
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, characterization and pharmaceutical/pharmacological applications of ibuprofen conjugates based on hydroxyethylcellulose†
Khawar Abbasa,
Muhammad Amina,
Muhammad Ajaz Hussain *a,
Muhammad Shera,
Syed Nasir Abbas Bukharib and
Kevin J. Edgarc
*a,
Muhammad Shera,
Syed Nasir Abbas Bukharib and
Kevin J. Edgarc
aDepartment of Chemistry, University of Sargodha, Ibn-e-Sina Block, Sargodha 40100, Pakistan. E-mail: majaz172@yahoo.com
bDepartment of Pharmaceutical Chemistry, College of Pharmacy, Aljouf University, Aljouf, Sakaka 2014, Saudi Arabia
cDepartment of Sustainable Biomaterials, Macromolecules Innovation Institute, Virginia Tech, 230 Cheatham Hall, Blacksburg, Virginia 24061, USA
First published on 31st October 2017
Abstract
We synthesized polymeric prodrugs in which ibuprofen (IBU) was conjugated in a one-pot preparation to the hydrophilic polysaccharide hydroxyethylcellulose (HEC). The amphiphilic conjugates formed nanoparticles of 450–700 nm diameter at a water/DMSO interface. Synthesized HEC-IBU conjugates showed significant anti-inflammatory (44–77% inhibition) and immunomodulatory potential in paw-edema and cytokine release assays, respectively. Cell viability studies on L929 cell lines were performed for HEC-IBU prodrugs. Pharmacokinetic studies in rabbit models revealed that IBU from the conjugate reaches a peak plasma concentration (Cmax) of 20 μg mL−1 in 4 h while the plasma half-life was noted to be 5.82 h μg mL−1.
1 Introduction
Ibuprofen (IBU) belongs to the class of propanoic acid derivatives of non-steroidal anti-inflammatory drugs (NSAIDs)1 and is widely used to treat pain, fever and inflammation. The carboxyl acid group of these NSAIDs is thought to be responsible for side effects including superficial stomach erosion, vomiting, dyspepsia, abdominal discomfort, serious bleeding, or activation of peptic ulcer and diarrhea,2–4 especially upon prolonged administration. IBU is also relatively poorly soluble, ca. 10 μg mL−1 even after carboxyl ionization at pH 6.8 (small intestine pH).5 So, it is desirable to modify the IBU carboxylic acid group6 to form bioreversible derivatives that may prevent IBU-associated side effects.Prodrug formation is a promising strategy for reducing the gastric side effects of NSAIDs. This concept involves conversion of drug to a therapeutically inactive form that is capable of releasing active drug at site of action after chemical and/or enzymatic transformation.7 In one realization of this concept, many investigators have appended drugs onto therapeutically inert polymeric backbones, thereby providing reservoirs for slow release of drug that are termed “polymeric prodrugs”.8 Researchers have explored polysaccharide frameworks to produce polymeric prodrugs because of their suitable characteristics, including low toxicity, low bioavailability of the polysaccharide itself (especially useful for oral administration), and the availability of multiple hydroxyl groups as “handles” for drug attachment. Polysaccharide-based prodrugs may possess advantages including increased drug bioavailability, improved distribution, and enhanced patient compliance. Properly designed polymeric prodrugs may also reduce exposure of the drug to the stomach contents, thereby reducing or eliminating stomach irritation or acidic and enzymatic drug degradation, in cases where these would otherwise be problematic.9–13
Recently, cellulose ethers have been used to synthesize polymeric prodrugs. Cellulose ethers are particularly well-suited to this task since they are frequently used in oral drug formulations already. Hydroxyalkyl ethers are especially suitable since they have DS(OH) of 3.0, and since the terminal alcohols of the oligo(hydroxyalkyl) chains are less sterically encumbered than the glucose hydroxyls.14,15 The hydroxyl functions of polysaccharide-based materials can be easily modified to synthesize ester prodrugs of NSAIDs and antibiotics16–18 which can act as substrates for endogenous esterase enzymes, making them suitably labile in vivo.19,20 Polysaccharide-based IBU prodrugs may also improve IBU bioavailability, and prevent stomach release, thereby reducing their ulcerogenic potential. Polymeric IBU prodrugs have been previously reported,21–23 but they have typically been appended to synthetic polymers, and typically by polymerization of an IBU-containing monomer. Such methods do not enjoy the benefits of a polysaccharide backbone, and could raise concerns about residual monomers, or side reactions during polymerization.
Our aim in this work was to attach IBU to the water-soluble, versatile cellulose ether hydroxyethyl cellulose (HEC). We hypothesize that HEC-IBU conjugates will afford enhanced therapeutic IBU levels and maintain IBU plasma level for a longer time period vs. unmodified drug. Therefore we also report results of bioavailability studies of the HEC-IBU conjugates in rabbit models. Moreover, pharmacological properties of the synthesized polymeric prodrugs were evaluated through anti-inflammatory, immunomodulatory and cytotoxicity studies to assess their potential for clinical efficacy.
