
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural changes in equimolar ceria–hafnia materials under solar thermochemical looping conditions: cation ordering, formation and stability of the pyrochlore structure†
Matthäus Rothensteiner ab,
Alexander Bonkcd,
Ulrich F. Vogtcd,
Hermann Emeriche and
Jeroen A. van Bokhoven*ab
ab,
Alexander Bonkcd,
Ulrich F. Vogtcd,
Hermann Emeriche and
Jeroen A. van Bokhoven*ab
aInstitute for Chemical and Bioengineering, ETH Zurich, Vladimir-Prelog-Weg 1-5/10, CH-8093 Zürich, Switzerland. E-mail: jeroen.vanbokhoven@chem.ethz.ch
bLaboratory for Catalysis and Sustainable Chemistry (LSK), Paul Scherrer Institute, CH-5232 Villigen PSI, Switzerland
cLaboratory Materials for Energy Conversion, Swiss Federal Laboratories for Materials Science and Technology, Überlandstrasse 129, CH-8600 Dübendorf, Switzerland
dAlbert-Ludwigs-University Freiburg, Crystallography, Institute for Geo- and Environmental Science, D-79098 Freiburg i.Br., Germany
eSwiss-Norwegian Beamlines at ESRF-The European Synchrotron, 71 Avenue des Martyrs, 38000 Grenoble, France
First published on 22nd November 2017
Abstract
Equimolar ceria–hafnia oxides form a pyrochlore structure Ce2Hf2O7, which exhibits an ordered arrangement of Ce3+ and Hf4+ cations under the reducing conditions of a solar thermochemical looping reactor for the two-step dissociation of water or carbon dioxide. The ceria–hafnia pyrochlore phase was prepared from oxidized ceria–hafnia powders by chemical reduction in a flow of H2/He and by auto-reduction in a flow of Ar at up to 1825 K. Full conversion of Ce4+ to Ce3+ was confirmed by thermogravimetric analysis and Ce K edge X-ray absorption spectroscopy. X-ray diffraction and Hf K edge X-ray absorption spectroscopy identified the pyrochlore phase. The dynamics of the structural changes were determined by time-resolved in situ Ce K edge X-ray absorption spectroscopy and in situ X-ray diffraction. Under the oxidizing conditions of the regeneration step of isothermal carbon dioxide splitting at 1800 K, the pyrochlore transformed to a mixture of fluorite-type or tetragonal ceria and monoclinic and orthorhombic hafnia phases. A κ-Ce2Hf2O8 phase, an oxidized form of the pyrochlore with an ordered arrangement of cations, was not detected.
1 Introduction
Solar energy can be converted and stored in chemical bonds by thermochemical dissociation of water and/or carbon dioxide in a two-step redox process. Eqn (1)–(3) describe the generation of solar fuels from water and/or carbon dioxide using ceria as a redox intermediate. Ceria-based solid solutions are the state-of-the-art oxygen storage material for this process.1–8 Typical process temperatures are 1773 K for the reduction (eqn (1)) and 1073 K for the regeneration steps (eqn (2) and (3)). Isothermal operation at typically 1773 K has been successfully demonstrated.
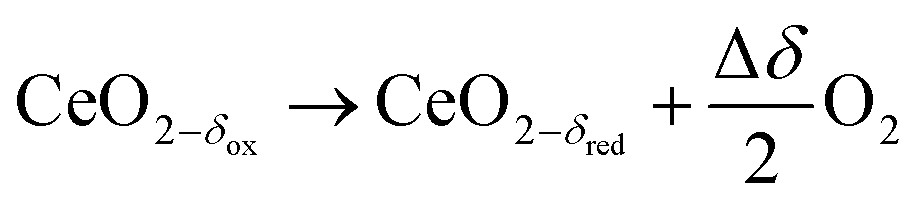 | (1) |
| CeO2−δred + ΔδH2O → CeO2−δox + ΔδH2 | (2) |
| CeO2−δred + ΔδCO2 → CeO2−δox + ΔδCO | (3) |
Considerable effort has been made to tune the oxygen-storage properties of ceria by introducing heterocations into the fluorite-type structure to increase the energy conversion efficiency of this process.9–17 The fuel yields of ceria–zirconia and ceria–hafnia solid solutions are very similar18–20 and higher than the fuel yield of pure ceria.21–28
Zirconium and hafnium have a similar chemistry and their oxides are isomorphs. Tabulated cation radii in crystals29 of Zr4+ and Hf4+ in 6-fold (0.72 and 0.71) and 8-fold coordination (0.84 and 0.83), respectively, are almost the same. The monoclinic–tetragonal phase transition of hafnia occurs at a temperature about 600 K higher than for zirconia.30,31 The crystal structure of ceria–zirconia and ceria–hafnia compounds depends on the composition and the oxidation state of cerium.32 The structure of bulk materials prepared from small particles can be controlled by the sintering conditions. Under oxidizing conditions, oxides of equimolar composition form solid solutions with cubic fluorite-type and tetragonal structures and a random arrangement of the cations. In contrast, under reducing conditions, ordering of the cations occurs.
