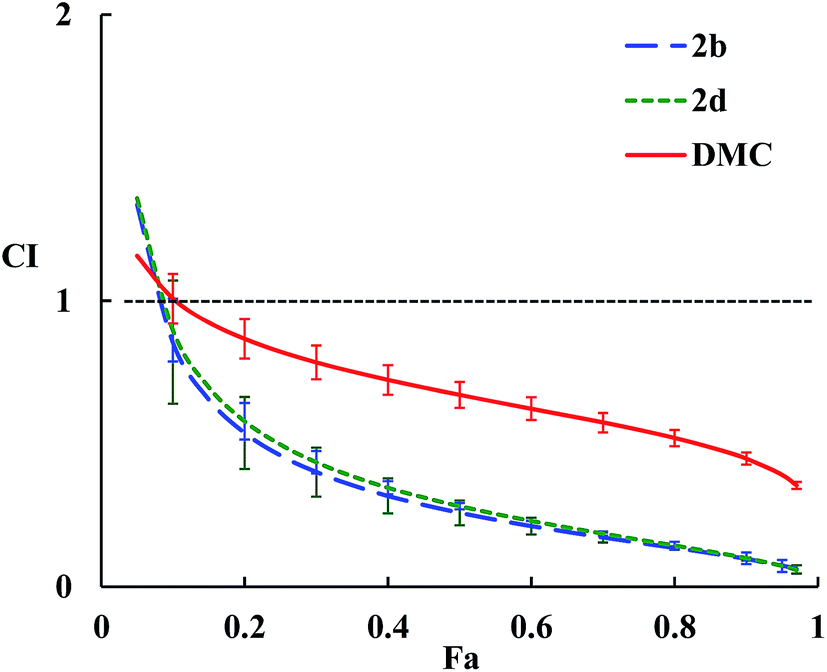 | ||
| Fig. 2 Fa–CI plots for drug combos of Taxol® with 2b, 2d, or DMC. The dashed horizontal line expresses CI = 1, and error bars show standard deviations. | ||
Structurally speaking, compound 2b with a di-butylamino substituent displays the highest synergistic effect with Taxol® among all the DMC derivatives tested. Compound 2b alone also shows the highest cytotoxicity against all cancer cells tested (Table 1). In contrast, compound 2d with a morpholine residue as the substituent exhibits a similar level of synergism with Taxol® as 2b does but the lowest cytotoxicity against all cancer cells tested including HeLa/Tax cells. Furthermore, compound 2f (with a triazole-moiety) is most cytotoxic against HeLa/Tax cells among all DMC derivatives but shows the lowest synergistic effect in combination with Taxol®. These observations indicate that the level of synergism for each drug combo does not correlate with the cytotoxic potency of individual DMC derivative. These data confirm the earlier findings that drug synergism is a complicated issue.32 The potential of these DMC derivatives to reverse multidrug resistance in HeLa/Tax cells might be a contributing factor for the strong synergism observed between Taxol® and individual DMC derivative.
2.3. Solubility
The solubility of DMC and selected synthetic derivatives in pure water with 0.1% formic acid (pH = 2.9) were determined by quantitative ultra-performance liquid chromatography (UPLC) analysis. The tested DMC derivatives bearing tertiary amine functional groups (2a and 2e) showed good solubility in acidic aqueous solution (2.27 and 1.04 μmol mL−1, respectively) while DMC is barely soluble (0.004 μmol mL−1). On the other hand, compound 2f (with a triazole moiety) shows a much lower solubility in the same medium (0.04 μmol mL−1), which correlates with the reduced basicity of the nitrogen atoms in the aromatic triazole ring.3. Conclusion
In summary, a group of DMC derivatives bearing different tertiary amine functionalities were readily synthesized. Their cytotoxicity and drug–drug interaction effects with Taxol® were investigated using cell based assays. All semi-synthetic DMC derivatives exhibited moderate to strong cytotoxicity against tested cell lines, with 2b being the most active compound which improved the potency of DMC by about 2-folds against HepG2 cells, 11-folds against MCF-7 cells, and 5-folds against HeLa cells. Compound 2b also showed considerable selectivity toward cancer cells over normal cells.The drug combination study uncovered that all the cytotoxic derivatives displayed moderate to strong drug synergism with Taxol®. The most cytotoxic derivative 2b also possesses the strongest synergistic effect with Taxol® with a combination index (CI) of 0.10 and a dose-reduction index (DRI) of 15.09 for Taxol® at 90% growth inhibition (GI90). Solubility study suggested that the tertiary amine derivatives were more soluble than DMC in pure water containing 0.1% formic acid (pH = 2.9). Overall, the study shows that the introduction of a tertiary amine functional group into the 4′-OH of DMC can improve the aqueous solubility, anticancer activity, and cancer selectivity of DMC. These findings may shed lights on the rational design of other chalcone or flavonoid derivatives aiming to improve their biological activities and physicochemical properties.
