DOI:
10.1039/C7RA06324E
(Paper)
RSC Adv., 2017,
7, 42819-42825
Enhanced photocatalytic CO2 reduction over Co-doped NH2-MIL-125(Ti) under visible light†
Received
6th June 2017
, Accepted 29th August 2017
First published on 5th September 2017
Abstract
Co-doped NH2-MIL-125(Ti) catalysts [Co/NH2-MIL-125(Ti)] are developed for the photocatalytic reduction of CO2 upon visible-light irradiation. Compared with NH2-MIL-125(Ti), Co/NH2-MIL-125(Ti) exhibits a significantly enhanced activity in CO2 reduction, due to the fact that the doping of Co nanoparticles onto NH2-MIL-125(Ti) can promote the visible-light harvesting and electron transfer. In addition, when benzylic alcohols as electron donors, instead of triethanolamine (TEOA) which is often used as a sacrificial agent, are added into the reaction system, Co/NH2-MIL-125(Ti) can simultaneously catalyze CO2 reduction to formic acid (HCOOH) and the selective oxidation of the benzylic alcohols to the corresponding aldehydes, making the proposed process more economical and environment-friendly.
1. Introduction
To solve the problems associated with both global warming and energy shortage, the conversion of CO2 into valuable organic products utilizing renewable solar energy is considered as a promising approach.1–3 Over the past four decades various photocatalysts, such as inorganic semiconductors, metal-incorporated zeolites, and metal complexes, have been developed for CO2 reduction.4–12 However, most of them are only active in an ultraviolet (UV) region and their efficiencies for CO2 reduction and selectivities for the desired products are still quite low. Therefore, it would be of great importance to develop visible-light-responsive and highly efficient, selective photocatalysts for CO2 reduction.
Metal–organic frameworks (MOFs), which have been extensively studied over the past two decades and shown a variety of applications in gas storage/separation, biomedicine, as well as heterogeneous catalysis,13–20 could be promising candidates for photocatalytic CO2 reduction. It has been reported that in MOFs the metal-oxo clusters could act as inorganic semiconducting quantum dots while the organic linkers serve as antennas to activate these semiconducting quantum dots by the ligand-to-metal charge transfer upon photoexcitation, thus making MOF-based photocatalysis possible.21 In addition, MOFs are good at CO2 capture due to their large surface area, high porosity, and tunable interactions with CO2,22 facilitating the photocatalytic reduction of CO2. In fact, some investigators have demonstrated the applications of MOF-based photocatalysts in CO2 reduction.23–32 For example, Jiang et al. reported that a very stable mesoporous zirconium-porphyrin MOF, PCN-222, could selectively capture CO2 and further photoreduce CO2 with triethanolamine (TEOA) as a sacrificial agent upon visible-light irradiation.23 Li et al. used series of photocatalysts based on UiO-66(Zr) in CO2 reduction to formates upon visible-light irradiation in the presence of TEOA.25–27 However, the activities of the reported MOF-based photocatalysts are still quite low and TEOA must be used as the sacrificial agent during CO2 reduction, which is not economical and environment-friendly, due to the following reasons: (1) TEOA cannot be converted to valuable products, just as a sacrificial hole scavenger; (2) the excessive emission of TEOA will cause the environmental pollution; and (3) the use of TEOA will lead to the laborious purification of the produced HCOOH due to the strong intermolecular interactions between the formed acid–base adducts. It is therefore indispensable to enhance the activities of MOF-based photocatalysts and to find alternatives to avoid using sacrificial agents such as TEOA in photocatalytic CO2 reduction. In our previous work, it was found that Ni-doped NH2-MIL-125(Ti) could effectively catalyze the aerobic oxidation of benzylic alcohols to the corresponding aldehydes upon visible-light irradiation.33 In addition, it is well known that benzylic alcohols can be used as electron donors.34 Thus, it is anticipated that the doping of some metallic nanoparticles (NPs) into MOFs such as NH2-MIL-125(Ti) might also enhance the photocatalytic activity in CO2 reduction and simultaneously promote the selective oxidation of the benzylic alcohols used as electron donors. It would be highly desirable that the benzylic alcohols can be selectively oxidized into the corresponding aldehydes during photocatalytic CO2 reduction, because the produced aldehydes and their derivatives are important building blocks for producing fine chemicals.
Herein, for the first time, we developed metallic Co NPs doped NH2-MIL-125(Ti) catalysts [Co/NH2-MIL-125(Ti)] for photocatalytic CO2 reduction using benzylic alcohols as electron donors upon visible-light irradiation. It's found that Co/NH2-MIL-125(Ti) catalysts exhibit significantly enhanced activities in CO2 reduction and benzylic alcohols could be oxidized to the corresponding aldehydes during photocatalytic CO2 reduction. The effects of the doped Co NPs on the photocatalysis were investigated in detail and the possible photocatalytic mechanism for CO2 reduction was proposed.
2. Experimental
2.1. Materials
All reagents with analytical reagent grade were used as received without further purification. Tetrabutyl titanate [Ti(OC4H9)4], potassium bicarbonate (KHCO3), triethanolamine (TEOA), acetonitrile (MeCN), acetonitrile-D3 (CD3CN) and cobalt(II) nitrate hexahydrate [Co(NO3)2·6H2O] were purchased from Shanghai Chemical Reagent Co. 2-Amino-benzenedicarboxylic acid (H2BDC-NH2), methanol (MeOH) and dimethylformamide (DMF) were purchased from Sigma Aldrich Co. Sodium borohydride (NaBH4), sodium carbonate (Na2CO3), sodium bicarbonate (NaHCO3), benzotrifluoride (BTF), benzyl alcohol, 4-methoxybenzyl alcohol, benzaldehyde, and 4-methoxybenzaldehyde were purchased from Aladdin Industrial Co.
2.2. Synthesis
2.2.1. NH2-MIL-125(Ti). Based on the recipe reported in the literature,30 NH2-MIL-125(Ti) was prepared via a hydrothermal treatment of H2BDC-NH2 (0.54 g, 3 mmol) and Ti(OC4H9)4 (0.26 mL, 0.75 mmol) in the solvent of DMF (9 mL) and dry MeOH (1 mL) at 150 °C for 3 days. After hydrothermal treatment, the resultant suspension was filtered, washed with DMF and MeOH, extracted by a Soxhlet extractor with MeOH, and finally vacuum-dried to obtain the product.
