
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtective effects of canolol against hydrogen peroxide-induced oxidative stress in AGS cells
Ling Han,
Xiaoyang Xia,
Xia Xiang *,
Fenghong Huang* and
Zhen Zhang
*,
Fenghong Huang* and
Zhen Zhang
Oil Crops Research Institute of the Chinese Academy of Agricultural Sciences, Oil Crops and Lipids Process Technology National & Local Joint Engineering Laboratory, Key Laboratory of Oilseeds Processing, Ministry of Agriculture, Hubei Key Laboratory of Lipid Chemistry and Nutrition, Key Laboratory of Biology and Genetic Improvement of Oil Crops, Ministry of Agriculture, Wuhan 430062, China. E-mail: xiangshi19850130@163.com; oiljgzx@gmail.com; Tel: +86-27-86711526
First published on 5th September 2017
Abstract
Oxidative stress, as mediated by ROS, plays a critical role during the occurrence and development of chronic diseases. Canolol is a phenolic compound recently isolated from crude canola oil with powerful antioxidative properties. In this paper, the effects of canolol on hydrogen peroxide (H2O2)-induced oxidative stress in AGS cells were investigated. Cell viability experiments showed negligible cytotoxicity of canolol to AGS cells within a dose of 100 μM. Additionally, it was found that canolol could effectively prevent the oxidative injury caused by H2O2. The pretreatment of AGS cells by canolol also resulted in a reduction in ROS and MDA levels, and LDH release. The survival of cells and the activities of superoxide dismutase (SOD), glutathione peroxidase (GSH) and catalase (CAT) were significantly increased with the pre-incubation of AGS cells with canolol before H2O2 exposure. Consequently, apoptosis and loss of mitochondrial membrane potential of the AGS cells were alleviated, and H2O2-induced caspase-3 activation was inhibited. Therefore, it was concluded that canolol effectively protected AGS cells against H2O2-induced oxidative stress and its antioxidative effect was significant.
1. Introduction
Canolol, 4-vinyl-2, 6-dimethoxyphenol, is a phenolic compound recently isolated from crude canola oil. A large amount of research has found that canolol could greatly improve the oxidation stability of rapeseed oil based on the unique property of strong lipid solubility.1–3 It has superior scavenging potency against alkylperoxyl radical (ROO˙), which is much higher than that of well-known antioxidants, such as quercetin, vitamin C, β-carotene, lutein, and α-tocopherol, etc.4 Cell model experiments demonstrated that canolol could provide a therapeutic effect on age-related macular degeneration.5 Animal experiments found that various inflammatory cytokines such as interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ), were also effectively suppressed by canolol.6 These studies suggested the huge potential of canolol for human nutrition and health, which is of great significance for enhancing the added value of oilseed products.As we known, the antioxidant system maintains the equilibrium between the generation and elimination of reactive oxygen species (ROS) in normal cell systems.7,8 Cellular redox balance in an organism could be broken by the overproduction of ROS, resulting in oxidative stress.9 Oxidative stress plays a critical role during the occurrence and development of chronic diseases, which induced injury or apoptosis to cause many human chronic diseases, including aging, diabetes, cardiovascular diseases and cancer.10–12 Generally, the use of naturally occurring compounds with antioxidant activities is considered to be a promising approach to reduce oxidative stress.13,14 Therefore, the protective effect against oxidative stress has been successfully used to explore the potential nutritional values of food or natural products.
Gastric cancer is one of the major public health issue and the second leading cause of cancer death worldwide, which significantly impacts human's health.15,16 It is closely related to the change of oxidant–antioxidant status, causing the increase of cell proliferation, angiogenesis and apoptosis.17,18 However, the detailed functions of canolol in oxidative stress of gastric carcinoma cell have not yet been investigated. Human gastric adenocarcinoma cell line AGS has been widely used for studying the potential of compounds compared to human gastric epithelial cell line GES-1.19–22 Hydrogen peroxide (H2O2) provides particularly important contributor to oxidative stress.23 Consequently, this study aimed to evaluate the role of canolol in modulating the oxidative stress of gastric carcinoma cell by treating AGS with hydrogen peroxide as a model. In addition, the protectively possible mechanisms were also explored by considering cytotoxicity and cell viability, intracellular ROS level, activities of antioxidant enzymes and protein, lipid peroxide level and apoptosis.