4. Experimental
4.1. General
All reagents and solvents were purchased from commercial sources, and further purification and drying by standard methods were employed when necessary. 1H and 13C NMR spectra were collected on a Bruker AVIII 500M instrument at 500 (1H) and 125 (13C) MHz or a Bruker Avance-600 instrument at 600 (1H) and 150 (13C) MHz. Chemical shifts are reported as ppm (δ units) using the residual solvent peak as reference and the coupling constants as J in hertz. The splitting pattern abbreviations are as follows: s = singlet, d = doublet, dd = double doublet, t = triplet, m = multiplet, brs = broad singlet. ESIMS data were obtained on an MDS SCIEX API 2000 LC/MS instrument. HRESIMS data were obtained on a Bruker maXis Q-TOF mass spectrometer. Preparative HPLC was run on a Waters 600 pump and a Waters 2487 dual λ absorbance detector. All separations were carried out with an SHIMADZU Shim-Pack Pro-ODS C18 column (20 mm × 25 cm) at a flow rate of 5 mL min−1. Reactions were monitored by TLC and spots were visualized by heating silica gel plates sprayed with 10% H2SO4 in EtOH.Our previous phytochemical study on the buds of Cleistocalyx operculatus demonstrated the isolation and structural characterization of 2′,4′-dihydroxy-6′-methoxy-3′,5′-dimethylchalcone (DMC), 5,7-dihydroxy-6,8-dimethylflavanone, 7-hydroxy-5-methoxy-6,8-dimethylflavanone, ethyl gallate, gallic acid, ursolic acid, β-sitosterol, and cinnamic acid.40 Similar process was applied herein to afford adequate amount of DMC for chemical modification. Briefly, 5 kg fresh buds of C. operculatus were soaked in 95% aqueous ethanol for 24 h. The ethanol solution was collected after filtration and then concentrated under vacuum to afford the crude extract. The crude extract was re-suspended in water and consecutively extracted with diethyl ether, ethyl acetate, and n-pentane (5 times each). The ethyl acetate extract was concentrated to provide a paste 13 g which was subjected to silica gel column chromatography eluted with gradient solution of chloroform–methanol (100:0, 99:1, 92:2, and 97:3, v/v). Orange precipitates were formed and collected from the 100:0 chloroform–methanol eluted fraction and then washed by methanol to afford DMC (0.69 g) in good purity.