2.2.2. Co/NH2-MIL-125(Ti). The Co-doped NH2-MIL-125(Ti) samples were prepared through solution infiltration of activated NH2-MIL-125(Ti) with MeOH solution (0.5 mL) containing Co(NO3)2·6H2O. The mixture was stirred for 12 h, then filtered and dried at 150 °C for 3 h, followed by treatment with a NaBH4 solution (20 mL, 0.05 M). The resulting solid powder was washed with MeOH and deionized water for several times and dried under N2 atmosphere at 60 °C for 10 h to get Co/NH2-MIL-125(Ti). The nominal doped amounts of Co in NH2-MIL-125(Ti) were 1.0, 2.0, and 3.0 wt%, denoted as 1.0 wt% Co/NH2-MIL-125(Ti), 2.0 wt% Co/NH2-MIL-125(Ti), and 3.0 wt% Co/NH2-MIL-125(Ti), respectively.
2.3. Characterization
X-ray powder diffraction (XRD) patterns were collected on a Philips PW3040/60 diffractometer using Cu Kα radiation (λ = 0.1541 nm) in a scanning range of 5–50° at 1° min−1. After digestion of Co/NH2-MIL-101(Ti) (0.05 g) in a solution of 2 mL HF (40 wt%) and 8 mL H2SO4 (98 wt%) diluted to 100 mL with deionized water, the Co loadings of the prepared catalysts were determined by an IRIS Intrepid IIXSP inductively coupling plasma-atomic emission spectrometer (ICP-AES). The Brunauer–Emmett–Teller (BET) surface areas of the prepared samples were determined by N2 adsorption/desorption at −196 °C using a Micromeritics ASAP 2020 instrument. The samples were degassed in vacuum at 150 °C for 12 h before the adsorption measurement. The UV-vis diffuse reflectance spectra (UV-vis DRS) of the samples were recorded on a Shimadzu UV-3600 spectrophotometer from 200 nm to 800 nm. The scanning electron microscope (SEM) was carried out on a Hitachi S-4800 apparatus equipped with a field emission gun. The high resolution transmission electron microscopy (HR-TEM) was conducted on a JEOL JEM-1200 working at 200 kV. The X-ray photoelectron spectroscopy (XPS) was carried out on Thermo Scientific EscaLab 250Xi using Al Kα radiation. The 13C nuclear magnetic resonance (NMR) was carried out on a Bruker AVANCE III 500M system (500 MHz). The isotopic 13CO2 reduction was performed in an NMR tube filled with photocatalyst in a CD3CN/TEOA (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) solution. The spectrum was recorded under the following conditions: acquisition time 1.0 s, 20000 times integration. The electron spin resonance (ESR) spectra were obtained over Bruker ESP 300 E electron paramagnetic resonance spectrometer at room temperature. The photoluminescence (PL) spectra were recorded on a Hitachi F-7000 spectrometer. The photocurrent (PC) analysis was carried out with a CHI440A workstation (Shanghai Chenhua Instruments Co.) in a conventional three-electrode cell using a Pt plate and an Ag/AgCl electrode as the counter electrode and reference electrode, respectively. The photocatalyst powder deposited on the fluoride tin oxide (FTO) substrate was employed as the working electrode and a quartz cell filled with 100 mL 0.2 M Na2SO4 electrolyte was used as the reaction system. A 300 W xenon lamp (Beijing Perfectlight, PLS-SXE 300c) system was applied as the excitation light source equipped with a UV cutoff filter, which was the same light source for the photocatalytic tests. The Mott–Schottky curves were measured using a ZENNIUM electrochemical analyzer (Zahner, Germany) in a three-electrode cell. Pt plate and Ag/AgCl electrode (3 M KCl) were used as the counter and reference electrode, respectively. The electrolyte was a 0.2 M aqueous solution of Na2SO4 without additive and was purged with N2 gas for 2 h prior to the measurements. The potential ranged from −0.3 V to 0.8 V (vs. Ag/AgCl), and the perturbation signal was 20 mV with the frequency from 500 Hz to 1500 Hz. The working electrodes were immersed in the electrolyte for 60 s before any measurement was taken.
:1 v/v) solution. The spectrum was recorded under the following conditions: acquisition time 1.0 s, 20000 times integration. The electron spin resonance (ESR) spectra were obtained over Bruker ESP 300 E electron paramagnetic resonance spectrometer at room temperature. The photoluminescence (PL) spectra were recorded on a Hitachi F-7000 spectrometer. The photocurrent (PC) analysis was carried out with a CHI440A workstation (Shanghai Chenhua Instruments Co.) in a conventional three-electrode cell using a Pt plate and an Ag/AgCl electrode as the counter electrode and reference electrode, respectively. The photocatalyst powder deposited on the fluoride tin oxide (FTO) substrate was employed as the working electrode and a quartz cell filled with 100 mL 0.2 M Na2SO4 electrolyte was used as the reaction system. A 300 W xenon lamp (Beijing Perfectlight, PLS-SXE 300c) system was applied as the excitation light source equipped with a UV cutoff filter, which was the same light source for the photocatalytic tests. The Mott–Schottky curves were measured using a ZENNIUM electrochemical analyzer (Zahner, Germany) in a three-electrode cell. Pt plate and Ag/AgCl electrode (3 M KCl) were used as the counter and reference electrode, respectively. The electrolyte was a 0.2 M aqueous solution of Na2SO4 without additive and was purged with N2 gas for 2 h prior to the measurements. The potential ranged from −0.3 V to 0.8 V (vs. Ag/AgCl), and the perturbation signal was 20 mV with the frequency from 500 Hz to 1500 Hz. The working electrodes were immersed in the electrolyte for 60 s before any measurement was taken.
2.4. Photocatalytic reaction
Prior to photocatalytic reactions, the catalysts were treated under vacuum at 150 °C to remove any adsorbed impurities. 50 mg of the photocatalyst was degassed and purged with CO2. A mixture of MeCN and TEOA with a volume ratio of 5 to 1 and a total volume of 60 mL, degassed by CO2 to remove dissolved O2, was injected into the reaction flask. The photocatalytic reaction was carried out under the irradiation of a 300 W Xe lamp with a UV-cut filter and an IR-cut filter (800 nm ≥ λ ≥ 420 nm). The formed HCOOH was detected by ion chromatography (930 Compact IC pro, Metrosep) with a Metrosep A Supp 5 250/4.0 column. A mixture of 3.2 mM Na2CO3 and 1.0 mM NaHCO3 was used as eluent. The gaseous reaction products were also analyzed using a GC-FID (Agilent 6890A) with a HP-5 capillary column.