2. Materials and methods
2.1 Materials
Ham's F-12K (Kaighn's) medium and fetal bovine serum (FBS) were obtained from Gibco (Life Technologies, Grand Island, NY). H2O2, Trypsin, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), propidium iodide (PI), dimethyl sulfoxide (DMSO), Western blot reagents, and 2,7-dichlorofluorescin diacetate (DCFH-DA) were purchased from Sigma-Aldrich Sigma (St. Louis, MO, USA). Lactate dehydrogenase (LDH), malondialdehyde (MDA), reduced glutathione (GSH), superoxide dismutase (SOD) and catalase (CAT) assay kits were obtained from Nanjing Jiancheng Bioengineering Institute (China). Annexin V-FITC Apoptosis Detection Kit was purchased from BD Biosciences (USA). Canolol was provided by our laboratory.2.2 Analysis of cell cytotoxicity and viability
2.3 Oxidative stress injury by H2O2
AGS cells were seeded in culture plates with a density of about 5 × 103 cells per well and incubated for 24 h before the experiment. AGS cells incubated in medium at 37 °C (control). Subsequently, the AGS cells were treated with H2O2 for another 4 h. The supernate was removed and treated with 10 μL of MTT (4 mg mL−1 final concentration in medium) for 4 h. After removing supernate, 100 μL of DMSO was added to dissolve the formed blue formazan crystals of live cells. The MTT measurement was the same as the described part of 2.2. The data were presented as the average standard deviation (n = 6).2.4 Measurement of intracellular reactive oxygen species (ROS)
The ROS level was determined using DCFH-DA assay kit. DCFH-DA was added to the cells and further oxidized to fluorescent 2,7-dichlorofluorescein (DCF) by ROS. In this work, AGS cells were incubated with canolol for 24 h and then treated by H2O2 for 4 h. After washing the cells with PBS three times, DCFH-DA was added and incubated with cells for 30 min at 37 °C in the dark. Then the cells were determined by on a microplate reader at the Ex/Em of 480 nm/525 nm and flow cytometry (BD Biosciences, San Jose, CA).2.5 Cell apoptosis assay
Annexin V-FITC/PI apoptosis detection kit was used to measure the apoptosis of AGS cells. Briefly, AGS cells were seeded in a 6-well culture plate and kept for overnight incubation at 37 °C in 5% CO2. Then, cells were treated with different canolol concentrations (5, 50, and 100 μM) for 24 h, and then incubated with H2O2 for 4 h. After incubation, cells were washed with PBS, harvested, and resuspended with binding buffer in tubes by Annexin-FITC and DAPI and kept at 4 °C for 20 min under dark conditions. Stained cells were analyzed using FACSCalibur™ Flow cytometry with Cell-Quest software.2.6 Measurements of LDH, MDA, GSH, SOD and CAT
The assays for LDH, MDA, GSH, SOD and CAT were carried out using commercial kits (Beyotime Biotechnology Co. Ltd, Shanghai, China). The treated cells were harvested and sonicated at 4 °C. The supernatants were collected for the analysis of LDH, MDA, SOD, GSH and CAT level by commercial kits following the manufacturer's instructions.2.7 Measurements of mitochondrial membrane potential
The mitochondrial membrane potential was measured using JC-1 dye with flow cytometry. The treated AGS cells were collected, washed, and resuspended in 500 μL PBS containing JC-1 solution at 37 °C for 20 min. After centrifugation, the cells were resuspended twice in PBS and analyzed by flow cytometry.2.8 Assay of caspase assay
The cell lysates of treated cells were extracted with lysis buffer containing 1% Triton X-100, 50 mM NaCl, 50 mM NaF, 20 mM Tris (pH 7.4), 1 mM EDTA, 1 mM EGTA, 1 mM sodium vanadate, 0.2 mM PMSF, and 0.5% NP-40. An equal amount of cell lysate (50 μg) was solubilized in sample buffer, boiled for 5 min, and electrophoresed on a 4–20% Tris–glycine gel (Invitrogen). Proteins were then transferred to polyvinylidine difluoride membrane (BioRad). Nonspecific binding was blocked with 20 mM Tris–HCl buffered saline (pH 7.6) plus 0.05% Tween-20 (TBS-T) containing 5% nonfat dry milk and 1% bovine serum albumin (BSA). After incubation with the appropriate primary antibody of caspase-3 (Cell Signaling), membranes were washed with TBS-T and then incubated with horseradish peroxidase antirabbit immunoglobulin G in the second reaction. Enhanced chemiluminescence reagent was used in accordance with the manufacturer's recommendations, and the resulting membranes were exposed to Kodak AR film and developed using a Kodak X-OM at processor.3. Results and discussions