2 Materials and methods
2.1 Materials
IBU was of US Pharmacopoeia standard. Imidazole and 4-methylbenzenesulfonyl chloride (syn: tosyl chloride) were obtained from Sigma-Aldrich. N,N′-Dimethylacetamide (DMAc), dimethylsulfoxide (DMSO) and diethyl ether were obtained from Fluka and used as received. HEC (DS 1.50, MS 2.50, Mw 3.6 × 105 g mol−1) was procured from Ashland Inc., Covington, KY, USA and vacuum dried at 110 °C overnight. Regenerated cellulose dialysis membrane (21.0 mm diameter; MWCO 12000–14000 Da) was from Cellu·Sep® T4, USA. Sterile disposable syringes (Injekt®, B. Braun) were used for blood sampling during bioavailability studies.2.2 Measurements
IR Prestige-21 (Shimadzu, Japan) instrument was used for recording FT-IR spectra. All spectra were recorded in transmittance mode using KBr pellets (3 mg sample/100 mg KBr). Structures of IBU and HEC-IBU conjugates were investigated by 13C (500 MHz, ≈5000 scans) and 1H (500 MHz, 16–64 scans) NMR spectroscopy on Bruker Avance II instruments. A Pharmaspec UV-1700 (Shimadzu, Japan) instrument was used for determination of drug contents (DC) of the HEC-IBU conjugates. Nano-assemblies of HEC-IBU conjugates were observed on Philips 420 transmission electron microscope operating at 120 kV. Bioavailability studies were carried out using an HPLC/UV technique on Agilent (1200 Series, Germany) instrument having Shim-Pack column (ODS 5 μm; 4.6 × 250 mm), pump (G1311A), degasser (G1322A) and UV-vis detector (G1315B DAD).2.3 Example synthesis: HEC-IBU conjugate 3
HEC (1.0 g, 3.62 mmol) was added to pre-heated DMAc (30 mL, 80 °C) and stirred for 30 min to obtain a clear solution. Imidazole (1.97 g, 28.96 mmol) was added to the HEC solution followed by tosyl chloride (2.76 g, 14.48 mmol). IBU (2.99 g, 14.48 mmol) was then added in portions and the reaction mixture was stirred at 80 °C for 8 h. HEC-IBU conjugate 3 was precipitated by adding the reaction mixture to diethyl ether (150 mL), and isolating the product by filtration. Impurities were removed by thorough washing of the precipitates using diethyl ether (100 mL) and products (off-white powder) were vacuum dried at 50 °C before further analysis.DS(IBU) = 1.85 by UV/vis spectrophotometry; yield: 1.80 g (79%); FTIR (KBr): 2964–3132 (OH), 2855 (CH), 1728 (COEster), 1445 (CH2), 1036 (COC) cm−1; 1H NMR (δ ppm, 500 MHz, DMSO-d6): 7.10–7.21 (H 12, 13, 15, 16), 3.25–4.49 (H 1–8), 3.59 (H 10), 2.38 (H 17), 1.76 (H 18), 1.31 (H 21), 0.82 (H 19, 20); 13C NMR (δ ppm, 500 MHz, DMSO-d6, NS 5000): 170.67 (C 9), 127.29–140.06 (C 11–16), 102.19–103.49 (C 1), 80.05–83.15 (C 2–4), 71.96–74.95 (C 5), 68.99–70.25 (C 6, 7), 60.67 (C 8), 44.57 (C 17), 30.02 (C 18), 22.56 (C 19, 20).
1H and 13C NMR spectra of HEC-IBU conjugates 1 and 2 showed chemical shift values comparable to those of conjugate 3. We selected HEC-IBU conjugate 3 for further study because it had the highest IBU content.
2.4 Determination of DC by UV/vis spectrophotometric analysis
DC of HEC-IBU conjugate 1–3 were calculated from the UV/vis spectrophotometric analysis. For this purpose, standard solutions of IBU were prepared in 0.1 N aq. NaOH and a calibration curve was plotted. HEC-IBU conjugates 1–3 (10 mg each) were then separately dissolved in 0.1 N aq. NaOH (10 mL) and heated at 80 °C for 10 h. After hydrolysis, sample volume was made up to 10 mL using 0.1 N aq. NaOH and absorbance was recorded at 222 nm (λmax) after suitable dilution (to stay within the range of calibration curve). DC was calculated by comparing absorbance with the standard calibration curve.2.5 Transmission electron microscopic (TEM) analysis
For recording of TEM images, HEC-IBU conjugate 3 (10 mg) was added to DMSO (10 mL) and dialysed against milli-Q water for 3 days, with frequent water replacement. The resultant slurry in the dialysis tube was drop casted on TEM grid (Cu) and its morphology was observed.2.6 High performance liquid chromatography (HPLC/UV) method development
An HPLC/UV method was developed and validated for determination of IBU in rabbit plasma samples following administration of HEC-IBU conjugate 3 according to a reported method.13 Results for HPLC/UV parameters of IBU are shown in the ESI (Table S1†).2.7 Pharmacokinetic studies
Experimental and postoperative care were followed as per protocols issued by the National Institute of Health's Guidelines for the Care and Use of Laboratory Animals. Approval of protocols for animal studies was granted by the Pharmacy Research Ethics Committee, University of Sargodha, Sargodha, Pakistan (Ref no. 17-2016/PREC).
Plasma concentration of IBU at different intervals of dose administration was plotted against time and area under the curve (AUC) was calculated. Kinetic parameters like clearance (Cl), half-life of drug (t1/2), peak drug concentration (Cmax) and volume of distribution (Vd) were calculated using the linear trapezoidal method.24
2.8 In vitro drug release study
A drug release study was carried out in a USP dissolution apparatus using simulated gastric fluid (SGF, pH 1.2) and simulated intestinal fluid (SIF, pH 7.2). The sample, i.e., HEC-IBU conjugate 3 (172 mg; equivalent to 50 mg IBU) was placed in a cellophane bag which was in turn placed in a dissolution vessel containing 900 mL release medium. The medium was stirred at 50 rpm and held at 37 °C. The experiment was performed at pH 1.2 for 2 h, then the medium was removed and replaced by pH 7.2 medium for 6 h, mimicking the gastro-intestinal tract (GIT) pH and residence time. Aliquots (5 mL) were withdrawn at specific time intervals, filtered and absorbance was measured at 221 (for pH 1.2) and 220 nm (for pH 7.2) using a UV-vis spectrophotometer. An amount of the appropriate release medium equal to the removed sample volume was added to the dissolution medium immediately after taking the sample.2.9 Bioassays
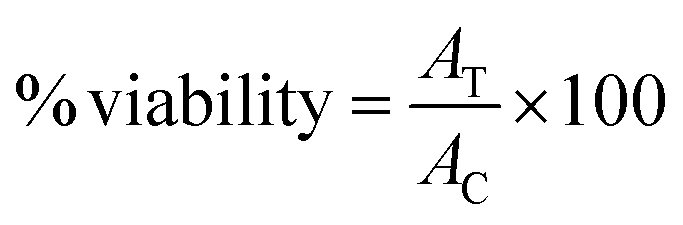 | (1) |
2.10 Statistical analyses
GraphPad Prism 5.0 (GraphPad Software Inc., La Jolla, CA, USA) was used for statistical analyses and data analysis was carried out using two-way ANOVA trailed by multiple-comparison test. A p value < 0.05 was considered statistically significant as denoted by asterisks in the respective Figures.3 Results and discussion
3.1 Synthesis and characterization of HEC-IBU prodrugs
A one pot and homogeneous reaction methodology was used for synthesis of HEC-IBU conjugates 1–3. The IBU carboxylic acid group was activated using tosyl chloride and further reacted with the hydroxyl groups of the HEC backbone in the presence of imidazole base to give conjugates 1–3. Mole ratios of drug to polymer were varied to tune DS(IBU). This synthesis strategy resulted in high yields (77–82%) with moderate to high DS(IBU) (0.51–1.85).Conditions and results of reactions are given in Table 1, while reaction methodology is depicted in Scheme 1.