When benzylic alcohols instead of TEOA were used as electron donors, KHCO3 (50 mg) was added to provide a basic environment for facilitating photocatalytic CO2 reduction. The volume ratio of MeCN to the benzylic alcohol was 10 to 1 with a total volume of 110 mL.
For the reusability test, the photocatalyst was separated from the reaction medium after the first run, repeatedly washed with MeOH three times, and then dried overnight in a vacuum oven at 150 °C. Then, the dried catalyst was used in the next run.
3. Results and discussion
3.1. Characterization
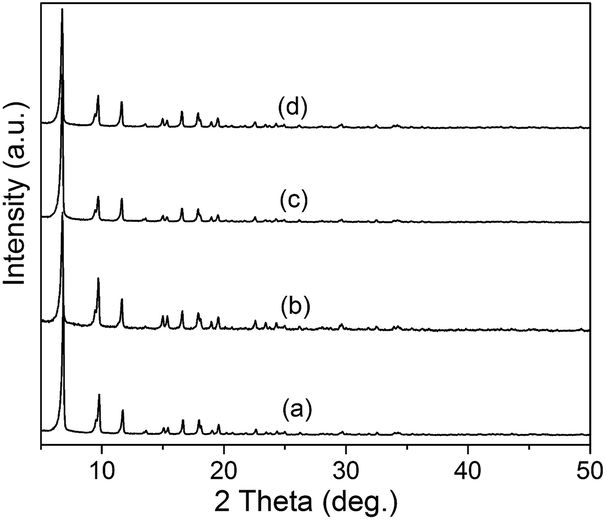
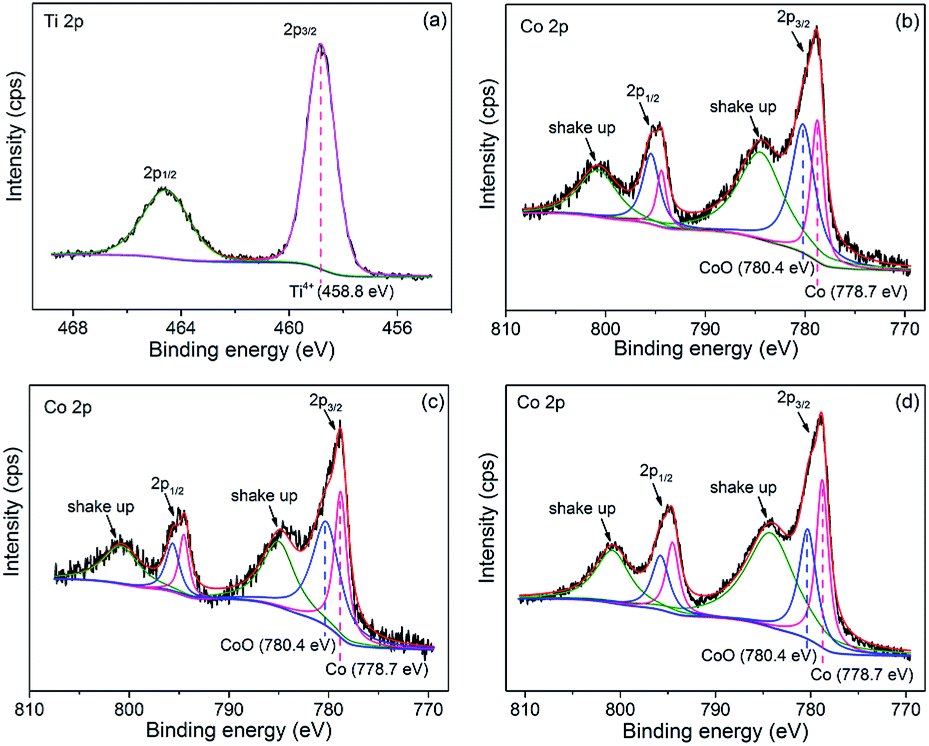
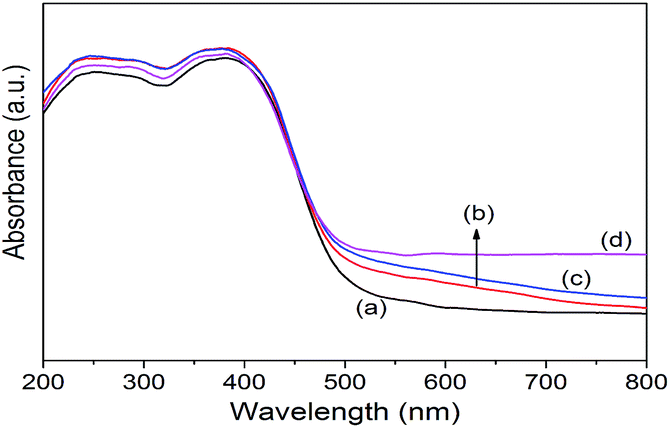
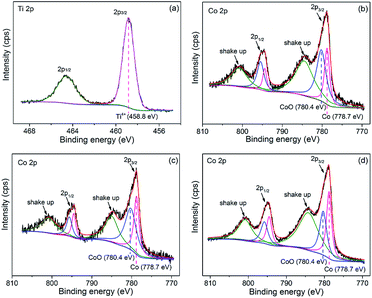
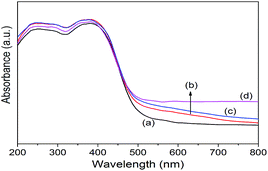
Co/NH2-MIL-125(Ti) samples were prepared by an impregnation of the activated NH2-MIL-125(Ti) with [Co(NO3)2·6H2O] solution, reduced by an aqueous NaBH4 solution. The physiochemical properties of the prepared catalysts are summarized in Table 1. The doped amounts of Co NPs on NH2-MIL-125(Ti) determined by the ICP-AES analysis were 0.91, 1.97, and 2.91 wt%, respectively, well corresponding to their nominal values in the synthesis. In comparison with NH2-MIL-125(Ti), the BET surface areas and the pore volumes of Co/NH2-MIL-125(Ti) tend to decrease gradually with increasing Co doping in the matrix of NH2-MIL-125(Ti), due to the fact that the cages of NH2-MIL-125(Ti) are occupied by the dispersed Co NPs and/or blocked by the Co NPs that could partially be deposited at the pore entrances of the MOF. Fig. 1 shows that the XRD patterns of Co/NH2-MIL-125(Ti) are almost the same as that of NH2-MIL-125(Ti), indicating that the doping of Co on NH2-MIL-125(Ti) does not influence its crystallinity. Additionally, no characteristic peak of Co species can be observed due to the low loading amount and high dispersion of Co on the MOF. The SEM images (Fig. 2) display that the morphologies of the Co-doped samples are the same as that of NH2-MIL-125, indicating that the structure of NH2-MIL-125 is well preserved. The sizes of the Co NPs doped on NH2-MIL-125(Ti) have a mean diameter of 5 nm from the TEM observation, as shown in Fig. 3. The HR-TEM images of Co/NH2-MIL-125(Ti) samples show the characteristic spacings of 0.177 and 0.205 nm for the (2 0 0) and the (1 1 1) lattice planes of cubic Co (JCPDS, no. 15-0806), and the characteristic spacings of 0.213 and 0.246 nm for the (2 0 0) and (1 1 1) lattice planes of CoO (JCPDS, no. 48-1719), respectively. In order to confirm the results from HR-TEM, the X-ray photoelectron spectroscopy was used to characterize the Co oxidation state (Fig. 4). The coexistence of the metallic and oxidized Co in Co/NH2-MIL-125(Ti) is evidenced by two peaks of Co 2p3/2 located at 778.7 and 780.4 eV, which are assigned to Co0 and Co2+.35 The oxidized Co could be ascribed to the partial oxidation when the Co NPs in Co/NH2-MIL-125(Ti) are exposed to the atmosphere since they are very active to be oxidized. It's generally accepted that their activity tends to increase with declining their particle size.36 As can be seen from Fig. 5, the UV-vis spectra of both NH2-MIL-125(Ti) and Co/NH2-MIL-125(Ti) show a similar absorption onset (ca. 505 nm), thereby suggesting an optical band gap of ∼2.5 eV as determined by the Kubelka–Munk function. However, the UV-vis spectra of Co/NH2-MIL-125(Ti) show an extra absorption band in the visible-light region ranging from 500 to 800 nm, compared with NH2-MIL-125(Ti), in accordance with their dark green color changed from yellow. The enhanced visible-light harvesting would be of great importance in photocatalysis and thus Co/NH2-MIL-125(Ti) catalysts are expected to possess better photocatalytic activities toward target reactions.