3.1 Cell viability and ROS content of AGS cells injured with H2O2
In order to accurately reflect the processing occurred in human body, a reliable in vitro cellular model was required.24 In this study, H2O2 was selected to induce injury on AGS cells for model construction. As shown in Fig. 1, with AGS cells exposed to increasing doses of H2O2 (60–1200 μM) for 4 h, dose-dependent decreases from 100.00 ± 0.93% to 46.45 ± 0.82% in cell viability were found by MTT assays (P < 0.05). Meanwhile, the ROS contents were enhanced in dose-dependent manner with the increase of H2O2 dose because of the increase of cell death. At 400 μM, H2O2 caused an approximate 50% loss in cell viability and 40% enhancement in ROS content. Notably, at 100, 200 and 400 μM H2O2, the cell viability were 88.83 ± 0.47%, 72.10 ± 1.28% and 53.28 ± 0.95%, respectively, which showed that there were large changes in cell viability from 100 to 200 μM and from 200 to 400 μM, which were approximate 17% and 19%, respectively. Because different cell lines have certain difference in susceptibility to H2O2-induced cell death, the dosage of 200 μM was used in our study to carry out the following experiments to obtain effective protection of canolol toward H2O2-induce cell death. | ||
| Fig. 1 The cell viability and ROS values in H2O2-injured AGS cells. The cells were treated with doses of 60–1200 μM for 4 h. The results were represented by mean ± SD (n = 3) from three independent experiments. Data was expressed as a percentage of the untreated control. The single asterisk indicated P < 0.05 versus the control group. | ||
3.2 Cytotoxicity and protective effects of canolol on AGS cells
In the last few decades, numerous bioactive natural products with potential health benefits were widely used as sources for food-health fields.25 The cytotoxicity of these natural products was important for their use. The cytotoxicity of canolol was first studied. The structure of canolol is shown in Fig. 2a. MTT assay shown in Fig. 2b demonstrated that there were hardly changes in cell viability when AGS cells incubated with 50 and 100 μM canolol. Significant decrease of cell viability was found with the increasing concentration of canolol from 200 to 1111 μM compared to the control group. The results indicated that there was no toxicity to AGS cells at the concentration of 100 μM used in this study. Then AGS cells were pre-treated with canolol over 24 h in different concentrations of canolol (5, 33, 50, 83, 100 μM) before H2O2 incubation (200 μM for 4 h). As shown in Fig. 2c, pretreatment with canolol prior to H2O2 exposure gradually enhanced the cell viability and decreased the ROS content of AGS cells with dose-dependent manner compared to solely H2O2-treated group (P < 0.05). Their cell viabilities were 71.39 ± 1.64% (H2O2-treated group), 73.67 ± 0.42% (5 μM canolol-treated group), 75.94 ± 1.37% (33 μM canolol-treated group), 83.27 ± 2.47% (50 μM canolol-treated group), 85.34 ± 0.41% (83 μM canolol-treated group), and 87.59 ± 1.77% (100 μM canolol-treated group). The ROS contents were 127.30 ± 1.92% (H2O2-treated group), 116.02 ± 5.88% (1 μM canolol-treated group), 101.17 ± 2.95% (33 μM canolol-treated group), 100.41 ± 3.88% (50 μM canolol-treated group), 94.63 ± 1.18% (83 μM canolol-treated group), and 92.23 ± 3.39% (100 μM canolol-treated group). At the dose of 100 μM, the cell surviving was up to 90% whereas cell viability was about 70% without canolol treatment, revealing the prominent inhibition of canolol toward H2O2-induced cell death. Consistent with these results, the morphology of AGS cells exposure to H2O2 was obviously damaged compared to that of the control group (Fig. 2d). Pretreatment with canolol, the morphology of the cells were similar to that of the cells without H2O2. Meanwhile, the morphology of cells recovered better at high concentration of canolol than that at low dose, showing positive effects against H2O2-induce AGS cell injury. | ||