Determination of the local structure of heterocations is a key to pinpointing oxygen vacancies and to understanding the relationship between the structure and the redox performance. Cation ordering in ceria-based materials for solar thermochemical looping processes has not been reported thus far. In most studies, the preparation of ceria-based oxides to obtain the porous structures required for thermochemical cycles were prepared by sintering in air.3,4,33,34 Cation ordering requires concentrations of the hetero-cation higher than those of ceria-based oxides which are typically tested for the thermochemical generation of solar fuel. Petkovich et al.25 discussed the effect of inhomogeneities of ceria–zirconia. However, they did not expose the materials to the very high temperatures of solar thermochemical cycling. Cation ordering may be difficult to detect by XRD.35,36
Fig. SI 1† displays the fluorite-type CeO2 and the pyrochlore structure A2B2O6O (space group Fd3m). It can be derived from a fluorite-type structure with an ordered arrangement of cations, such as Ce3+ and Zr4+, by removing and displacing oxygen atoms from their original lattice positions. A3+ ions are coordinated to eight oxygen atoms and B4+ ions to six. The ratio of the ionic radii of the A and B cations is typically in the range of 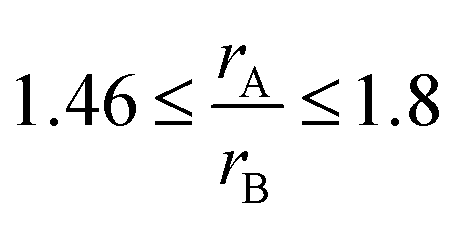 .37 Variations in the oxygen stoichiometry causes distortions as well as changes in symmetry.38 Upon oxidation, cation ordering is maintained and the cubic κ-Ce2Zr2O8 phase is formed. Intermediate oxygen-rich non-stoichiometric phases, such as the Ce2Zr2O7.5 phase,39–41 exhibit an ordered arrangement of cations. The extent of order/disorder in the arrangement of cations and oxygen vacancies strongly affects the capacity and dynamics of the release and uptake of oxygen.42–46 Similar to ceria–zirconia, relatively wide ranges of concentrations of solid solutions with pyrochlore structure are present in ceria–hafnia. Homogeneity ranges become narrower as the stability of the pyrochlore phase decreases with increasing temperature and decreasing cation radius of the rare-earth. Unlike the fluorite-type phases, the pyrochlore hafnates are more stable than the corresponding zirconates.47
.37 Variations in the oxygen stoichiometry causes distortions as well as changes in symmetry.38 Upon oxidation, cation ordering is maintained and the cubic κ-Ce2Zr2O8 phase is formed. Intermediate oxygen-rich non-stoichiometric phases, such as the Ce2Zr2O7.5 phase,39–41 exhibit an ordered arrangement of cations. The extent of order/disorder in the arrangement of cations and oxygen vacancies strongly affects the capacity and dynamics of the release and uptake of oxygen.42–46 Similar to ceria–zirconia, relatively wide ranges of concentrations of solid solutions with pyrochlore structure are present in ceria–hafnia. Homogeneity ranges become narrower as the stability of the pyrochlore phase decreases with increasing temperature and decreasing cation radius of the rare-earth. Unlike the fluorite-type phases, the pyrochlore hafnates are more stable than the corresponding zirconates.47
The case of ceria-containing pyrochlores such as hafnates, containing cerium in a 3+ oxidation state, has not been considered in many earlier studies, because their preparation requires vacuum or more reducing conditions and high temperature. Experimental evidence of structural changes in ceria–hafnia materials under reducing conditions at elevated temperatures is scarce. Andrievskaya et al.48,49 determined ceria–hafnia–zirconia phase equilibria at 1773 K in air. Stanek et al.50 had no data on ceria–hafnia on hand for their predictive study of hafnia-containing pyrochlore phase fields. Baidya et al.51 compared the chemical reduction of fluorite-type Ce0.5Hf0.5O2 and Ce0.5Zr0.5O2 prepared by solution combustion. Different hydrogen uptake profiles were obtained by temperature-programmed reduction in dry hydrogen up to 1073 K. Hydrogen uptake of ceria–hafnia shifted to higher temperature compared to that of ceria–zirconia. While ceria–zirconia was reduced to Ce0.5Zr0.5O1.75 and even beyond that, ceria–hafnia could be reduced only to a composition of Ce0.5Hf0.5O1.77 and the pyrochlore crystal structure was not detected. Bonk et al.20 prepared ceria–hafnia powders containing up to 20 mol% hafnia (Ce0.8Hf0.2O2) by means of a Pechini-type synthesis and obtained ceramic bodies by sintering at 1973 K in air. The lattice expansion was related to the non-stoichiometry of fluorite-type phases using in situ XRD during a switch from vacuum to 2 bar hydrogen at 873 K. Weak reflections of other phases emerged during chemical reduction in hydrogen at 873 K. However, there were no indications that a pyrochlore phase had formed. The pyrochlore structure and cation ordering have not been reported for ceria–hafnia nanoparticles. Zhou and Gorte52 reported single-phase Ce0.5Hf0.5O2 nanoparticles and Raitano et al.53 reported the phase boundary of the cubic domain of nanocrystals, which was close to x = 0.5 in Ce1−xHfxO2. Sharma et al.54 performed XANES (at the O K and Ce M5 edges) and EXAFS (at the Ce K, Zr K and Hf L3 edges) of nanoparticles with the composition Ce0.5Zr0.5O2, Ce0.5Hf0.5O2 and Ce0.5Hf0.25Zr0.25O2. Fujimori et al.55 confirmed the existence of the tetragonal “t” phase in ceria-rich compounds with up to 20 mol% hafnia by means of Raman scattering and synchrotron XRD.
We report cation ordering in equimolar ceria–hafnia materials as well as the formation of cation-ordered ceria–hafnia pyrochlore and describe its stability under realistic solar thermochemical looping conditions. In addition to thermogravimetric analysis (TGA) and X-ray diffraction (XRD), we used our high-temperature in situ cells56,57 for X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) measurements at the Ce K and Hf K edges, and high-energy in situ synchrotron XRD with a focused beam from RT to 1823 K.
2 Experimental
2.1 Material synthesis
The ceria–hafnia materials were synthesized by a polymerized complex method (Pechini-type synthesis). (NH4)2Ce(NO3)6 (Alfa Aesar, 99.9%) and ZrO(NO3)2, (Alfa Aesar, 99.9%) or HfCl4, (Alfa Aesar, 99.9%) were dissolved in deionized water. Anhydrous citric acid (CA, Sigma Aldrich, 99.5%) was added to 1,4-butanediol (BD) (Sigma Aldrich, 99%) at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 molar ratio (with [CA]:[M4+] = 4:1) and stirred at 373 K until all citric acid dissolved. The aqueous solution containing the dissolved metal salts was added to the CA/BD mixture and heated to 423 K to promote the esterification reaction. The resulting highly viscous polymer was dried at 353 K for 4 h and fired for 10 h at 973 K in a constant air flow to remove organic matter.19 Sintering of samples was performed in a Carbolite HTF 17/10 furnace at 1873 K for 5 h in air.