4.2. Preparation of (E)-1-(4-(2-bromoethoxy)-2-hydroxy-6-methoxy-3,5-dimethylphenyl)-3-phenylprop-2-en-1-one (1)
1,4-Dibromobutane (1.45 g, 6.72 mmol) was added to a mixture of DMC (500 mg, 1.68 mmol) and anhydrous K2CO3 (927 mg, 6.72 mmol) in dry acetone (150 mL) and stirred under reflux for 6 h. After removing the solvent under reduced pressure, the residue was dissolved in EtOAc and sequentially washed with 1% aqueous HCl solution, distilled water and dried over anhydrous Na2SO4. The crude product was purified by silica gel column chromatography eluting with petroleum ether/acetone (v/v, from 99:1 to 85:15) to afford compound 1 as an orange waxy solid; 59% yield; 1H NMR (500 MHz, CDCl3) δ: 13.11 (s, 1H, 2′-OH), 7.97 (d, J = 15.7 Hz, 1H, H-β), 7.86 (d, J = 15.7 Hz, 1H, H-α), 7.64 (m, 2H, H-2, H-6), 7.41 (m, 3H, H-3, H-4, H-5), 3.84 (t, J = 6.2 Hz, 2H, H-1′′), 3.66 (s, 3H, 6′-OCH3), 3.52 (t, J = 6.6 Hz, 2H, H-4′′), 2.16 and 2.15 (s, each 3H, 3′-CH3 and 5′-CH3), 2.13 (m, 2H, H-2′′), 1.98 (m, 2H, H-3′′); 13C NMR (125 MHz, CDCl3) δ: 194.10 (CO), 162.65 (C-6′), 161.74 (C-4′), 158.79 (C-2′), 143.51 (C-β), 135.29 (C-1), 130.48 (C-4), 129.06 (C-3, C-5), 128.60 (C-2, C-6), 126.63 (C-α), 115.78 and 115.74 (C-3′ and C-5′), 111.91 (C-1′), 71.87 (C-1′′), 62.41 (6′-OCH3), 33.57 (C-4′′), 29.61 and 29.07 (C-2′′ and C-3′′), 9.19 and 9.06 (3′-CH3 and 5′-CH3); HRESIMS m/z: 455.0844 [M + Na]+ (calcd for C22H25BrNaO4, 455.0828).
4.3. General procedure for the synthesis of compounds 2a–2f
Different secondary amine (0.48 mmol) was added into a mixture of compound 1 (50 mg, 0.12 mmol) and anhydrous K2CO3 (0.48 mmol) in CHCl3 (20 mL) and stirred under reflux for 10 h. The reaction mixture was washed with distilled water three times and dried over anhydrous Na2SO4. The crude products were subjected to preparative HPLC using CH3OH/H2O (v/v, from 50:50 to 100:0, linear gradient, 60 min) as eluents to afford compounds 2a–2f.
4.4. Cytotoxic activity
4.5. Solubility
The solubility of selected compounds (2a, 2e, and 2f) were evaluated using UPLC quantitative analysis with DMC as reference. The UPLC runs were carried out on a Waters ACQUITY UPLC H-Class system (Waters, Milford, MA, USA) equipting a Waters ACQUITY UPLC BEH C18 column (1.7 μm, 2.1 × 50 mm). Samples were eluded with gradient acetonitrile/water (A/W) solvent system (initial A/W = 30/70, 10 min linear gradient to A/W = 80/20) at a flow rate of 0.5 mL min−1. Six standard samples of each test compounds with gradient concentrations were prepared by dilution of a methanol stock solution of which the exact concentration was known. Prepared samples were injected into the UPLC system and the standard curves were automatically generated by the software Empower3 (Waters) with r2 no less than 0.995. Excess amount of the tertiary amine derivatives 2a, 2e, and 2f were suspended in pure water containing 0.1% formic acid (pH = 2.9). The mixtures were sonicated for 5 min at room temperature and followed by centrifugation at 12000 rpm for 2 min using a Thermo Fisher microcentrifuge (Thermo Fisher Scientific, Boston, MA, USA). An aliqout of the clear supernates was injected into the UPLC system, and the peak area values of each sample was substituted into the equation, which was generated by the corresponding standard curve, to calculate the solubility.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Mr Yunfei Yuan, South China Botanical Garden, Chinese Academy of Sciences, for NMR spectroscopic measurements, and Ms Aijun Sun, South China Sea Institute of Oceanology, Chinese Academy of Sciences, for HRESIMS measurements. This work was supported by National Science Foundation of China (NSFC) (Grant no. 31470423 and 31528002).References
- A. J. Leon-Gonzalez, N. Acero, D. Munoz-Mingarro, I. Navarro and C. Martin-Cordero, Curr. Med. Chem., 2015, 22, 3407–3425 CrossRef CAS PubMed.