| Fig. 2 (a) The structure of canolol. (b) The cytotoxicity of canolol on AGS cells. AGS cells incubated with different concentrations of canolol for 24 h by MTT assay. The curves shown in each graph were from a single experiment (mean ± SD, n = 6). The single asterisk indicated P < 0.05 versus the control group. (c) Effects of canolol on the cell viability and ROS content in AGS cells. The cells were pretreated with canolol (5–100 μM) for 24 h. The cell viability was determined by MTT assay. The ROS value was determined by ROS kits. The results were represented by mean ± SD (n = 3) from three independent experiments. Data was expressed as a percentage of the untreated control. The single asterisk indicated P < 0.05 versus the injury group. (d) Morphology of AGS cells exposed to H2O2 (200 μM) for 4 h with and without canolol pretreatment. | ||
3.3 The inhibition of canolol on ROS production in AGS cells
It has been reported that ROS production is closely related with cell damage and death,26 providing insights into the antioxidative activity of canolol. Therefore, the level of intracellular ROS was assessed in canolol-treated AGS cells by using H2DCFDA, a fluorescent ROS indicator. As shown in Fig. 3, there were significant differences in the fluorescents signals of canolol pretreatment AGS cells. The mean fluorescence of ROS indicator was increased by about 1.5 fold in H2O2-treated AGS cells relative to the untreated control cells (P < 0.05). The signals for canolol pretreated groups were gradually decreased in dose-dependent manner (P < 0.05). At the canolol concentration of 100 μM, the fluorescent signal was greatly decreased to 1.2 fold compared to H2O2 injury group, which was close to the normal level without H2O2 treatment. The results showed that canolol could effectively protect the cells from intracellular ROS damage induced by H2O2. It has been reported that exogenous treatment with H2O2 could induce abnormal accumulation of intracellular ROS and the damage of cellular antioxidant defenses.27 Excess ROS caused protein and lipid oxidation as well as the destruction of nuclear DNA and mitochondrial integrity to ultimately induce cell death. The pre-treatment of cells with canolol could effectively eliminate these negative impacts by reducing the ROS levels. | ||
| Fig. 3 The inhibition of canolol on H2O2-induced intracellular ROS production in AGS cells. (a) The intracellular ROS using H2DCFDA by flow cytometric analysis. (b) The relative fluorescent intensity of ROS indicator compared to control group. The AGS cells were incubated with canolol for 24 h, and then treated with H2O2 for 4 h. The results were represented by a mean ± SD (n = 3). #P < 0.05 versus the control group. *P < 0.05 versus H2O2-induced cells without canolol pretreatment. | ||
Oxidative stress and ROS accumulation have been involved in many human chronic diseases, including aging, diabetes, cardiovascular diseases and cancer. In normal cell physiological process, the antioxidant system maintains the equilibrium between the generation and elimination of reactive oxygen species, whereas ROS levels dramatically increase under environmental stress, such as oxidative stress.28 Previous study reported that canolol treatment could obviously reduce the generation of ROS production of t-butyl hydroxide-damaged HEK-293 cells.5 Similarly, in the present study, canolol could remarkably diminish the ROS content generated by H2O2-induced oxidative stress in AGS cells.