:4 molar ratio (with [CA]:[M4+] = 4:1) and stirred at 373 K until all citric acid dissolved. The aqueous solution containing the dissolved metal salts was added to the CA/BD mixture and heated to 423 K to promote the esterification reaction. The resulting highly viscous polymer was dried at 353 K for 4 h and fired for 10 h at 973 K in a constant air flow to remove organic matter.19 Sintering of samples was performed in a Carbolite HTF 17/10 furnace at 1873 K for 5 h in air.
2.2 Thermogravimetric analysis
Thermogravimetric analysis (TGA) experiments were carried out using a Netzsch STA 449F3 thermogravimetric analyzer. Samples were placed inside an alumina crucible supported by an alumina rod which houses a thermocouple. After oxidation at 973 K in air, the temperature was reduced to 573 K and the gas flow in the analyzer was switched to argon 5.0. The heating and cooling rate was 30 K min−1. A blank run was used to correct buoyancy effects. Between the TGA and XRD measurements samples were ground in a mortar and stored for five months under ambient conditions.2.3 X-ray diffraction
X-ray diffraction measurements of pressed pellets, after calcination and after sintering, respectively, were performed using a Bruker D8 diffractometer with a Cu Kα source. The step size was 0.014° 2θ and the scan speed 1° min−1. For X-ray diffraction measurements after TGA, the pellets were ground in a mortar and a powder-ethanol slurry was deposited onto a sample holder. Coupled theta–two-theta scans were performed on another Bruker D8 instrument with a Cu source on a 280 mm measurement cycle, 2.5° axial Soller slits, a Ni filter, and a Lynx-eye detector. The step size was 0.0296° 2θ and dwell times were 96 (Hf10 and Hf20) and 384 ms (Hf50). The Kα2 signal was stripped by means of the Bruker Diffrac.da vinci software.2.4 In situ X-ray absorption spectroscopy
In situ XAS experiments were carried out at BM01B, Swiss-Norwegian Beam Lines at the European Synchrotron Radiation Facility in Grenoble, France. BM01B, a multi-technique beam line provided access to very hard X-ray photons.58 The storage ring (6 GeV) was operated in 7/8 multibunch filling mode at a maximum ring current of 200 mA. Measurements were performed in transmission mode. Fresh ceria–hafnia powder (31 mg) was pressed at 1 ton to obtain a thin pellet with a diameter of 5 mm. The pellet was mounted on an alumina sample holder and placed inside a laboratory-constructed high-temperature in situ XAS cell56 consisting of an alumina tube heated by an infrared furnace (Ulvak VHT E44). The temperature was measured by a Pt/Rh thermocouple, and the cold ends of the tube were sealed by PTFE windows. The composition of the product gases was monitored by a quadrupole mass spectrometer (MS, Pfeiffer Omnistar GSD320). X-ray intensities were monitored by ionization chambers filled with 1 bar krypton/argon (20/80 vol%) and 1.2 bar krypton before and after the in situ cell, respectively. A ceria or hafnia/cellulose pellet was placed as a reference between the second and third ionization chamber. At both the Ce K and Hf K edges, the Si-111 monochromator was tuned to maximum intensity because rejection of higher order harmonics is unnecessary. Owing to their high energy (ca 125 keV at the Ce K edge) and their low intensity in the spectrum of the source, a bending magnet with critical energy of 20 keV, their interaction with the ion chambers was negligible. Energy scans were carried out at a step size of 1 eV. Ce K edge XANES and EXAFS scans were carried out with a dwell time of 50 and 200 ms, respectively. Hf K edge XANES and EXAFS scans with a dwell time of 100 and 200 ms, respectively, and 3–5 scans were merged (in μ(E)) for XAS data analysis. The flow rate of argon 6.0 (BIP, Air Products) was 100 mL min−1.The absolute value of the Hf K edge energy (tabulated value: 65.351 keV) was not determined, because Hf metal was unavailable as a standard. About 25% of the incoming intensity was absorbed at 65 keV in the ion chambers that were filled with Kr at 1.1 bar. Hf K edge spectra were normalized by fitting a linear background in the pre-edge range and a second order polynomial in the post-edge region, −230 to −150 and 70 to 350 (XANES) or 1500 eV (EXAFS) relative to the maximum of the first derivative, respectively. Hafnia (Alfa Aesar, 99.95%, see ESI†) was the reference material. Data processing and analysis were performed with MATLAB code and the Athena and Artemis software packages.59 Fitting of Ce K and Hf K edge EXAFS data was performed by exchanging Zr by Hf in the feff6 input file which was generated by the ‘atoms’ code of Demeter 0.9.21 and crystallographic data of Ce2Zr2O7 determined by neutron diffraction (Raison et al., ICSD collection code 168595).60 Fitting was performed in R-space at 1.15 to 4.4 Å with a Hanning window function for the Fourier transform.
2.5 In situ X-ray diffraction
During in situ XRD measurements, the storage ring was operated in 16 bunch mode with a ring current of 90 mA and a half-life of 10 h. The combined XAS/XRD measurements were carried out in the high temperature in situ cell.57 The beam was focused at a photon energy of 41.507 keV, which corresponds to a wavelength of 0.2987 Å. The wavelength was calibrated against a Si NIST standard using the beamline's high-resolution diffractometer. The intensity of diffracted photons was collected with a Dexela 2923 CMOS 2D pixel detector. In this configuration, a resolution of typically 0.005° 2θ is achievable. Sets of five dark (background) and five exposed images were recorded with an exposure time of four seconds and a fast shutter. From each set, one background-corrected diffraction pattern was obtained. The time resolution of the measurements was 0.876 min−1. The position of the sample relative to the pixel detector was determined by means of a LaB6 660c NIST standard in a 0.4 mm quartz capillary mounted on top of the inner ceramic tube. To ensure that the sample was exactly 3 mm inside the tube, the whole cell was moved 3 mm towards the source. Data reduction by azimuthal integration was performed with the PyFAI software package61 and background subtraction was performed in MATLAB and the baseline estimation and denoising using sparsity (BEADS) algorithm.62The auto-reduced pellet was decorated with platinum by dipping it into hydrogen hexachloroplatinate (H2PtCl6(6H2O), 99.9%, (38–40% Pt), Acros) to increase the reaction rate on the surface before the isothermal carbon dioxide splitting experiment. Gases for in situ XRD experiments were argon 6.0 (BIP, Air Products), 5% hydrogen 5.0 in helium 5.0 (Messer gases), and carbon dioxide 4.8, (O2 ≤ 2 vpm, CO ≤ 1 vpm, H2O ≤ 3 vpm, CH4 ≤ 2 vpm, N2 ≤ 8 vpm). The flow rates of argon and hydrogen/helium were 200 mL min−1, and the flow rate of carbon dioxide was 100 mL min−1.