| HEC-IBU conjugate | Molar ratioa | DCb | DSc | Yield (%) | Solubility |
|---|---|---|---|---|---|
a HEC repeating unit (HRU)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :IBU:tosyl chloride:imidazole.b DC (mg drug/100 mg of conjugate) calculated by UV/vis spectroscopy.c DS assessed in terms of substituted groups per HRU from DC. :IBU:tosyl chloride:imidazole.b DC (mg drug/100 mg of conjugate) calculated by UV/vis spectroscopy.c DS assessed in terms of substituted groups per HRU from DC. |
|||||
| 1 | 1:1:1:2 |
29 | 0.51 | 82 | DMSO, DMAc, H2O |
| 2 | 1:2:2:4 |
43 | 1.10 | 77 | DMF, DMSO, DMAc |
| 3 | 1:4:4:8 |
56 | 1.85 | 79 | DMF, DMSO, DMAc |
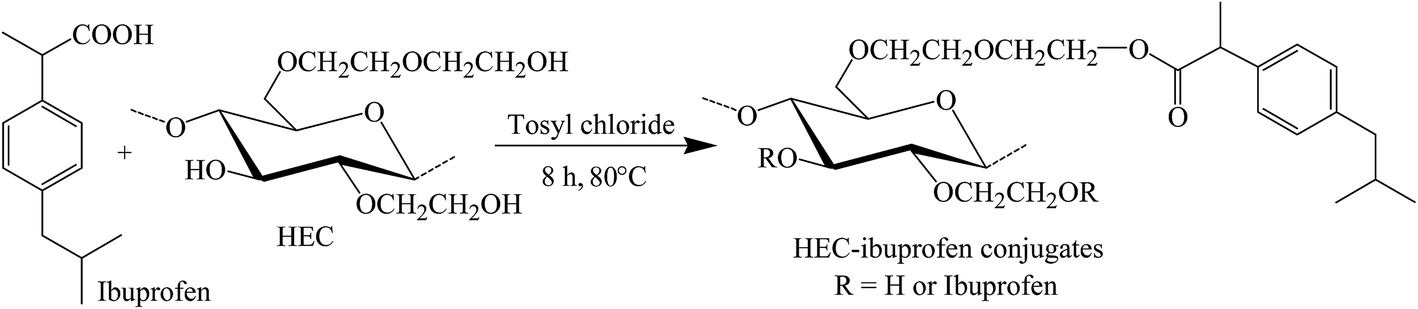 | ||
| Scheme 1 Synthesis of HEC-IBU conjugates using tosyl chloride. | ||
FT-IR spectra of HEC-IBU conjugates 1–3 showed ester signals in the range of 1725–1736 cm−1 which indicated acylation of HEC by IBU. For example, the FT-IR spectrum of HEC-IBU conjugate 3 displays shifting of the carbonyl stretch from 1711 (IBU carboxyl) to 1728 cm−1 (ester carbonyl of 3). Methylene signals of the conjugates were detectable at 1445 cm−1. The COC group of the HEC backbone was detected at 1036 cm−1. As a typical example, overlay spectra of IBU and HEC-IBU conjugate 3 are shown in Fig. 1.
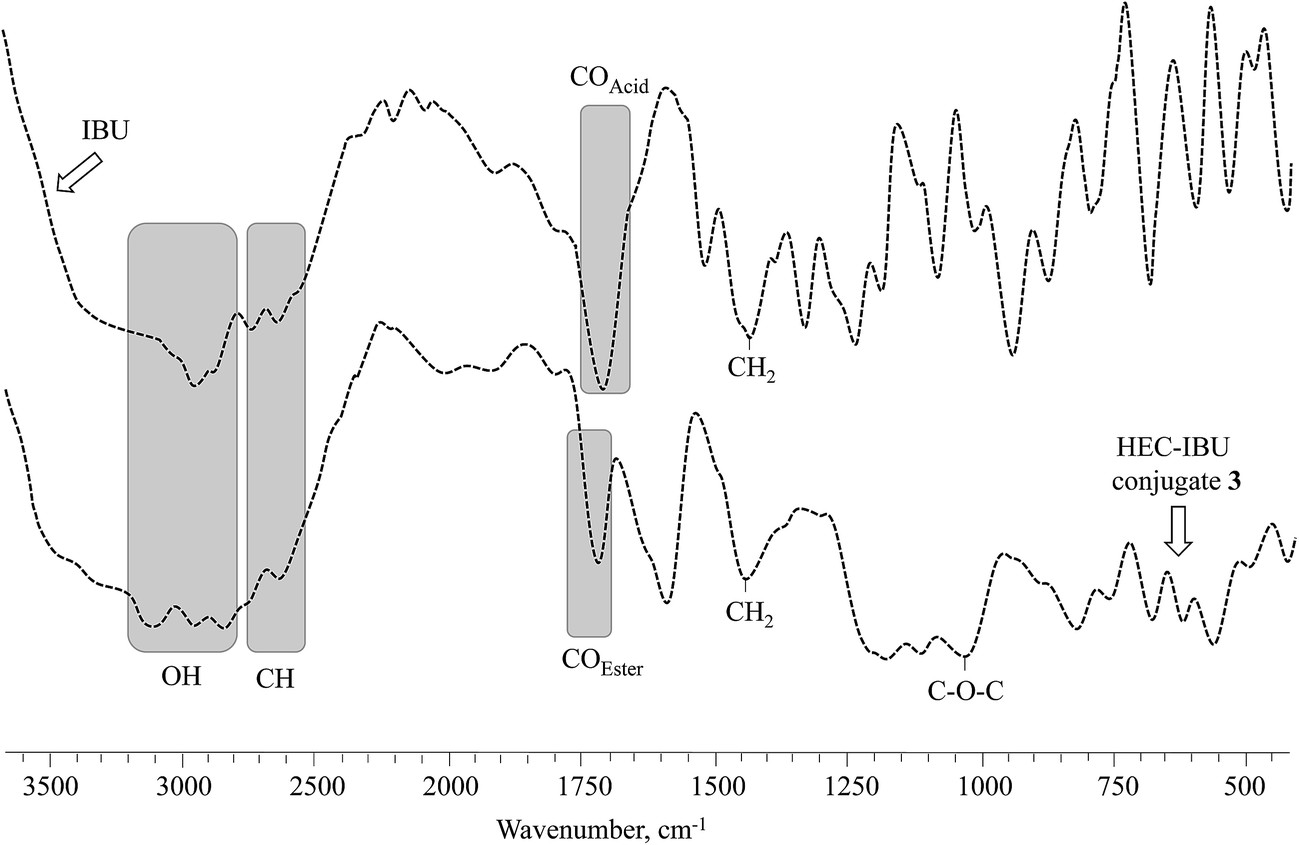 | ||
| Fig. 1 FT-IR (transmittance, KBr) spectra of IBU and HEC-IBU conjugate 3. | ||