3.4 Effects of canolol on H2O2-induced apoptosis in AGS cells
Excessive ROS readily results in damage of the biomolecules within the cell,29 causes proteins to denature and aggregate along with the collapse of the cell membrane, and eventually leads to cell apoptosis.30 Therefore, cell apoptosis is the major consequence for oxidation-induced change. In the present study, the anti-apoptosis effect of canolol on AGS cells was evaluated using the Annexin V-FITC/PI assay based on flow cytometry. In Fig. 4, the lower left quadrant represented normal cells. Cells in the lower right quadrant were considered as early apoptotic, and cells in the upper right quadrant were described as advanced apoptotic or necrotic. Compared to control group, H2O2-treated group increased the apoptosis percentage from 3.6% to 10.8%, resulting in the fact that H2O2 diffused freely into and out of cells and tissues, destroying the intracellular environment, and ultimately leading to cell apoptosis. However, the apoptosis percentage gradually decreased in a dose-dependent manner when the cells were pretreated by canolol before they were subjected to H2O2, which indicated that canolol provided a protection toward AGS cells against apoptosis caused by H2O2. Combination with the results of canolol toward the ROS production in AGS cells, the anti-apoptosis properties of canolol were related to ROS scavenging.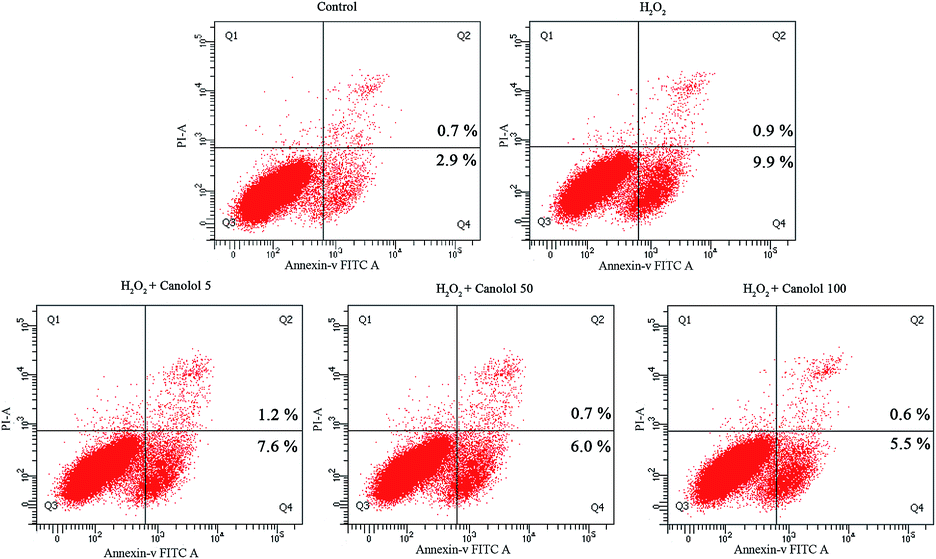 | ||
| Fig. 4 Effects of canolol on H2O2-induced apoptosis in AGS cells. The AGS cells were preincubated with different concentrations of canolol (5, 50 and 100 μM) for 24 h and then treated with H2O2 for 4 h. | ||
3.5 The effect of canolol on the activities of biomolecules and enzymes levels in H2O2-injured AGS cells
Due to the elimination of ROS was related to biomolecules and oxidant enzymes,31 the effects of canolol on the activities of biomolecules and enzymes levels in H2O2-injured AGS cells were investigated. As shown in Fig. 5a and b, after AGS cells were exposed to H2O2, the levels of LDH release and MDA were significantly increased compared to control group. Compared to H2O2-injured group, canolol-treated group significantly decreased the levels of LDH release and MDA, and displayed in dose-dependent manner with the increasing dose of canolol, suggesting that canolol prevented the damage of fragile cell membrane from H2O2, inhibiting the lipid peroxidation and reactive oxygen from pouring into the cells. | ||
| Fig. 5 The effect of canolol on the expression levels of LDH, MDA, GSH, SOD and CAT in H2O2-injured AGS cells. (a) Quantitative analysis of the value of LDH release level. (b) Quantitative analysis of the relative value of MDA. (c–e) Quantitative analysis of the values of GSH, SOD and CAT. The AGS cells were incubated with different concentrations of canolol for 24 h and then incubated with H2O2 for 4 h, which were collected to detect the expression levels of oxidative-stress-related biomolecules and enzymes. The results were represented by a mean ± SD (n = 3). #P < 0.05 versus the control group. *P < 0.05 versus H2O2-induced cells without canolol pretreatment. | ||
Meanwhile, H2O2-injured group obviously destroyed the activities and decreased the levels of GSH, SOD and CAT compared to the untreated group (Fig. 5c and e). With the pretreated AGS cells with canolol for 24 h and then incubation with H2O2, the levels of GSH, SOD and CAT were elevated in dose-dependent manner compared to injury group. The results clearly showed that canolol activated the intracellular antioxidant system, enhanced the expressions of antioxidant enzymes to protect AGS cells against H2O2-induced damage by scavenging intracellular ROS.
Lipid peroxidation is one of the primary events in free radical-mediated cell injury. MDA as a by-product of lipid peroxidation induced by excessive ROS is widely used as a biomarker of oxidative stress.32 In normal cell systems, there exists a balance level between generation of ROS and their elimination by intercellular antioxidant systems composed of some enzymes, such as SOD, GSH, and CAT. With the occurrence of stimuli, the balance is broken and generates excessive ROS, resulting in the lipid peroxidation. It has been reported that natural compounds trigger the detoxification enzymes that boost cellular defenses against oxidative stress.33 From the results, the modulation of SOD, GSH, and CAT activities by pretreatment with canolol compared to treatment with H2O2, indicates an ability of the cell defense system to respond to an oxidative insult.