3 Results
3.1 Sample preparation
Samples of Hf10C (Ce0.9Hf0.1O2), Hf20C (Ce0.8Hf0.2O2) and Hf50C (Ce0.5Hf0.5O2) were synthesized by the polymerized complex method and calcination (C) at 973 K in air. Fig. 1 presents XRD patterns of samples that were collected after calcination (C) at 973 K as well as after sintering (S) pressed pellets in air at 1873 K. The XRD patterns of the calcined materials indicate the fluorite-type crystal structure of ceria. The peaks were substantially broadened due to the small crystallite size that is obtained with the polymerized-complex method. Reflections of phases other than the fluorite-type were not detected. The positions of the peaks were very similar in the diffraction pattern of calcined Hf10C and Hf20C, while the peak positions in the diffraction pattern of calcined Hf50C shifted to higher angles, indicating a contraction of the lattice with increasing hafnia content. The diffraction peaks of calcined Hf20C were more asymmetric than those of calcined Hf10C and Hf50C. Peak broadening was most pronounced in the diffraction pattern of calcined Hf50C. In the diffraction patterns of calcined Hf10C and Hf20C, the widths of the reflections were very similar.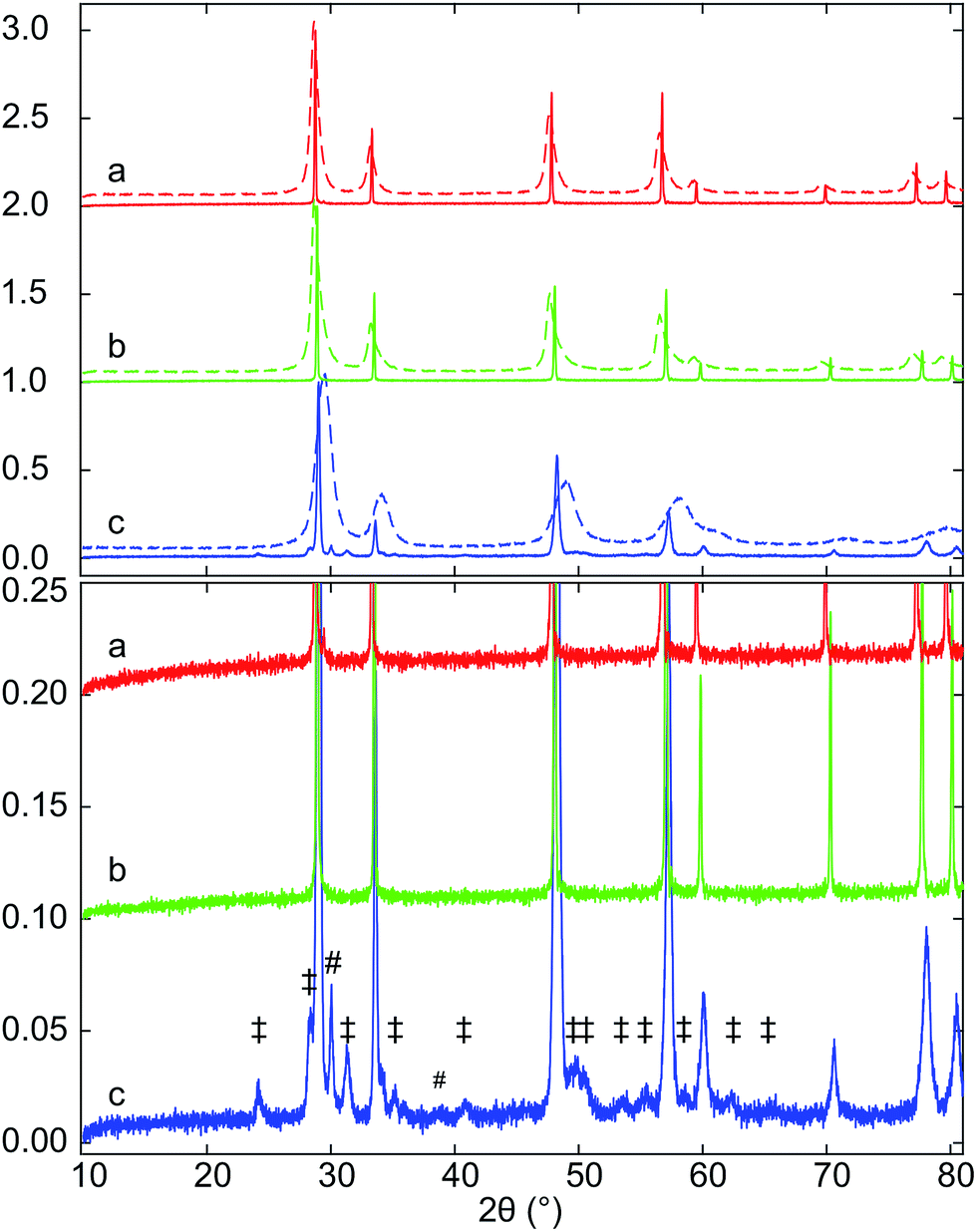 | ||
| Fig. 1 XRD patterns of (a) Hf10C and Hf10CS, (b) Hf20C and Hf20CS, (c) Hf50C and Hf50CS prepared by polymerized complex method, calcination (C) at 973 K in air (dashed lines) and after subsequent sintering (S) of a pressed pellet at 1873 K in air (solid lines). Diesis (‡) indicate reflections of monoclinic hafnia and octothorps (#) peaks assigned to monoclinic and/or orthorhombic hafnia. | ||
While all reflections of sintered Hf10CS and Hf20CS were assigned to the fluorite-type crystal structure, a large number of weak reflections below 10% normalized intensity were observed in the diffraction pattern of sintered Hf50CS, all of which were assigned to monoclinic and orthorhombic hafnia. In the diffraction pattern of sintered Hf50CS, the normalized intensity of the reflections indicating the cubic ceria lattice were asymmetric, smaller and significantly broader than those in the patterns of sintered Hf10CS and Hf20CS. Reflections indicating the pyrochlore structure were not detected.