- D. K. Mahapatra, S. K. Bharti and V. Asati, Eur. J. Med. Chem., 2015, 98, 69–114 CrossRef CAS PubMed.
- D. K. Mahapatra, S. K. Bharti and V. Asati, Eur. J. Med. Chem., 2015, 101, 496–524 CrossRef CAS PubMed.
- Z. Nowakowska, Eur. J. Med. Chem., 2007, 42, 125–137 CrossRef CAS PubMed.
- C. Kontogiorgis, M. Mantzanidou and D. Hadjipavlou-Litina, Mini-Rev. Med. Chem., 2008, 8, 1224–1242 CrossRef CAS PubMed.
- J. R. Dimmock, D. W. Elias, M. A. Beazely and N. M. Kandepu, Curr. Med. Chem., 1999, 6, 1125–1149 CAS.
- P. Singh, A. Anand and V. Kumar, Eur. J. Med. Chem., 2014, 85, 758–777 CrossRef CAS PubMed.
- N. K. Sahu, S. S. Balbhadra, J. Choudhary and D. V. Kohli, Curr. Med. Chem., 2012, 19, 209–225 CrossRef CAS PubMed.
- B. Zhou and C. Xing, Med. Chem., 2015, 5, 388 Search PubMed.
- K. E. Malterud, T. Anthonsen and G. B. Lorentzen, Phytochemistry, 1977, 16, 1805–1809 CrossRef CAS.
- R. Srivastava, A. K. Shaw and D. K. Kulshreshtha, Phytochemistry, 1995, 38, 687–689 CrossRef CAS.
- M. M. Salem and K. A. Werbovetz, J. Nat. Prod., 2005, 68, 108–111 CrossRef CAS PubMed.
- C.-L. Ye, Y.-H. Lu and D.-Z. Wei, Phytochemistry, 2004, 65, 445–447 CrossRef CAS PubMed.
- C.-L. Ye, J.-W. Liu, D.-Z. Wei, Y.-H. Lu and F. Qian, Pharmacol. Res., 2004, 50, 505–510 CrossRef CAS PubMed.
- C.-L. Ye, J.-W. Liu, D.-Z. Wei, Y.-H. Lu and F. Qian, Cancer Chemother. Pharmacol., 2005, 56, 70–74 CrossRef CAS PubMed.
- F. Qian, C.-L. Ye, D.-Z. Wei, Y.-H. Lu and S.-L. Yang, J. Chemother., 2005, 17, 309–314 CrossRef CAS PubMed.
- H.-Y. Huang, J.-L. Niu and Y.-H. Lu, J. Sci. Food Agric., 2012, 92, 135–140 CrossRef CAS PubMed.
- H.-Y. Huang, J.-L. Niu, L.-M. Zhao and Y.-H. Lu, Phytomedicine, 2011, 18, 1086–1092 CrossRef CAS PubMed.
- Y. Sone, J. K. Moon, T. T. Mai, N. Thu, E. Asano, K. Yamaguchi, Y. Otsuka and T. Shibamoto, J. Sci. Food Agric., 2011, 91, 2259–2264 CAS.
- W.-G. Yu, J. Qian and Y.-H. Lu, J. Agric. Food Chem., 2011, 59, 12821–12829 CrossRef CAS PubMed.
- Y.-C. Hu, D.-M. Hao, L.-X. Zhou, Z. Zhang, N. Huang, M. Hoptroff and Y.-H. Lu, J. Agric. Food Chem., 2014, 62, 1602–1608 CrossRef CAS PubMed.
- X.-F. Zhu, B.-F. Xie, J.-M. Zhou, G.-K. Feng, Z.-C. Liu, X.-Y. Wei, F.-X. Zhang, M.-F. Liu and Y.-X. Zeng, Mol. Pharmacol., 2005, 67, 1444–1450 CrossRef CAS PubMed.