Table 1 Physiochemical properties of the prepared catalysts
| Catalyst |
Co loadinga wt% |
SBET m2 g−1 |
SLangmuir m2 g−1 |
Vtotal cm3 g−1 |
| The doped amounts of Co in NH2-MIL-125(Ti) were determined by the ICP-AES analysis. |
| NH2-MIL-125 |
— |
1132 |
1586 |
0.65 |
| 1.0 wt% NH2-MIL-125 |
0.91 |
926 |
1360 |
0.57 |
| 2.0 wt% NH2-MIL-125 |
1.97 |
824 |
1214 |
0.54 |
| 3.0 wt% NH2-MIL-125 |
2.98 |
765 |
1123 |
0.53 |
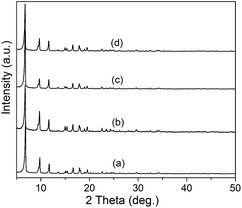 |
| | Fig. 1 XRD patterns of NH2-MIL-125(Ti) (a), 1.0 wt% Co/NH2-MIL-125(Ti) (b), 2.0 wt% Co/NH2-MIL-125(Ti) (c), and 3.0 wt% Co/NH2-MIL-125(Ti) (d). | |
 |
| | Fig. 2 SEM images of NH2-MIL-125(Ti) (a), 1.0 wt% Co/NH2-MIL-125(Ti) (b), 2.0 wt% Co/NH2-MIL-125(Ti) (c), and 3.0 wt% Co/NH2-MIL-125(Ti) (d). | |
 |
| | Fig. 3 HR-TEM images of NH2-MIL-125(Ti) (a), 1.0 wt% Co/NH2-MIL-125(Ti) (b), 2.0 wt% Co/NH2-MIL-125(Ti) (c), and 3.0 wt% Co/NH2-MIL-125(Ti) (d). | |
 |
| | Fig. 4 High-resolution XPS spectra for Co 2p of NH2-MIL-125(Ti) (a), 1.0 wt% Co/NH2-MIL-125(Ti) (b), 2.0 wt% Co/NH2-MIL-125(Ti) (c), and 3.0 wt% Co/NH2-MIL-125(Ti) (d). | |
 |
| | Fig. 5 UV-vis diffuse reflectance spectra of NH2-MIL-125(Ti) (a), 1.0 wt% Co/NH2-MIL-125(Ti) (b), 2.0 wt% Co/NH2-MIL-125(Ti) (c), and 3.0 wt% Co/NH2-MIL-125(Ti) (d). | |
3.2. Photocatalytic performance
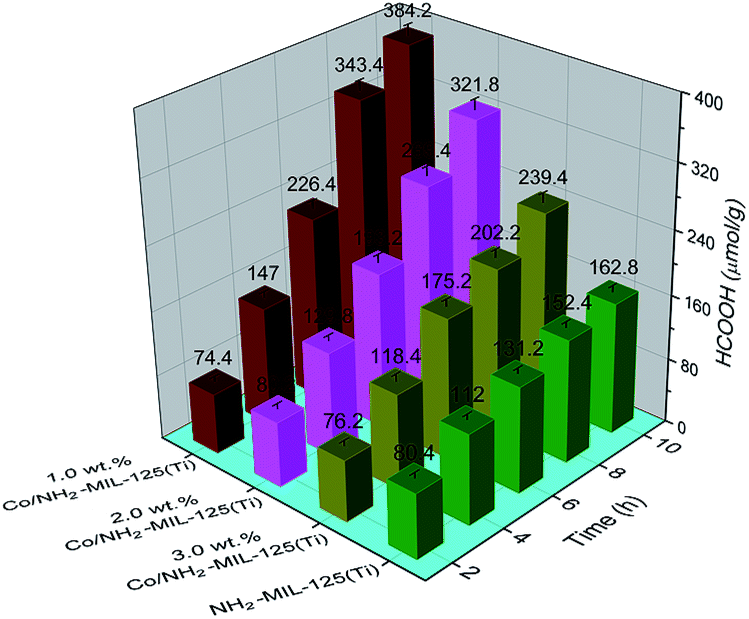
3.2.1. Photocatalytic CO2 reduction using TEOA as the sacrificial agent. To investigate the effects of the doped Co NPs on the photocatalytic performance, photocatalytic CO2 reduction was carried out using TEOA as the sacrificial agent upon visible-light irradiation. No reaction took place in dark or without catalyst. Both NH2-MIL-125(Ti) and Co/NH2-MIL-125(Ti) can catalyze the reduction of CO2 into HCOOH upon visible-light irradiation. A concentration change of HCOOH with irradiation time over the prepared photocatalysts is shown in Fig. 6. Compared to the photocatalytic activity of NH2-MIL-125(Ti), Co/NH2-MIL-125(Ti) catalysts are more active in CO2 reduction. The effects of the Co content in the MOF on the photocatalytic performance were also examined. One can see that the optimum content of Co in NH2-MIL-125(Ti) is about 1.0 wt% for photocatalytic CO2 reduction with about 38.4 μmol gcat−1 h−1 of the HCOOH produced in 10 h irradiation, since the excessive Co in the MOF may cause the blocking of the MOF pores to reduce the photoexciting capacity of NH2-MIL-125(Ti). The isotopic 13CO2 reaction was used to confirm that the produced HCOOH originated from CO2. The 13C NMR spectrum (Fig. S1 in the ESI†) shows three peaks at 125.76, 159.56, and 165.29 ppm, which can be assigned to dissolved CO2, HCO3−, and HCOOH, respectively.24 This indicates that the produced HCOOH originates from CO2. In addition, the control experiments using cobalt(II) salt or CoO mixed with NH2-MIL-125(Ti) as the photocatalyst were carried out in order to verify which Co species (metallic or Co2+) would play a role in the reaction, see Table S1 in the ESI.† The amount of the HCOOH produced in the presence of cobalt(II) salt or CoO-mixed with NH2-MIL-125(Ti) (entries 2 and 3) is almost the same as that over NH2-MIL-125(Ti) (entry 1), indicating that the enhanced activity of Co/NH2-MIL-125(Ti) results from the Co NPs but not from the Co oxidized species.