3.6 The effect of canolol on mitochondrial membrane potential (Δψm) in H2O2-injured AGS cells
Mitochondrion plays a significant role in the apoptotic process, and the level of mitochondrial membrane potential (Δψm) is considered to be an indicator of apoptosis.34 JC-1 dye and flow cytometric analysis were used to assess the change of Δψm during apoptosis induced by H2O2 and protection of canolol in AGS cells. The decrease of red/green ratio indicated dissipation of the mitochondrial Δψm. As shown in Fig. 6, with H2O2-injured AGS cells, the red/green ratio decreased from 49.0% to 4.7%, indicating that the H2O2 caused a depolarization in AGS cells. Pretreated with various concentration of canolol made the red/green ratio enhanced from 4.7% to 15.4% compared to H2O2 injury group, indicating that canolol partly suppressed the change by H2O2 to inhibit the apoptosis and necrosis. | ||
| Fig. 6 Effects of canolol on Δψm in H2O2-injured AGS cells by JC-1 assay. (a) Flow cytometric analysis. (b) The values of red/green ratio. | ||
3.7 Effects of canolol on caspase-3 activation in H2O2-injured AGS cells
It has been widely reported that caspases are important mediators of apoptosis induced by various apoptotic stimuli.35 In order to explore the effects of canolol on apoptosis, the cleaved caspase-3 was investigated by western blotting analysis to examine the enzymatic activity of the caspase-3 with respect to changes in protein expression. As shown in Fig. 7, H2O2-injured group significantly increased the activity of leaved caspase-3 (17 KD) compared to control group, which indicated that H2O2 activated caspase-3 to induce apoptosis. However, canolol-treated group attenuated the increase in cleaved caspase-3 levels induced by H2O2 in AGS cells with dose-dependent manner, suggesting that canolol protected AGS cells against apoptosis by attenuation of caspase-3 activation. | ||
| Fig. 7 Effects of canolol on the caspase-3 activation in AGS cells. AGS cells were treated with canolol and then exposed to H2O2 for 4 h. The density of caspase-3 was determined. | ||
The relationship between oxidative stress and apoptosis has been widely explored.36,37 Current results confirmed that the anti-apoptosis properties of canolol were related to ROS scavenging. It has been reported that H2O2-induced cells exhibited high expression levels of caspase-3 to cause cell apoptosis.38 Bai and his co-workers reported that D-limonene-treated group could inhibit the elevation of caspase-3 expression level compared with the H2O2-treated group in human lens epithelial cells.39 Consistent with these results, our study also exhibited that canolol-treated group similarly prevented the increase of caspase-3 expression level compared with the H2O2-treated group. The short-term effect of canolol is mainly antiapoptotic. Bcl-2 family members play an important role in regulating apoptosis in H2O2-induced oxidative stress. These proteins are either antiapoptotic (e.g., Bcl-2, Bcl-XL, and Mcl-1) or proapoptotic (e.g., Bax, Bak, and Bad), and the interactions among them may influence cell fate.40 MAPKs pathways including ERK1/2, JNK1/2/3 and p38 MAPKs are also important regulators of apoptosis.41 It has been reported that Y-27632, a selective ROCK inhibitor could effectively inhibit H2O2-induced cell apoptosis by the inhibition of activated JNK1/2/3 and p38 MAPKs. Therefore, the antiapoptotic properties of canolol might also directly be related to the expression of Bcl-2 family members and MAPKs pathways. However, the exact mechanism underlying complexation response by canolol is still not fully understood. Thus it is necessary to investigate whether canolol has antioxidant effect by the above signal pathways in the future.