3.2 Thermogravimetric analysis
Fig. 2 shows the mass loss of the three samples determined by thermogravimetric analysis during heating to 1773 K in a flow of argon. The oxygen release profiles of Hf10C and Hf20C were very similar: auto-reduction started at about 1173 K and weight loss occurred in two steps. Before and at the first decrease in weight at intermediate temperature, the auto-reduction profiles of Hf10C and Hf20C overlapped. The mass change in Hf20C was higher than that in Hf10C in the second step at higher temperature. Hf50C also showed that oxygen was released in two steps; however, the onset of auto-reduction started already at about 573 K. The weight loss profile of Hf50C was similar to Hf10C and Hf20C at temperatures up to about 1173 K but at higher temperature there was a substantially higher rate of oxygen release. The mass loss of all three samples leveled out when they were kept at 1773 K for about 100 minutes and did not change during subsequent cooling to 573 K. Table 1 reports the weight changes and composition of the auto-reduced materials assuming complete oxidation of the pristine material and that weight loss was solely caused by the release of oxygen. While the composition of Hf10CT Hf20CT is typical of nonstoichiometric fluorite-type ceria, that of Hf50CT suggests changes in the crystal structure and complete reduction of cerium from Ce4+ to Ce3+.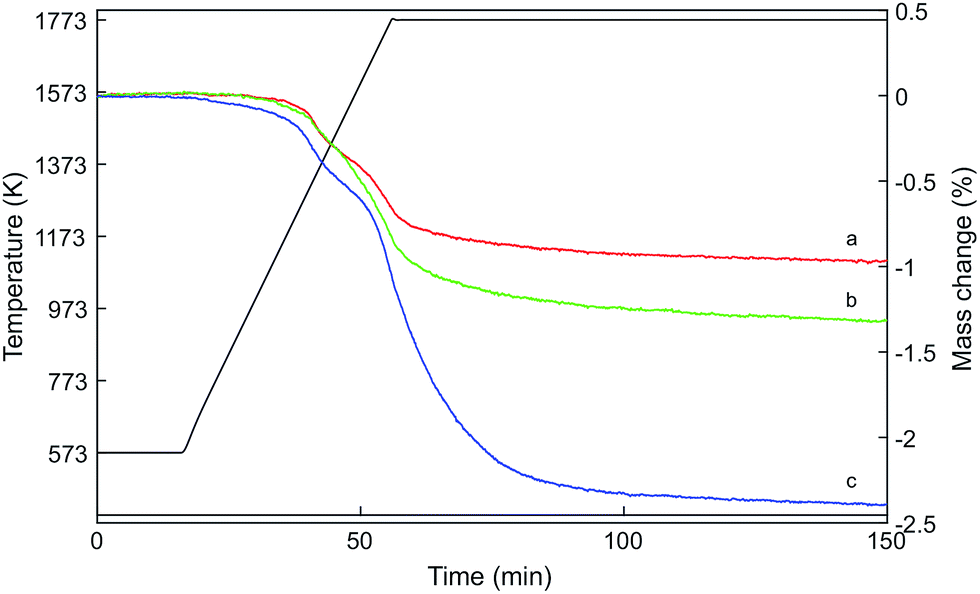 | ||
| Fig. 2 Relative mass change of (a) Hf10, (b) Hf20, and (c) Hf50 by heating to 1773 K in a flow of argon. The samples were oxidized in air at 973 K before thermal auto-reduction. The mass losses leveled out after about 100 min at 1773 K. | ||
| Sample | Weight change (%) | Composition |
|---|---|---|
| Hf10CT | −0.97 | Ce0.9Hf0.1O1.89 |
| Hf20CT | −1.32 | Ce0.8Hf0.2O1.85 |
| Hf50CT | −2.38 | Ce0.5Hf0.5O1.72 |
Fig. 3 displays XRD patterns of the ceria–hafnia materials after thermal auto-reduction (T). Auto-reduced Hf10 showed only the reflections of the fluorite-type phase, which indicates that hafnium ions were well dispersed/randomly distributed and that they substitute cerium ions in the fluorite-type lattice. Thermal reduction of the materials with a higher hafnium content gave rise to additional very weak reflections with normalized intensity below 0.03, which indicate the ordering of cations in pyrochlore-type and pyrochlore-related crystal structures. The peak positions were located at slightly higher angles than that in literature data based on XRD40 and neutron diffraction60 of pyrochlore-type ceria–zirconia. The main component of Hf20CT is a fluorite-type solid solution; however also some reflections indicating the presence of a cation-ordered phase were found. The fluorite-type phase in Hf20CT had a slightly smaller lattice parameter than that of Hf10CT, indicated by a shift to higher diffraction angles.