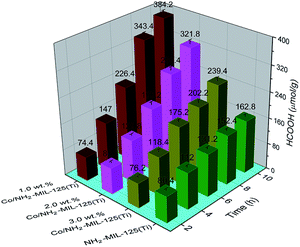 |
| | Fig. 6 Amount of the produced HCOOH as a function of irradiation time over NH2-MIL-125(Ti), 1.0 wt% Co/NH2-MIL-125(Ti), 2.0 wt% Co/NH2-MIL-125(Ti), and 3.0 wt% Co/NH2-MIL-125(Ti). The solutions were irradiated with a Xe lamp and filters producing light in a range of 420–800 nm. Photocatalyst: 50 mg, MeCN/TEOA volume ratio: 5/1, total solution volume: 60 mL. | |
3.2.2. Aerobic oxidation of benzylic alcohols. In our previous study, it was found that Ni-doped NH2-MIL-125(Ti) shows an enhanced photocatalytic activity in the selective oxidation of benzylic alcohols to the corresponding aldehydes.33 Herein, the aerobic oxidation of benzylic alcohols over Co/NH2-MIL-125(Ti) was also investigated, and these results are summarized in Table 2. Remarkably, compared with NH2-MIL-125(Ti), Co/NH2-MIL-125(Ti) shows an enhanced activity for the oxidation of benzyl alcohol and 4-methoxybenzyl alcohol with high selectivity (>99%). The conversions of the benzylic alcohols over Co/NH2-MIL-125(Ti) are even higher than those over Ni/NH2-MIL-125(Ti), implying that the electron transfer is more effective for the doping of the Co NPs into NH2-MIL-125(Ti).
Table 2 Aerobic photocatalytic oxidation of benzylic alcohols to the corresponding aldehydes over 1 wt% Co/NH2-MIL-125(Ti) upon visible-light irradiationa
| Entry |
Catalyst |
Benzylic alcohol |
Conversion (%) |
Selectivity (%) |
| Reaction conditions: 50 mg of catalyst and 0.3 mmol of benzylic alcohol in 6 mL of BTF at room temperature for 10 h. |
| 1 |
NH2-MIL-125(Ti) |
Benzyl alcohol |
12.5 |
>99 |
| 2 |
4-Methoxybenzyl alcohol |
42.5 |
>99 |
| 3 |
1 wt% Co/NH2-MIL-125(Ti) |
Benzyl alcohol |
23.9 |
>99 |
| 4 |
4-Methoxybenzyl alcohol |
50.6 |
>99 |
3.2.3. Photocatalytic CO2 reduction coupled with oxidation of benzylic alcohols. Because TEOA as the sacrificial electron donor is not economical and environment-friendly while benzylic alcohols can be used as electron donors, we therefore carried out photocatalytic CO2 reduction with the oxidation of benzylic alcohols. These results are summarized in Table 3. KHCO3 was added into the reaction medium in order to provide a basic environment for facilitating the photocatalytic reduction. When benzyl alcohol was used as the electron donor, no product was detected over NH2-MIL-125(Ti) upon visible-light irradiation for 10 h. On the contrary, the yields of benzaldehyde and HCOOH over 1.0 wt% Co/NH2-MIL-125(Ti) under the same conditions were 2.2 and 2.4 μmol gcat−1 h−1 (entries 1 and 3), respectively. In case of 4-methoxybenzyl alcohol, 4-methoxybenzaldehyde and HCOOH were formed over both NH2-MIL-125(Ti) and 1.0 wt% Co/NH2-MIL-125(Ti) catalysts (entries 2 and 4), and the yields of 4-methoxybenzaldehyde and HCOOH over 1.0 wt% Co/NH2-MIL-125(Ti) were up to 5.8 and 6.0 μmol gcat−1 h−1. It is undoubted that the electron donating substituent on the phenyl ring is favorable for the photocatalytic reaction. It has been reported that the photocatalytic activity can be enhanced if the catalyst is made more electron conductive.37,38 Although there is still the lack of thorough understanding on the photocatalytically active sites, it is proposed that the enhanced catalytic activity might come from the electronic interactions between the Co NPs and NH2-MIL-125(Ti).