- V. J. Stella and K. W. Nti-Addae, Adv. Drug Delivery Rev., 2007, 59, 677–694 CrossRef CAS PubMed.
- J.-F. Shi, P. Wu, Z.-H. Jiang and X.-Y. Wei, Eur. J. Med. Chem., 2014, 71, 219–228 CrossRef CAS PubMed.
- A. Subarnas, A. Diantini, R. Abdulah, A. Zuhrotun, Y. E. Hadisaputri, I. M. Puspitasari, C. Yamazaki, H. Kuwano and H. Koyama, Oncol. Lett., 2015, 9, 2303–2306 Search PubMed.
- M. Figul, A. Söling, H. Dong, T.-C. Chou and N. Rainov, Cancer Chemother. Pharmacol., 2003, 52, 41–46 CrossRef CAS PubMed.
- M. Balzarotti, E. Ciusani, C. Calatozzolo, D. Croci, A. Boiardi and A. Salmaggi, Oncol. Res., 2004, 14, 325–330 CAS.
- A. Martins, N. Tóth, A. Ványolós, Z. Béni, I. Zupkó, J. Molnár, M. Báthori and A. Hunyadi, J. Med. Chem., 2012, 55, 5034–5043 CrossRef CAS PubMed.
- X. Kong, Q. Zhu, R. M. Ruprecht, K. A. Watanabe, J. M. Zeidler, J. Gold, B. Polsky, D. Armstrong and T.-C. Chou, Antimicrob. Agents Chemother., 1991, 35, 2003–2011 CrossRef CAS PubMed.
- K. Y. Hostetler, J. L. Hammond, G. D. Kini, S. E. Hostetler, J. R. Beadle, K. A. Aldern, T.-C. Chou, D. D. Richman and J. W. Mellors, Antiviral Chem. Chemother., 2000, 11, 213–219 CrossRef CAS PubMed.
- K. L. Hartshorn, M. W. Vogt, T.-C. Chou, R. S. Blumberg, R. Byington, R. Schooley and M. Hirsch, Antimicrob. Agents Chemother., 1987, 31, 168–172 CrossRef CAS PubMed.
- T.-C. Chou, Pharmacol. Rev., 2006, 58, 621–681 CrossRef CAS PubMed.
- A. Goldin and N. Mantel, Cancer Res., 1957, 17, 635–654 CAS.
- W. R. Greco, G. Bravo and J. C. Parsons, Pharmacol. Rev., 1995, 47, 331–385 CAS.
- T.-C. Chou and P. Talalay, Trends Pharmacol. Sci., 1983, 4, 450–454 CrossRef CAS.
- T.-C. Chou and P. Talalay, Adv. Enzyme Regul., 1984, 22, 27–55 CrossRef CAS PubMed.
- T.-C. Chou and M. Hayball, CalcuSyn for windows: multiple drug dose-effect analyzer and manual, Biosoft, Cambridge, UK, 1997 Search PubMed.
- T.-C. Chou and N. Martin, CompuSyn for drug combinations: PC software and user's guide: a computer program for quantitation of synergism and antagonism in drug combinations, and the determination of IC50 and ED50 and LD50 values, ComboSyn, Paramus, NJ, 2005 Search PubMed.
- J. Chou and T.-C. Chou, Computerized simulation of dose reduction index (DRI) in synergistic drug combinations, Pharmacologist, 1988, 30, A231 Search PubMed.
- F.-X. Zhang, M.-F. Liu and R.-R. Lu, Acta Bot. Sin., 1990, 32, 469–472 CAS.
- X. Peng, F. Gong, Y. Chen, Y. Jiang, J. Liu, M. Yu, S. Zhang, M. Wang, G. Xiao and H. Liao, Cell Death Dis., 2014, 5, e1367 CrossRef CAS PubMed.
- K. Takara, Y. Obata, E. Yoshikawa, N. Kitada, T. Sakaeda, N. Ohnishi and T. Yokoyama, Cancer Chemother. Pharmacol., 2006, 58, 785–793 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08639c |
| This journal is © The Royal Society of Chemistry 2017 |