1H NMR (500 MHz, DMSO-d6) spectra were recorded to establish the structure of HEC-IBU conjugates 1–3. For example, with HEC-IBU conjugate 3 protons of the aromatic system were detected in the range of δ 7.10–7.21 (H 12, 13, 15, 16) ppm. Protons of CH2 groups of IBU were observed at δ 2.38 (H 17) while its CH3 protons were detected at 0.82 (H 19, 20) and 1.31 (H 21) ppm. Protons of the isobutyl CH groups appeared at δ 1.76 (H 18) and 3.59 (H 10). Broad signals of the HEC backbone were observed in the range of 3.25–4.49 (H 1–8) ppm. Overlay 1H NMR spectra of IBU and HEC-IBU conjugate 3 are shown in Fig. 2a and b. For comparison, FT-IR (Fig. S1†) and 1H NMR (Fig. S2†) spectra of HEC are given in ESI.†
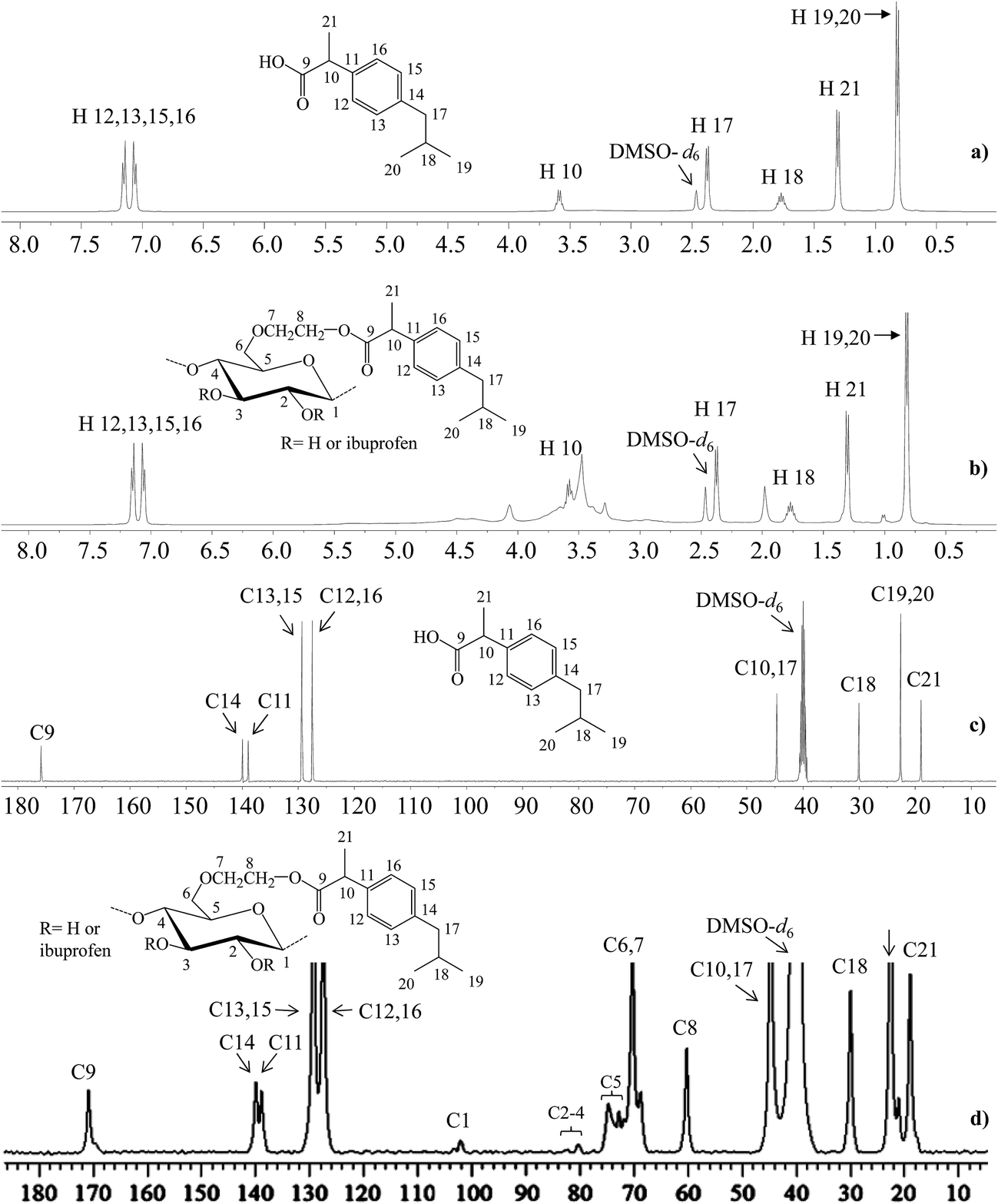 | ||
| Fig. 2 1H NMR (500 MHz, DMSO-d6, ppm) and 13C NMR (500 MHz, 5000 scans, DMSO-d6, ppm) spectra: (a) 1H and (c) 13C NMR spectra of IBU, (b) 1H and (d) 13C NMR spectra of HEC-IBU conjugate 3. | ||
In the 13C NMR (500 MHz, DMSO-d6) spectrum of HEC-IBU conjugate 3, the resonance at δ 170.67 ppm was assigned to the ester carbonyl moiety (C 9). Carbons of the aromatic system were observed at δ 127.29–140.06 (C 11–16) while carbons of the isobutyl group were present at 44.57 (C 17), 30.02 (C 18) and 22.56 (C 19, 20) ppm. The IBU carbon adjacent to the carbonyl group (C 10) resonated at δ 44.57 and the IBU methyl carbon (C 21) resonated at 18.91 ppm. Polymer backbone carbons appeared at δ 102.19–103.49 (C 1), 80.05–83.15 (C 2–4), 71.96–74.95 (C 5), 68.99–70.25 (C 6, 7) and 60.67 (C 8) ppm. The presence of characteristic drug and polymer signals along with the new ester signals further confirmed successful synthesis of HEC-IBU conjugate 3. Overlay 13C NMR spectra of IBU and HEC-IBU conjugate 3 are shown in Fig. 2c and d.