Table 3 Photocatalytic reduction of CO2 coupled with oxidation of different benzylic alcoholsa
| Entry |
Catalyst |
Benzylic alcohol |
Benzylic aldehyde (μmol gcat−1 h−1) |
HCOOH (μmol gcat−1 h−1) |
| Reaction conditions: 50 mg of photocatalyst, a MeCN/benzylic alcohol volume ratio of 10 to 1, a total solution volume of 110 mL upon visible-light irradiation (800 nm ≥ λ ≥ 420 nm) for 10 h. n.d.: not detect. |
| 1 |
NH2-MIL-125(Ti) |
Benzyl alcohol |
n.d.b |
n.d. |
| 2 |
4-Methoxybenzyl alcohol |
4.6 |
4.8 |
| 3 |
1 wt% Co/NH2-MIL-125(Ti) |
Benzyl alcohol |
2.2 |
2.3 |
| 4 |
4-Methoxybenzyl alcohol |
5.8 |
6.0 |
3.2.4. Reusability. A six-run experiment of photocatalytic CO2 reduction with TEOA as the sacrificial agent was carried out to check the reusability of 1 wt% Co/NH2-MIL-125(Ti) as an illustrative example, as shown in Fig. S2 in the ESI.† Obviously, the recycling use of the photocatalyst for six runs shows no obvious decrease of the photocatalytic activity. In addition, the XRD, FT-IR, N2 adsorption–desorption, and XPS characterization results for the photocatalyst after the 6th-run reaction are almost identical to those for the fresh one (Fig. S3 in the ESI†), suggesting that the prepared catalyst is stable during the photocatalytic reaction. In addition, the photocatalytic reduction of CO2 upon UV irradiation was carried out to further check the stability of 1 wt% Co/NH2-MIL-125(Ti), and the corresponding results are shown in Fig. S4 in the ESI,† in which there is no obvious decrease in the photocatalytic activity for six runs. Moreover, the results from the XRD, N2 adsorption–desorption, and FT-IR characterizations of the photocatalyst after the 6th-run reaction are almost the same as those for the fresh one (Fig. S5 in the ESI†), indicating that the photocatalyst is stable upon UV-light irradiation.
3.3. Mechanism for photocatalytic CO2 reduction
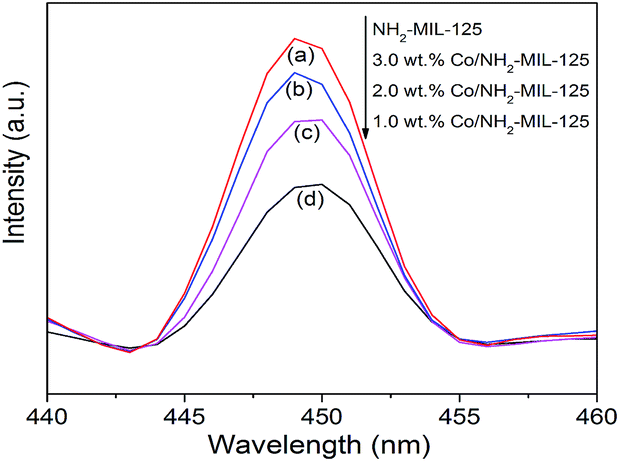
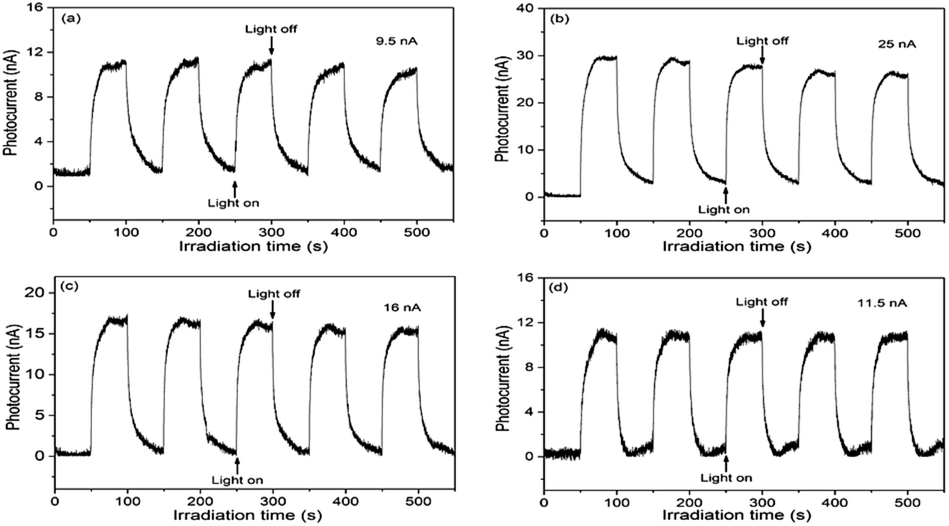
To elucidate the semiconducting properties of NH2-MIL-101(Ti) upon light excitation, the Mott–Schottky method was applied to estimate the flat band positions of the photocatalysts, because it is a generally accepted method in the determination of the flat band positions of semiconductors.39,40 The flat band positions for NH2-MIL-125(Ti) and 1.0 wt% Co/NH2-MIL-125(Ti) were determined to be −0.40 V and −0.43 V vs. NHE, respectively (Fig. S6 in the ESI†). As observed from the Mott–Schottky measurements, both NH2-MIL-125(Ti) and 1.0 wt% Co/NH2-MIL-125(Ti) have characteristics of n-type semiconductors, therefore their conduction band (CB) positions should be close to their flat band ones. A comparable position observed for the conduction bands of NH2-MIL-125(Ti) and Co/NH2-MIL-125(Ti) indicates that the introduction of Co NPs does not influence the conduction band, implying that the photo-generated electrons could be located at the metal centers in these MOFs. The CB position is more negative than the reduction potential of CO2 to form HCOOH (−0.28 V vs. NHE),41 indicating that Co/NH2-MIL-101(Ti) can reduce CO2 to form HCOOH (Fig. S7 in the ESI†). Although the doping of Co NPs into NH2-MIL-125(Ti) could not affect its energy band structure, it could promote the photo-generated charge separation efficiency, which is confirmed by the PL and PC experiments. Fig. 7 shows the PL spectra of NH2-MIL-125(Ti) and Co/NH2-MIL-125(Ti) excited at 303 nm. NH2-MIL-125(Ti) exhibits a PL peak at around 449 nm, and such a peak is greatly weakened upon being doped with Co NPs, indicating that Co/NH2-MIL-125(Ti) can significantly prohibit the photo-generated charge recombination. The interactions between Co NPs and NH2-MIL-125(Ti) can create a new way to extend the service life of photo-generated charge carriers via facilitating the electron transfer through Co NPs.42 Electron cannot return from the excited state to the ground state, thus directly weakens the PL intensity of NH2-MIL-125(Ti). The transient photocurrent density responses of the catalysts in an on–off cycle mode are shown in Fig. 8. Upon visible-light irradiation, all Co/NH2-MIL-125(Ti) catalysts show much higher photoelectric currents in comparison with NH2-MIL-125(Ti), among which 1.0 wt% Co/NH2-MIL-125(Ti) exhibits the best photocurrent response. Based on these results, it can be inferred that the doping of Co NPs into NH2-MIL-125(Ti) could facilitate photo-induced charge transfer and restrain the recombination of photo-generated charge efficiently, resulting in the higher photocatalytic activity in CO2 reduction. The in situ ESR experiment was also carried out to detect the active species involved in the photocatalytic reaction, as shown in Fig. S8 in the ESI.† A signal assignable to Ti3+ species is observed in the ESR spectrum of Co/NH2-MIL-101(Ti) after visible-light irradiation.34 When CO2 is introduced into the reaction system, the signal in the ESR spectrum corresponding to Ti3+ species disappears, indicating the reoxidation of generated Ti3+ species to Ti4+ species.