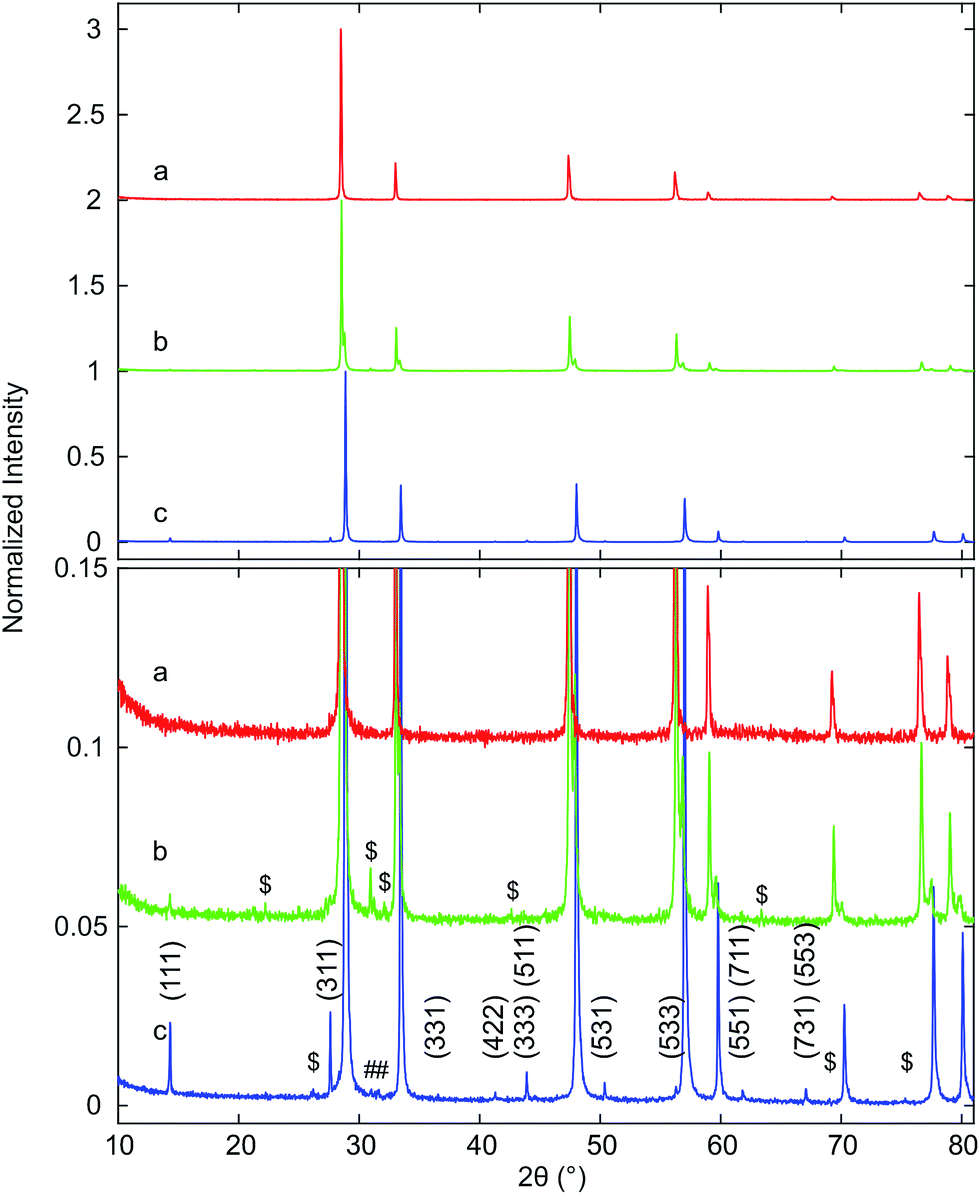 | ||
| Fig. 3 XRD patterns of (a) Hf10CT, (b) Hf20CT, and (c) Hf50CT after thermal auto-reduction (T) of the calcined materials in a thermogravimetric analyzer at 1773 K. The close-up highlights weak reflections that were assigned to lattice planes with Miller indices (hkl) of the pyrochlore phase, based on the crystal structure of the ceria–zirconia system.60 Octothorps (#) mark peaks close to the position of the (111) reflection of monoclinic hafnia and the (121) reflection of orthorhombic hafnia. Dollar signs ($) indicate peaks without clear assignment. | ||
Reflections assigned to the cation-ordered phase of auto-reduced Hf20CT were less intense and appeared at lower angles than corresponding reflections in the diffraction pattern of Hf50CT. The reflections arising from cation ordering in the diffraction pattern of Hf50CT were sharp and, in contrast to the diffraction pattern of Hf20CT, most of the reflections of the pyrochlore crystal structure were visible. At the scattering angles of the reported40 (220), (620), (642), (733), (660), (822), (751), (555) reflections of Ce2Zr2O7 at 23.39, 53.91, 64.86, 71.85, 74.9 and 76.73° 2θ peaks could not be distinguished from noise. In the diffraction pattern of Hf50CT, the intensity of the (331) reflection was 90% more intense and that of the (331) reflection 98% less intense than the corresponding reflections40 in the diffraction pattern of Ce2Zr2O7. Reflections close to those of the (200) and (600) reflections Ce2Zr2O7.5 phase40 reported at 16.65 and 51.49° 2θ were not detected. The asymmetry of the reflections related to the cubic lattice planes might indicate partial re-oxidation to Ce2Zr2O7+x. There were no reflections of the hexagonal Ce2O3 phase. Very weak reflections at about 31° 2θ indicated the presence of a very small amount of monoclinic or orthorhombic hafnia. Corresponding reflections of cubic lattice planes of the cation-ordered structure appeared at higher angles relative to the fluorite-type lattice of Hf10CT and Hf20CT, indicative of a smaller lattice parameter in auto-reduced Hf50CT. Peaks in the pattern of Hf50CT were asymmetric with a tail at the higher 2θ side.