4. Conclusions
This study demonstrated that the cytotoxicity and protective effects of canolol toward H2O2-induced oxidative damage in AGS cells. MTT assays showed that there was no toxicity for canolol to AGS cells within the concentration of 100 μM. Canolol might protect H2O2-induced AGS cells death via attenuation of apoptosis and overproduction of ROS by its effect on redox system. A schematic model was designed to describe the cellular mechanism that canolol ameliorated ROS generation and apoptosis under H2O2 conditions in AGS cells. Briefly, canolol significantly inhibited ROS overproduction, mitochondrial damage, caspase activation and apoptosis in AGS cells response to H2O2 injury. Meanwhile, these effects of canolol were related to the increase of enzymatic activities and the decrease of LDH release and MDA levels. These findings suggested that this rapeseed polyphenol might be an important compound to be used in the development of new food agents for human health.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (31601438), the Agricultural Science and Technology Innovation Project of Chinese Academy of Agricultural Sciences (CAAS-ASTIP-2013-OCRI), the Earmarked Fund for China Agriculture Research System (CARS-13) and the Director Fund of Oil Crops Research Institute (1610172016003).References
- K. Shrestha and B. D. Meulenaer, J. Agric. Food Chem., 2014, 62, 5412–5419 CrossRef CAS PubMed.
- K. Shrestha, C. V. Stevens and M. B. De, J. Agric. Food Chem., 2012, 60, 7506–7512 CrossRef CAS PubMed.
- C. Zheng, M. Yang, Q. Zhou, C. S. Liu and F. H. Huang, Eur. J. Lipid Sci. Technol., 2015, 116, 1675–1684 CrossRef.
- D. Wakamatsu, S. Morimura, T. Sawa, K. Kida, C. Nakai and H. Maeda, Biosci., Biotechnol., Biochem., 2005, 69, 1568–1574 CrossRef CAS PubMed.
- X. Dong, Z. Li, W. Wang, W. Zhang, S. Liu, X. Zhang, J. Fang, H. Maeda and M. Matsukura, Mol. Vision, 2011, 17, 2040–2048 CAS.
- X. Cao, T. Tsukamoto, T. Seki, H. Tanaka, S. Morimura, L. Cao, T. Mizoshita, H. Ban, T. Toyoda and H. Maeda, Int. J. Cancer, 2010, 122, 1445–1454 CrossRef PubMed.
- N. Jallali, H. Ridha, C. Thrasivoulou, C. Underwood, P. E. Butler and T. Cowen, Osteoarthritis Cartilage, 2005, 13, 614–622 CrossRef CAS PubMed.
- Z. Z. Xie, Y. Liu and J. S. Bian, Oxid. Med. Cell. Longevity, 2016, 16, 6043038 Search PubMed.
- X. Qin, M. Cao, F. Lai, F. Yang, W. Ge, X. Zhang, S. Cheng, X. Sun, G. Qin and W. Shen, PLoS One, 2015, 10, e0127551 Search PubMed.
- D. F. Dai and P. S. Rabinovitch, Trends Cardiovasc. Med., 2009, 19, 213–220 CrossRef CAS PubMed.
- M. Dhanasekaran and J. Ren, Curr. Neurovasc. Res., 2005, 2, 447–459 CrossRef CAS PubMed.
- T. V. Fiorentino, A. Prioletta, P. Zuo and F. Folli, Curr. Pharm. Des., 2013, 19, 5695–5703 CrossRef CAS PubMed.
- V. L. Alfonso, B. Pedro, G. Francesca and J. L. Quiles, Antioxidants, 2015, 4, 447–481 CrossRef PubMed.
- H. Kim, B. S. Park, K. G. Lee, C. Y. Choi, S. S. Jang, Y. H. Kim and S. E. Lee, J. Agric. Food Chem., 2005, 53, 8537–8541 CrossRef CAS PubMed.
- M. Orditura, G. Galizia, V. Sforza, V. Gambardella, A. Fabozzi, M. M. Laterza, F. Andreozzi, J. Ventriglia, B. Savastano and A. Mabilia, World J. Gastroenterol., 2014, 20, 1635–1649 CrossRef CAS PubMed.
- T. D. Sablet, M. B. Piazuelo, C. L. Shaffer, B. G. Schneider, M. Asim, R. Chaturvedi, L. E. Bravo, L. A. Sicinschi, A. G. Delgado and R. M. Mera, Gut, 2011, 60, 1189–1195 CrossRef PubMed.
- P. Correa, G. Malcom, B. Schmidt, E. Fontham, B. Ruiz, J. C. Bravo, L. E. Bravo, G. Zarama and J. L. Realpe, Aliment. Pharmacol. Ther., 1998, 12, 73–82 CAS.
- M. Serafini, R. Bellocco, A. Wolk and A. M. Ekström, Gastroenterology, 2002, 123, 985–991 CrossRef CAS.