3.2 Transmission electron microscopic (TEM) analysis
HEC-IBU conjugates were synthesized with a range of DS(IBU) values in order to tune the hydrophilic/hydrophobic balance (amphiphilic character). Previous investigators have shown that colloidal forms of drugs and prodrugs could be highly advantageous drug delivery systems to achieve defined drug release and improved pharmacokinetic profiles.29 Among the three polymeric prodrugs prepared, only HEC-IBU-conjugate 3 showed self assembly behaviour due to its balanced hydrophobic/hydrophilic character. Dialysis of a DMSO solution of 3 against milli Q water afforded nanoparticles of the HEC-IBU conjugate, with diameter ranging from 450–700 nm (Fig. 3). Conjugates 1 and 2 did not form well-defined morphologies; their lower DS(IBU) values apparently were insufficient to provide balanced hydrophobicity and hydrophilicity that would lead to self-assembly. | ||
| Fig. 3 TEM images of the HEC-IBU conjugate 3. | ||
3.3 Pharmacokinetic studies
A validated reversed phase HPLC/UV method was used for determination of plasma levels of IBU from HEC-IBU conjugate 3. Results of method validation and values of different parameters are shown in the ESI (see Table S1†).A single oral dose (206 mg, equivalent to 120 mg IBU) of HEC-IBU conjugate 3 was given to albino rabbits and blood samples were collected at specific time intervals. The plasma concentration vs. time curve showed AUC0–∞ of 256.43 h μg mL−1 and Cmax value of 20.12 μg mL−1. HEC-IBU conjugate 3 showed significantly delayed IBU release, with tmax of 4.0 h vs. unmodified IBU with tmax of 2.1 h. This is also reflected in the plasma half-life (t1/2) values, which are ca. 3× longer for HEC-IBU conjugate 3 (5.82 h) than for IBU itself (1.73 h). The relative AUC values indicated that bioavailability of IBU from conjugate 3 was approximately 1.5× higher than that from unmodified drug. Moreover, the HEC-IBU conjugate had delayed release properties; these show potential to be exploited to achieve sustained plasma levels of IBU and reduced dosage frequency. Plasma concentration of IBU at different time points is shown in Fig. 4 while pharmacokinetic parameters are summarized in Table 2.
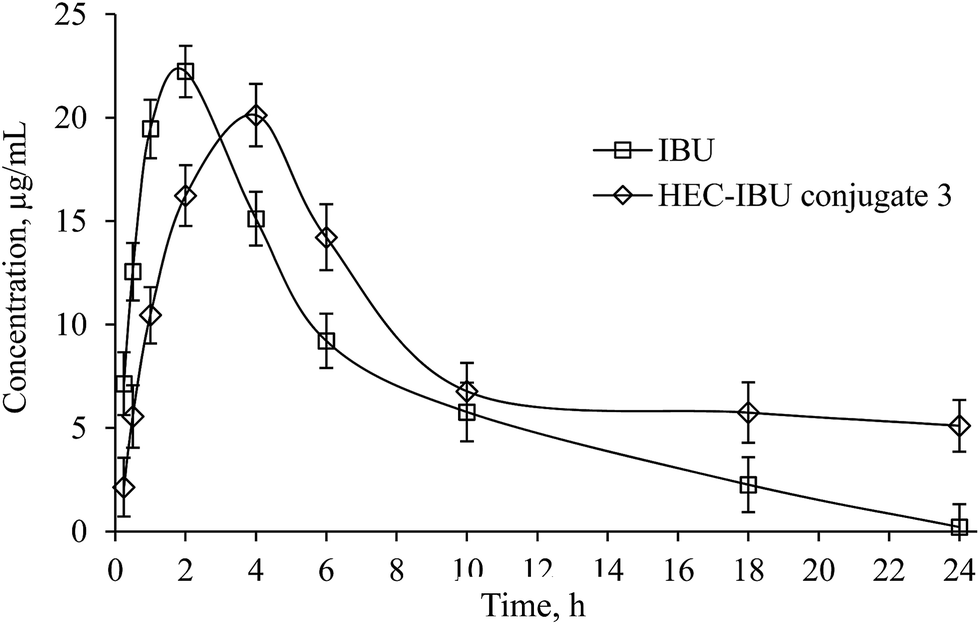 | ||
| Fig. 4 Plasma concentration vs. time curve of IBU from HEC-IBU conjugate 3 following single oral dose of 206 mg. | ||
| Parameter | IBU | IBU from HEC-IBU conjugate 3 |
|---|---|---|
| tmax (h) | 2.0 | 4.0 |
| Cmax (μg mL−1) | 22.23 ± 2.29 | 20.12 ± 3.41 |
| t1/2 (h) | 1.73 ± 1.21 | 5.82 ± 0.87 |
| AUC0–∞ (h μg mL−1) | 163 ± 4.23 | 256.43 ± 6.77 |
| Vd (L kg−1) | 0.61 ± 0.41 | 1.31 ± 0.1 |
| Cl (L h−1) | 0.24 ± 0.07 | 0.16 ± 0.04 |
3.4 In vitro drug release