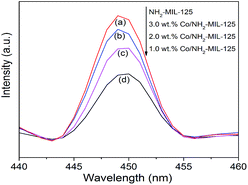 |
| | Fig. 7 Photoluminescence spectra of NH2-MIL-125(Ti) (a), 3.0 wt% Co/NH2-MIL-125(Ti) (b), 2.0 wt% Co/NH2-MIL-125(Ti) (c), and 1.0 wt% Co/NH2-MIL-125(Ti) (d) excited at 303 nm. | |
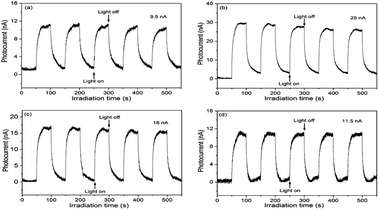 |
| | Fig. 8 Transient photocurrent responses of NH2-MIL-125(Ti) (a), 1.0 wt% Co/NH2-MIL-125(Ti) (b), 2.0 wt% Co/NH2-MIL-125(Ti) (c), and 3.0 wt% Co/NH2-MIL-125(Ti) (d) in 0.2 M Na2SO4 aqueous solution without bias versus Ag/AgCl upon visible-light irradiation (800 nm ≥ λ ≥ 420 nm). | |
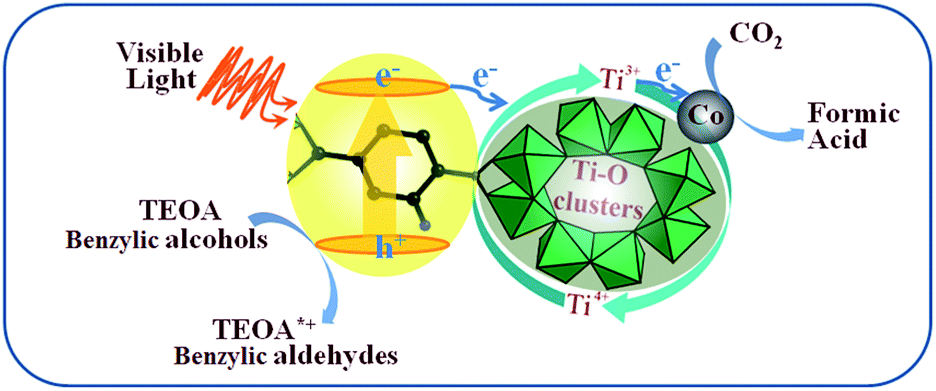
Herein, a mechanism for the enhancement of the photocatalytic reduction over Co/NH2-MIL-125(Ti) was proposed, see Scheme 1. The excited ligands can transfer electrons to Ti oxo-clusters to form Ti3+ moieties upon visible-light irradiation, as termed linker-to-cluster charge-transfer (LCCT). The as-formed Ti3+ can reduce CO2 into HCOOH in the presence of TEOA or benzylic alcohols acting as electron and hydrogen donors to achieve a complete photocatalytic cycle. When Co NPs are introduced into NH2-MIL-125(Ti), they can also accept the electrons from the excited ligands, because Co NPs are usually good at electron traps. Meanwhile, the as-produced H atoms from TEOA or benzylic alcohols can spill over from the Co NPs to the bridging oxygen next to the Ti–O oxo-clusters and donate electrons to reduce Ti4+ to form Ti3+. TEOA or the benzylic alcohols would be converted into TEOA*+ or the aldehydes by a de-protonation process during CO2 reduction. Thus, the more efficient light-harvesting and charge-transfer of Co/NH2-MIL-125(Ti) lead to a higher photocatalytic activity.
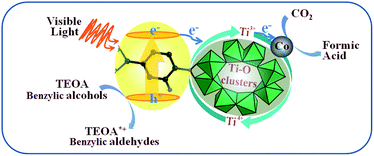 |
| | Scheme 1 Proposed mechanism for photocatalytic CO2 reduction over Co/NH2-MIL-125(Ti) upon visible-light irradiation. | |
4. Conclusions
The photocatalytic reduction of CO2 over Co/NH2-MIL-125(Ti) upon visible-light irradiation has been fully investigated. Compared with NH2-MIL-125(Ti), Co/NH2-MIL-125(Ti) catalysts exhibit significantly enhanced activities for the photocatalytic reduction. In addition, the benzylic alcohols instead of TEOA used as elector donors can be oxidized to the corresponding aldehydes during the photocatalytic reduction of CO2 over Co/NH2-MIL-125(Ti), making the proposed process more economical and environment-friendly. Although the photocatalytic activity for CO2 reduction coupled with the oxidation of benzylic alcohols over Co/NH2-MIL-125(Ti) is still low, the current study provides a novel way to convert CO2 into valuable chemicals coupled with other organic transformations. Additionally, the elucidation of the mechanism on photocatalytic CO2 reduction over Co/NH2-MIL-125(Ti) provides some guidance in the development of high-efficient and visible-light-responsive photocatalysts based on MOF materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for the financial support by the National Natural Science Foundation of China (21303166, 21476214, and 21576243).