3.3 In situ XAS at the Ce K edge
Fig. 4 shows time-resolved Ce K edge XANES spectra that were collected during the auto-reduction (R) of Hf50C in a flow of argon. The temperature was first ramped to 1773 K at 33.3 K min−1, kept constant for 30 minutes before cooling to room temperature at the same rate, similar to the TGA experiment described above (see Fig. 5 for more details). With increasing temperature, thermal damping lowered the amplitude of the absorption fine structure, which smeared spectral features related to structural changes. Except for a discontinuity at about 873 K, the XANES features changed gradually. Fig. 4a shows the drop in the intensity of the white line during heating. The white line intensity of the auto-reduced material changed only slightly upon cooling to room temperature (Fig. 4c and d). During cooling, there was only a minute shift in the energy of the Ce K edge position to higher energy. In the absorption fine structure of Hf50CR, oscillations of higher frequency compared to those in the spectrum of pristine oxidized Hf50C emerged and the edge position shifted to lower energy, indicating significant changes in the local geometrical and electronic structure of cerium. Fig. 5 presents the temperature profile, the mass spectrometer signals of oxygen, and carbon dioxide of the temperature-programmed auto-reduction of Hf50 in the in situ XAS experiment. The oxygen trace (m/z = 32) was in good agreement with the derivative of the mass change in the TGA experiment (2). In both experiments, two maxima in the rate of oxygen release were observed: a maximum at intermediate temperature and then the absolute maximum at 1773 K. The carbon dioxide signal indicated desorption from the sample and reactor walls and, to some extent also was due to carbon that was burned and hindered the detection of oxygen. In contrast to the TGA experiment, there was no pre-treatment of oxidation in air at 973 K and in argon at 573 K before heating. Thus, the release or desorption of carbon dioxide from the sample and reactor started at very low temperature. When the temperature decreased, all the oxygen released from the sample had been removed from the reactor because there was a very small drop in the oxygen signal to its baseline.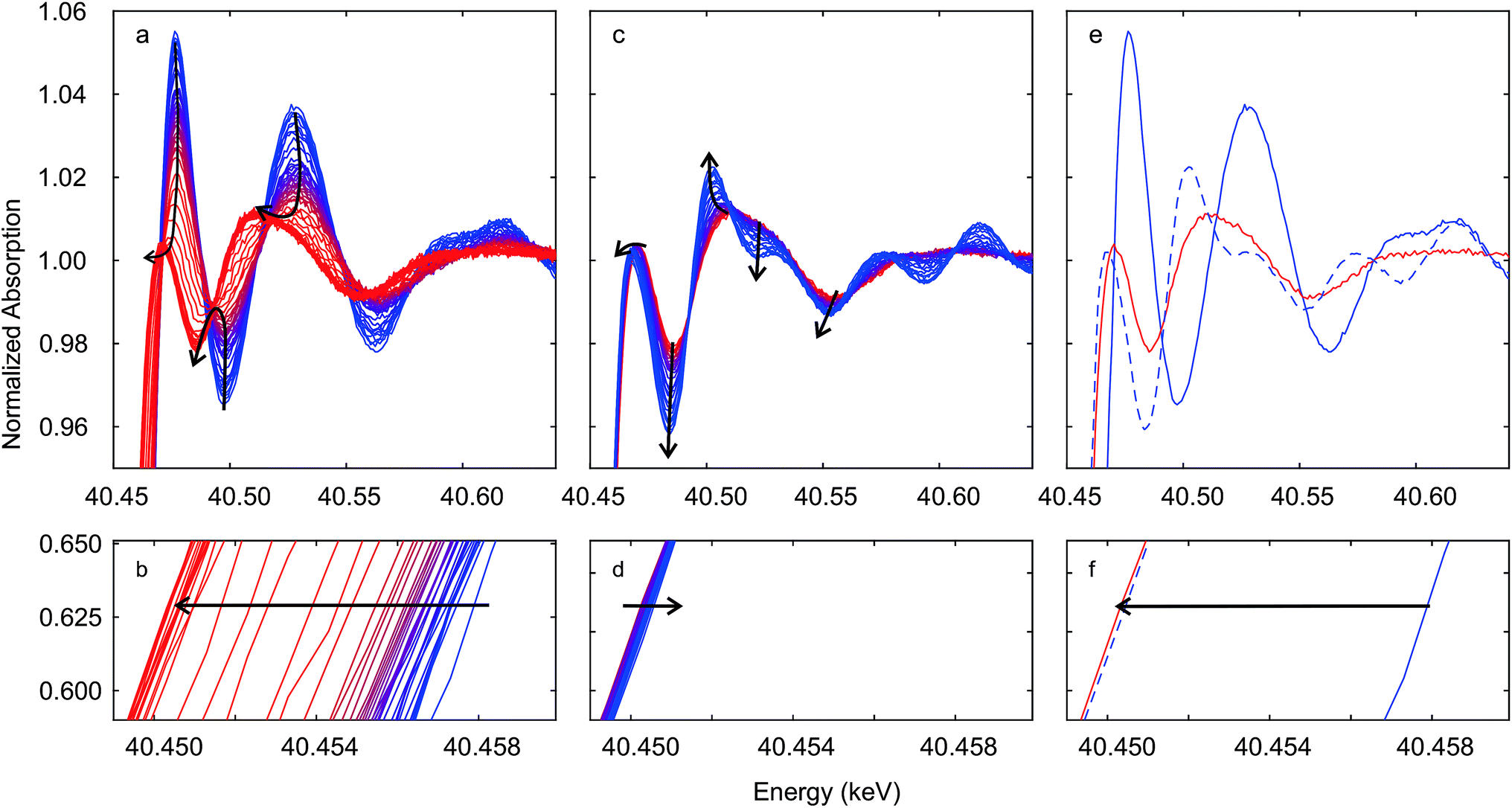 | ||
| Fig. 4 Time-resolved Ce K edge XAS recorded during auto-reduction of Hf50C by heating in a flow of argon (see Fig. 5). Black arrows indicate changes in spectral features. Spectra in (a and b) were recorded during heating from room temperature (blue line) to 1773 K and at 1773 K (red lines); spectra in (c and d) were recorded during cooling from 1773 K (red line) to room temperature (blue line). Spectra in (e and f) were recorded at room temperature before (blue solid line) and after (blue dashed line) auto-reduction and at 1773 K before cooling (red line). Correction by aligning the simultaneously recorded reference spectra was not carried out. | ||