We carried out an in vitro drug release study in a manner mimicking the GIT environment that would be experienced by the conjugate in vivo; first exposure to gastric pH for 2 h, then changing dissolution media to intestinal pH for 6 h (Fig. 5). It was observed that only 8% drug was released at pH 1.2 in 2 h from HEC-IBU conjugate 3. The minimal release indicated that the HEC-IBU conjugate is relatively stable to acid-catalyzed hydrolysis, thereby supporting our concept that the prodrug might minimize IBU-stomach exposure. After 6 h at pH 7.2, 85% IBU was released. This is consistent with the fact that polysaccharide ester hydrolysis is much faster at alkaline than at acidic pH.30 For comparison, a physical mixture of HEC and IBU (50 mg each) was used as a control and IBU was completely released from this mixture within 1.5 h at pH 1.2.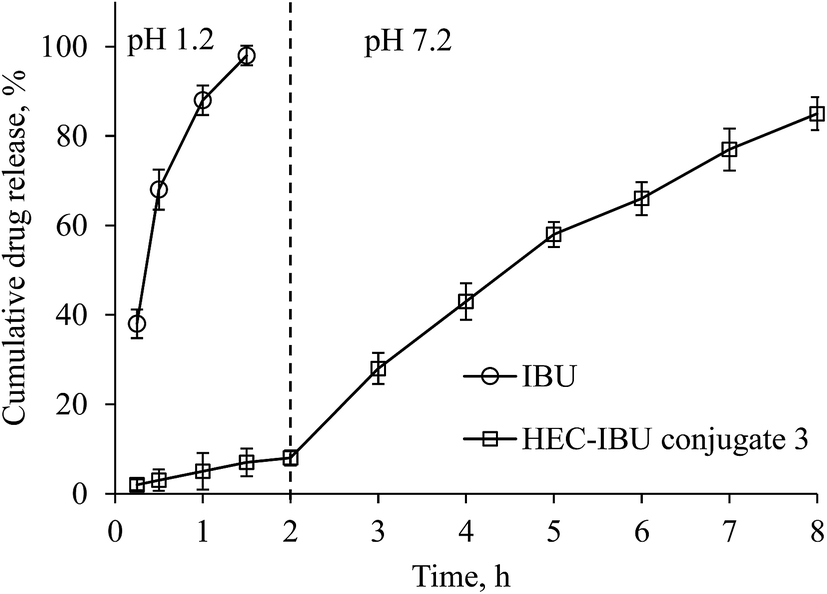 | ||
| Fig. 5 In vitro release of IBU and HEC-IBU conjugate 3 in SGF and SIF. | ||
3.5 Carrageenan-induced paw edema
Inhibition of carrageenan-induced paw edema was used to evaluate the anti-inflammatory potential of HEC-IBU conjugate 3. The results are plotted in Fig. 6 as % inhibition of swelling for animals orally administered with HEC-IBU conjugate 3 and compared with % inhibition of unmodified IBU. Although the volume of the injected paws of the animals dosed with HEC-IBU conjugate 3 increased gradually up to 4 h, significantly less swelling (expressed as % inhibition, reaching 77% after 4 h) was observed. Unmodified IBU showed higher % inhibition in the first 2 h than HEC-IBU conjugate 3, but lower inhibition was observed from 3 h onwards, consistent with slower release from the polymeric prodrug. These results indicated that anti-inflammatory properties of IBU were retained and sustained effects were observed after its attachment to HEC as HEC-IBU conjugate 3.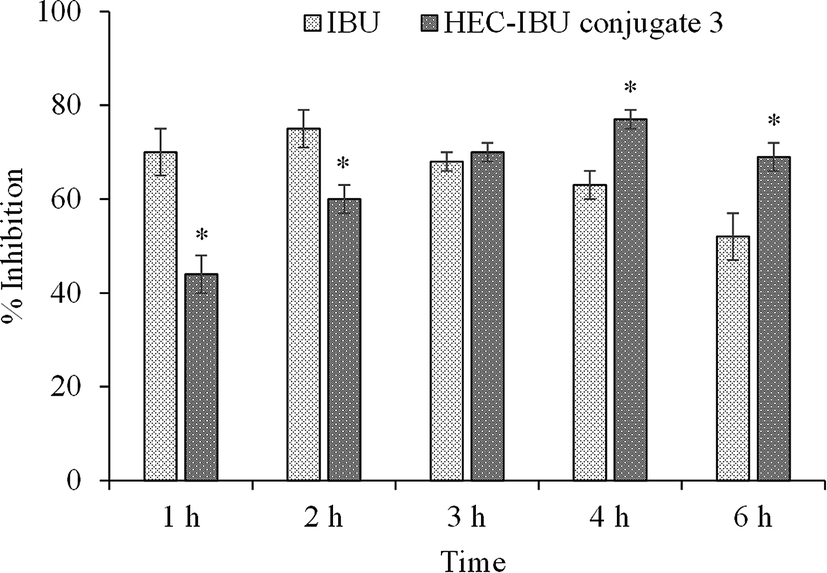 | ||
| Fig. 6 Carrageenan induced paw edema inhibition vs. time for orally administered IBU and HEC-IBU conjugate 3 (% inhibition vs. water-only control). Statistically significant when compared to control indicated with *p < 0.05. | ||
3.6 In vitro cytotoxicity assay
The MTT assay was used to evaluate cytotoxicity of HEC-IBU conjugate 3 up to 72 h exposure (Fig. 7) and showed no toxicity against L929 cell lines up to 24 h in the concentration range of 2–10 mM. However, there was significant reduction in the mitochondrial enzymes on prolonged exposure (48 and 72 h). Since these time ranges are higher than clearance time of IBU, the prodrug was considered to be non-cytotoxic, hence biocompatible within the time it is likely to remain in the human body.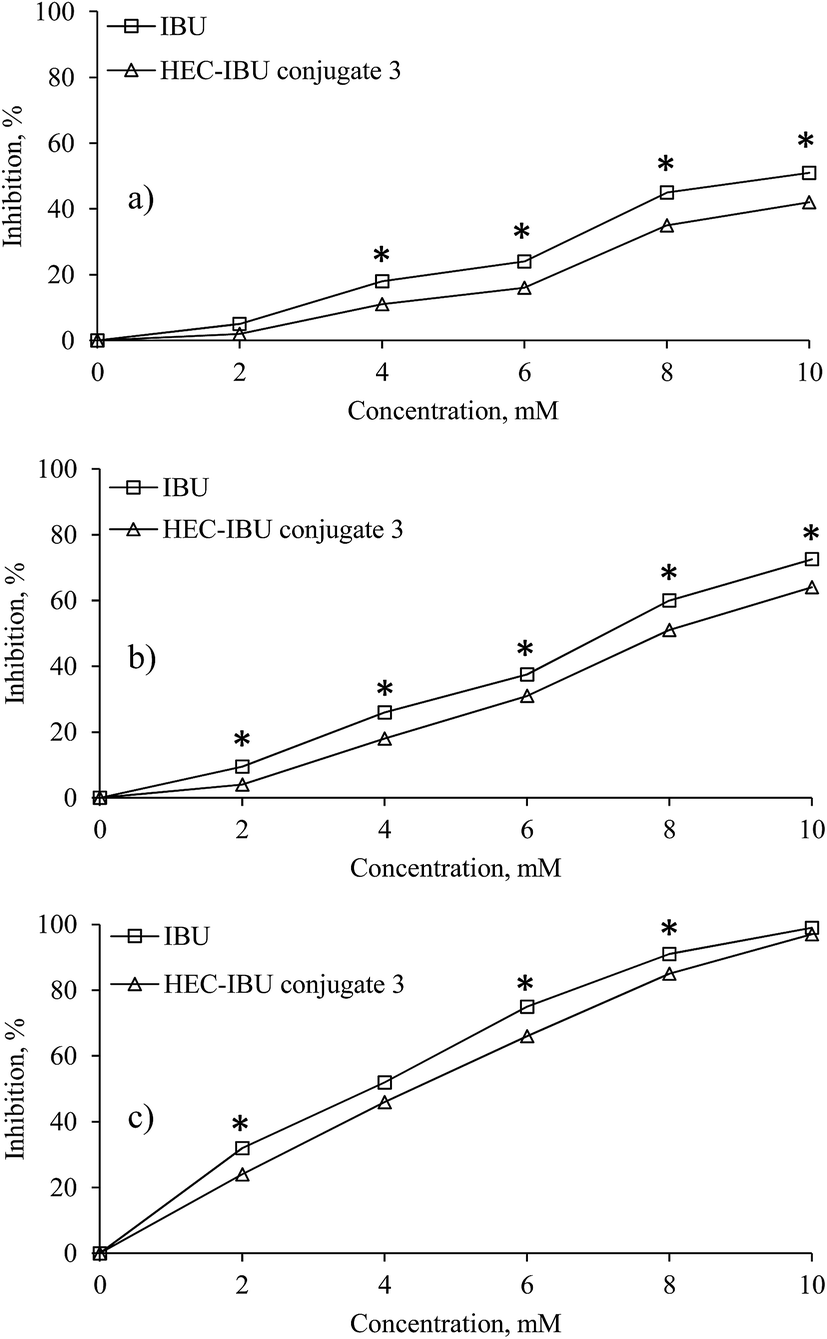 | ||
| Fig. 7 Cytotoxic effects (percent inhibition) by different concentrations of HEC-IBU conjugate 3 and IBU control on the L929 cell line after 24 (a), 48 (b) and 72 h (c). *p < 0.05 is considered significant when compared to control. | ||
3.7 Cytokine release assay