References
- W. Wang, M. O. Tadé and Z. Shao, Chem. Soc. Rev., 2015, 44, 5371–5408 RSC.
- P. Lanzafame, G. Centi and S. Perathoner, Chem. Soc. Rev., 2014, 43, 7562–7580 RSC.
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473–486 RSC.
- G. Qin, Y. Zhang, X. Ke, X. Tong, Z. Sun, M. Liang and S. Xue, Appl. Catal., B, 2013, 129, 599–605 CrossRef CAS.
- Y. Wang, Q. Lai, F. Zhang, X. Shen, M. Fan, Y. He and S. Ren, RSC Adv., 2014, 4, 44442–44451 RSC.
- Y. He, L. Zhang, B. Teng and M. Fan, Environ. Sci. Technol., 2015, 49, 649–656 CrossRef CAS PubMed.
- Y. He, Y. Wang, L. Zhang, B. Teng and M. Fan, Appl. Catal., B, 2015, 168–169, 1–8 CAS.
- N. Linares, A. M. Silvestre-Albero, E. Serrano, J. Silvestre-Albero and J. García-Martínez, Chem. Soc. Rev., 2014, 43, 7681–7717 RSC.
- J. Rongé, T. Bosserez, D. Martel, C. Nervi, L. Boarino, F. Taulelle, G. Decher, S. Bordiga and J. A. Martens, Chem. Soc. Rev., 2014, 43, 7963–7981 RSC.
- W. Tu, Y. Zhou and Z. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed.
- W. Lin and H. Frei, J. Am. Chem. Soc., 2005, 127, 1610–1611 CrossRef CAS PubMed.
- S. Sato, T. Morikawa, T. Kajino and O. Ishitani, Angew. Chem., Int. Ed., 2013, 125, 1022–1026 CrossRef.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- H.-C. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed.
- L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- A. Corma, H. García, F. X. Llabrés and I. Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- T. Tachikawa, J. R. Choi, M. Fujitsuka and T. Majima, J. Phys. Chem. C, 2008, 112, 14090–14101 CAS.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- H.-Q. Xu, J. Hu, D. Wang, Z. Li, Q. Zhang, Y. Luo, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2015, 137, 13440–13443 CrossRef CAS PubMed.
- D. Wang, R. Huang, W. Liu, D. Sun and Z. Li, ACS Catal., 2014, 4, 4254–4260 CrossRef CAS.
- D. Sun, Y. Fu, W. Liu, L. Ye, D. Wang, L. Yang, X. Fu and Z. Li, Chem.–Eur. J., 2013, 19, 14279–14285 CrossRef CAS PubMed.
- D. Sun, W. Liu, M. Qiu, Y. Zhang and Z. Li, Chem. Commun., 2015, 51, 2056–2059 RSC.
- Y. Lee, S. Kim, J. K. Kang and S. M. Cohen, Chem. Commun., 2015, 51, 5735–5738 RSC.
- S. Zhang, L. Li, S. Zhao, Z. Sun and J. Luo, Inorg. Chem., 2015, 54, 8375–8379 CrossRef CAS PubMed.
- C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- L. Shi, T. Wang, H. Zhang, K. Chang and J. Ye, Adv. Funct. Mater., 2015, 25, 5360–5367 CrossRef CAS.
- D. Sun, W. Liu, Y. Fu, Z. Fang, F. Sun, X. Fu, Y. Zhang and Z. Li, Chem.–Eur. J., 2014, 20, 4780–4788 CrossRef CAS PubMed.
- Y. Fu, L. Sun, H. Yang, L. Xu, F. Zhang and W. Zhu, Appl. Catal., B, 2016, 187, 212–217 CrossRef CAS.
- M. Dan-Hardi, C. Serre, T. Frot, L. Rozes, G. Maurin, C. Sanchez and G. Férey, J. Am. Chem. Soc., 2009, 131, 10857–10859 CrossRef CAS PubMed.
- H. Jin, J. Wang, D. Su, Z. Wei, Z. Pang and Y. Wang, J. Am. Chem. Soc., 2015, 137, 2688–2694 CrossRef CAS PubMed.
- P. Bazylewski, D. W. Boukhvalov, A. I. Kukharenko, E. Z. Kurmaev, A. Hunt, A. Moewes, Y. H. Lee, S. O. Cholakh and G. S. Chang, RSC Adv., 2015, 5, 75600–75606 RSC.
- H. Fei, Y. Yang, Z. Peng, G. Ruan, Q. Zhong, L. Li, E. L. G. Samuel and J. M. Tour, ACS Appl. Mater. Interfaces, 2015, 7, 8083–8087 CAS.
- L. Wu, Q. Li, C. H. Wu, H. Zhu, A. Mendoza-Garcia, B. Shen, J. Guo and S. Sun, J. Am. Chem. Soc., 2015, 137, 7071–7074 CrossRef CAS PubMed.
- A. Ishikawa, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2002, 124, 13547–13553 CrossRef CAS PubMed.
- A. Wolcott, W. A. Smith, T. R. Kuykendall, Y. Zhao and J. Z. Zhang, Small, 2009, 5, 104–111 CrossRef CAS PubMed.
- V. P. Indrakanti, J. D. Kubicki and H. H. Schobert, Energy Environ. Sci., 2009, 2, 745–758 CAS.
- G. Li, L. Wu, F. Li, P. Xu, D. Zhang and H. Li, Nanoscale, 2013, 5, 2118–2125 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06324e |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab,
Huan Yanga,
Rongfei Dua,
Gaomei Tua,
Chunhui Xua,
Fumin Zhanga,
Maohong Fan
ab,
Huan Yanga,
Rongfei Dua,
Gaomei Tua,
Chunhui Xua,
Fumin Zhanga,
Maohong Fan