- D. Kuck, B. Kolmerer, C. Ikingkonert, P. H. Krammer, W. Stremmel and J. Rudi, Infect. Immun., 2001, 69, 5080–5087 CrossRef CAS PubMed.
- M. T. Lin, C. Y. Juan, K. J. Chang, W. J. Chen and M. L. Kuo, Carcinogenesis, 2001, 22, 1947–1953 CrossRef CAS PubMed.
- H. Shirin, E. M. Sordillo, S. H. Oh, H. Yamamoto, T. Delohery, I. B. Weinstein and S. F. Moss, Cancer Res., 1999, 59, 2277–2281 CAS.
- H. Teng, Q. Huang and L. Chen, Food Funct., 2016, 7, 4605–4613 CAS.
- Z. S. Xu, X. Y. Wang, D. M. Xiao, L. F. Hu, M. Lu, Z. Y. Wu and J. S. Bian, Free Radicals Biol. Med., 2011, 50, 1314–1323 CrossRef CAS PubMed.
- F. Cheli and A. Baldi, J. Food Sci., 2011, 76, R197–R205 CrossRef CAS PubMed.
- B. David, J. L. Wolfender and D. A. Dias, Phytochem. Rev., 2015, 14, 299–315 CrossRef CAS.
- P. Newsholme, E. P. Haber, S. M. Hirabara, E. L. Rebelato, J. Procopio, D. Morgan, H. C. Oliveira-Emilio, A. R. Carpinelli and R. Curi, J. Physiol., 2007, 583, 9–24 CrossRef CAS PubMed.
- R. S. Sohal and W. C. Orr, Free Radicals Biol. Med., 2012, 52, 539–555 CrossRef CAS PubMed.
- C. Liu, J. Hong, H. Yang, J. Wu, D. Ma, D. Li, D. Lin and R. Lai, Free Radicals Biol. Med., 2010, 48, 1173 CrossRef CAS PubMed.
- C. M. Quinzii, L. C. López, R. W. Gilkerson, B. Dorado, J. Coku, A. B. Naini, C. Lagier-Tourenne, M. Schuelke, L. Salviati and R. Carrozzo, FASEB J., 2010, 24, 3733–3743 CrossRef CAS PubMed.
- A. Srivastava, R. Harish and T. Shivanandappa, J. Agric. Food Chem., 2006, 54, 790–795 CrossRef CAS PubMed.
- S. A. Comhair and S. C. Erzurum, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2002, 283, 246–255 CrossRef PubMed.
- X. Lu, H. Guo and Y. Zhang, Int. J. Biol. Macromol., 2012, 50, 50–58 CrossRef CAS PubMed.
- Y. Kim, Y. Choi, H. Ham, H. S. Jeong and J. Lee, Food Chem., 2013, 137, 136–141 CrossRef CAS PubMed.
- M. Leist and M. Jäättelä, Nat. Rev. Mol. Cell Biol., 2001, 2, 589–598 CrossRef CAS PubMed.
- K. F. Tonissen, G. D. Trapani, S. J. Ralph and J. Neuzil, Mol. Nutr. Food Res., 2009, 53, 87–103 CAS.
- M. G. Manuele, M. L. Barreiro Arcos, R. Davicino, G. Ferraro, G. Cremaschi and C. Anesini, Cancer Invest., 2010, 28, 135–145 CrossRef CAS PubMed.
- D. Roberto, P. Micucci, T. Sebastian, F. Graciela and C. Anesini, Basic Clin. Pharmacol. Toxicol., 2010, 106, 38–44 CAS.
- H. P. Yao, X. J. Tang, X. T. Shao, F. Lei, N. P. Wu and Y. Ke, Cell Res., 2007, 17, 565–571 CrossRef CAS PubMed.
- J. Bai, Y. Zheng, G. Wang and P. Liu, Oxid. Med. Cell. Longevity, 2016, 14, 1–12 Search PubMed.
- M. G. VanderHeiden, N. S. Chandel, E. K. Williamson, P. T. Schumacker and C. B. Thompson, Cell, 1997, 91, 627–637 CrossRef CAS.
- X. Wang, B. Wang, Z. Li, G. Zhu, L. Heng, X. Zhu, Q. Yang, J. Ma and G. Gao, RSC Adv., 2016, 6, 49187–49197 RSC.
| This journal is © The Royal Society of Chemistry 2017 |