NSAIDs are known to execute their anti-inflammatory effects through inhibition of cyclooxygenase (COX) enzymes. NSAIDs that can regulate cytokine release are known to be effective COX inhibitors. Therefore, the effect of HEC-IBU conjugate 3 on release of tumor necrosis factor-α (TNF-α) and interleukin 6 (IL-6) from stimulated macrophages was investigated to determine its immunomodulatory potential. Levels of cytokines like TNF-α and IL-6 were taken from supernatant collected from THP-1 cultures. HEC-IBU conjugate 3 showed a 30 and 31% inhibition in the level of TNF-α and IL-6, respectively which is comparable with IBU (29 and 31%, respectively). These results confirmed that HEC-IBU conjugate 3 is as effective an immunomodulatory agent as is IBU. It is inferred from this data that the activity of IBU after conjugate formation with HEC was retained, as expected.4 Conclusions
Amphiphilic prodrugs of IBU appended through ester linkages to HEC were synthesized in a simple, efficient, one-pot process. The pharmacokinetic profile of an HEC-IBU prodrug confirms the original hypothesis that ester hydrolysis from the prodrug can afford slow IBU release and higher bioavailability, while delaying release long enough to minimize stomach exposure. The prodrugs showed significant anti-inflammatory properties along with immunomodulatory potential. It is important to underscore this result; there was no guarantee that the prodrug ester linkage would hydrolyze quickly enough under gastrointestinal conditions, but these in vivo results clearly prove that the combination of attachment at the more accessible, distal end of the oligohydroxyethyl chain and the relatively labile ester linkage are sufficient to get rather complete release (since bioavailability actually goes up for the conjugate vs. an IBU dose containing an equivalent amount of drug). The enhanced bioavailability is very interesting and worthy of further study. It may indicate that these amphiphilic conjugates are effective at preventing IBU crystallization after release, thus permitting supersaturation in the small intestine; however, further study is needed to confirm or refute this or any other mechanistic hypothesis. Preliminary acute toxicity assessment by MTT assay was encouraging, indicating no significant excess toxicity vs. IBU over the normal exposure time to IBU. Minimization of stomach exposure to IBU may reduce NSAID associated gastric side-effects; higher bioavailability may also permit dose reduction, potentially further reducing NSAID side effects. It was inferred from the study that cellulose ethers like HEC could be exploited to produce polymeric prodrugs for achieving extended release of NSAIDs (and likely other drugs), thereby avoiding degradation of acid sensitive drugs in the acidic environment of the stomach, as well as exposure of the stomach to drugs that may cause stomach irritation or other problems. In conclusion, synthesized MPDs of IBU appear to be robust systems that are easy to manufacture and capable of modulating drug dose, kinetics of drug release, and administration frequency.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
K. Abbas gratefully acknowledges the financial support provided by Higher Education Commission (HEC), Pakistan under “HEC Indigenous 5000 Fellowships Scheme”. IBU was a generous gift from Candid Pharmaceuticals Sialkot, Pakistan.References
- S. S. Adams, K. F. McCullough and J. S. Nicholson, Arch. Int. Pharmacodyn. Ther., 1969, 178, 115–129 CAS.
- Ibuprofen, in Meyler's Side Effects of Drugs, ed. J. K. Aronson, Elsevier, Oxford, 16th edn, 2016, pp. 5–12 Search PubMed.
- G. D. Champion, P. H. Feng, T. Azuma, D. E. Caughey, K. H. Chan, S. Kashiwazaki, H.-C. Liu, A. R. Nasution, M. Hobunaga, S. Prichanond, T. P. Torralba, V. Udom and M. C. Yoo, Drugs, 1997, 53, 61–69 CrossRef.
- E. Autret, R. J. Marty, B. Henry, C. Laborde, S. Courcier, J. M. Goehrs, G. Languillat and R. Launois, Eur. J. Clin. Pharmacol., 1997, 51, 367–371 CrossRef CAS PubMed.
- M. C. Shelton, J. D. Posey-Dowty, L. Lingerfelt, S. K. Kirk, S. Klein and K. J. Edgar, Enhanced dissolution of poorly soluble drugs from solid dispersions in carboxymethylcellulose acetate butyrate matrices, in Polysaccharide Materials: Performance by Design, ACS Symposium Series, American Chemical Society, Washington, DC, 2010, pp. 93–113 Search PubMed.
- Y. S. Peng, S. C. Lin, S. J. Huang, Y. M. Wang, Y. J. Lin, L. F. Wang and J. S. Chen, Eur. J. Pharm. Sci., 2006, 29, 60–69 CrossRef CAS PubMed.
- V. J. Stella and K. W. Nti-Addae, Adv. Drug Delivery Rev., 2007, 59, 677–694 CrossRef CAS PubMed.
- B. Louage, L. Nuhn, M. D. P. Risseeuw, N. Vanparijs, R. De Coen, I. Karalic, S. Van Calenbergh and B. G. De Geest, Angew. Chem., Int. Ed., 2016, 55, 11791–11796 CrossRef CAS PubMed.
- H. Bundgaard, Drugs Future, 1991, 16, 443–458 Search PubMed.
- W. Xu, J. Ding, C. Xiao, L. Li, X. Zhuang and X. Chen, Biomaterials, 2015, 54, 27–86 Search PubMed.
- D. Li, J. Han, J. Ding, L. Chen and X. Chen, Carbohydr. Polym., 2017, 161, 33–41 CrossRef CAS PubMed.
- D. Li, J. Ding, X. Zhuang, L. Chen and X. Chen, J. Mater. Chem. B, 2016, 4, 5167–5177 RSC.
- M. A. Hussain, K. Abbas, M. Sher, M. N. Tahir, W. Tremel, M. S. Iqbal, M. Amin and M. Badshah, Macromol. Res., 2011, 19, 1296–1302 CrossRef CAS.
- E. Bingham, B. Cohrssen and C. H. Powell, Patty's Toxicology Volumes 1–9, John Wiley & Sons, New York, 5th edn, 2001, p. V5 949 Search PubMed.
- M. A. Hussain, K. Abbas, S. N. A. Bukhari and I. Jantan, Int. Mater. Rev., 2017, 62, 78–98 CrossRef CAS.
- M. Amin, N. S. Abbas, M. A. Hussain, K. J. Edgar, M. N. Tahir, W. Tremel and M. Sher, Cellulose, 2015, 22, 2011–2022 CrossRef CAS.
- N. S. Abbas, M. Amin, M. A. Hussain, K. J. Edgar, M. N. Tahir and W. Tremel, Carbohydr. Polym., 2016, 136, 1297–1306 CrossRef CAS PubMed.
- M. A. Hussain, K. Abbas, M. Amin, B. A. Lodhi, S. Iqbal, M. N. Tahir and W. Tremel, Cellulose, 2015, 22, 461–471 CrossRef CAS.
- H. Bundgaard, in Design of Prodrugs, ed. H. Bundgaard, Elsevier, Plenum Press, New York, 1986, pp. 49–68 Search PubMed.
- H. Bundgaard, Adv. Drug Delivery Rev., 1989, 3, 39–65 CrossRef CAS.
- S. Davaran and A. A. Entezami, J. Controlled Release, 1997, 47, 41–49 CrossRef CAS.
- C. Y. Lin, H. H. Peng, M. H. Chen, J. S. Sun, C. J. Chang, T. Y. Liu and M. H. Chen, J. Biomater. Appl., 2016, 30, 1589–1600 CrossRef CAS PubMed.
- M. Babazadeh, M. Sheidaei, S. Abbaspour and L. Edjlali, Sci. Pharm., 2013, 81, 281–296 CrossRef CAS PubMed.
- L. Shargel, S. Wu-Pong and A. C. Yu, Applied Biopharmaceutics and Pharmacokinetics, ed. L. Shargel, et al., McGraw-Hill, New York, 6th edn, 2012 Search PubMed.
- C. A. Winter, E. A. Risley and G. W. Nuss, Proc. Soc. Exp. Biol. Med., 1962, 111, 544–547 CrossRef CAS PubMed.
- Pharmacology and Toxicology of Ibuprofen, in Ibuprofen: Discovery, Development and Therapeutics, ed. K. D. Rainsford, John Wiley-Blackwell, 2015, p. 150 Search PubMed.
- M. V. Berridge and A. N. Tan, Arch. Biochem. Biophys., 1993, 303, 474–482 CrossRef CAS PubMed.
- R. R. Rosario-Meléndez, W. Yu and K. E. Uhrich, Biomacromolecules, 2013, 14, 3542–3548 CrossRef PubMed.
- S. R. Mudshinge, A. B. Deore, S. Patil and C. M. Bhalgat, Saudi Pharm. J., 2011, 19, 129–141 CrossRef CAS PubMed.
- X. Zheng, R. F. Gandour and K. J. Edgar, Biomacromolecules, 2013, 14, 1388–1394 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08502h |
| This journal is © The Royal Society of Chemistry 2017 |