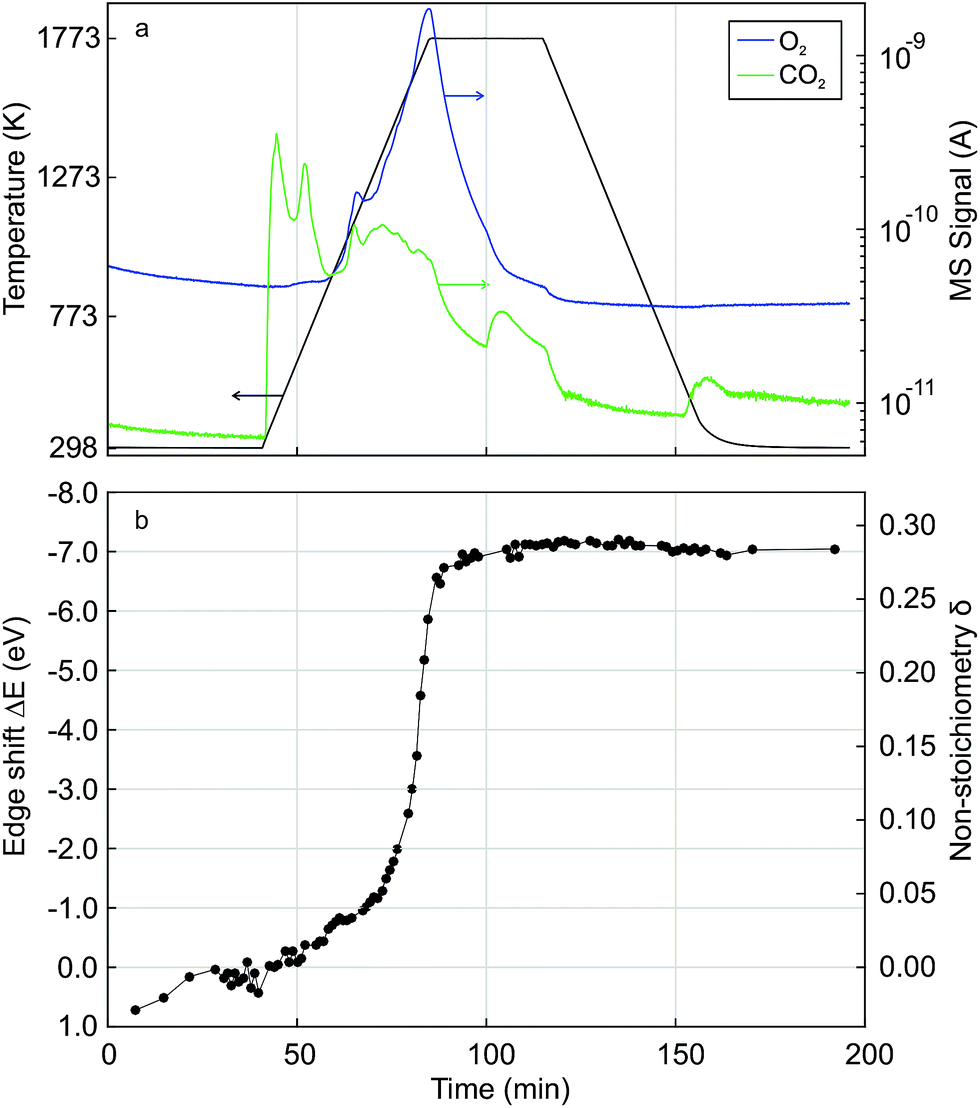 | ||
| Fig. 5 Time-resolved in situ Ce K edge XAS during temperature-programmed auto-reduction of Hf50C in a flow of argon. (a) Temperature profile and the MS signals of oxygen (m/z = 32) and carbon dioxide (m/z = 44); (b) edge shift and non-stoichiometry δ in Ce0.5Hf0.5O2−δ determined by quantification of the energy shift in the edge position. Corrections by alignment of reference spectra was not performed. | ||
Fig. 5 also shows the energy shift in the Ce K edge position that was quantified by interpolation at a constant normalized absorption56 of 0.633. The corresponding value of the non-stoichiometry δ in Ce0.5Hf0.5O2−δ was calculated assuming a linear relationship between the change in the energy of the edge position and the Ce3+ concentration. The energy of the edge position changed63 by −6.2 eV upon full conversion of Ce4+ to Ce3+. In agreement with the MS signals, XAS indicated that auto-reduction to Ce3+ was virtually complete when the temperature reached 1773 K. The non-stoichiometry reached a maximum of 0.29 at 1773 K, and the mean value determined from the 10 last spectra was 0.28 ± 0.04. This clearly indicated that, during auto-reduction, cerium changed from Ce4+ to Ce3+, which is required for the formation of the ceria–hafnia pyrochlore structure. In contrast to the MS oxygen signal, there were not two inflection points in the curve of δ determined by XAS. The non-stoichiometry δ corresponds to the cumulative oxygen release and thus its temporal derivative gives the oxygen release rate, which is proportional to the MS signal of oxygen.
3.4 Hf K edge XANES
Immediately after measurements at the Ce K edge during auto-reduction of Hf50CR (Fig. 4 and 5), spectra were recorded at the Hf K edge at room temperature. Fig. 6 presents Hf K edge spectra of Hf50C and Hf50CR, recorded before and after the in situ XAS auto-reduction experiment. Furthermore, it shows spectra of the reference material, hafnia powder that consisted of a mixture of monoclinic and orthorhombic phases (see ESI†). Spectral features were less pronounced in the spectrum of pristine Hf50C compared to the auto-reduced material, mainly due to lower crystallinity and homogeneity of the powders compared to the dense ceramic body of auto-reduced Hf50CR. A spectral feature close to the absorption edge at a normalized absorption of 0.92 was most intense in Hf50CR, less pronounced in the hafnia reference and weak in pristine Hf50C. The energy at the first maximum (white line) was highest in the spectrum of Hf50CR. At energy higher than that of the white line, the largest amplitude of the fine structure was found in the spectrum of auto-reduced Hf50CR. The differences in the energy and intensity of the Hf K edge XANES features of Hf50C and Hf50CR suggest changes in the oxygen coordination geometry.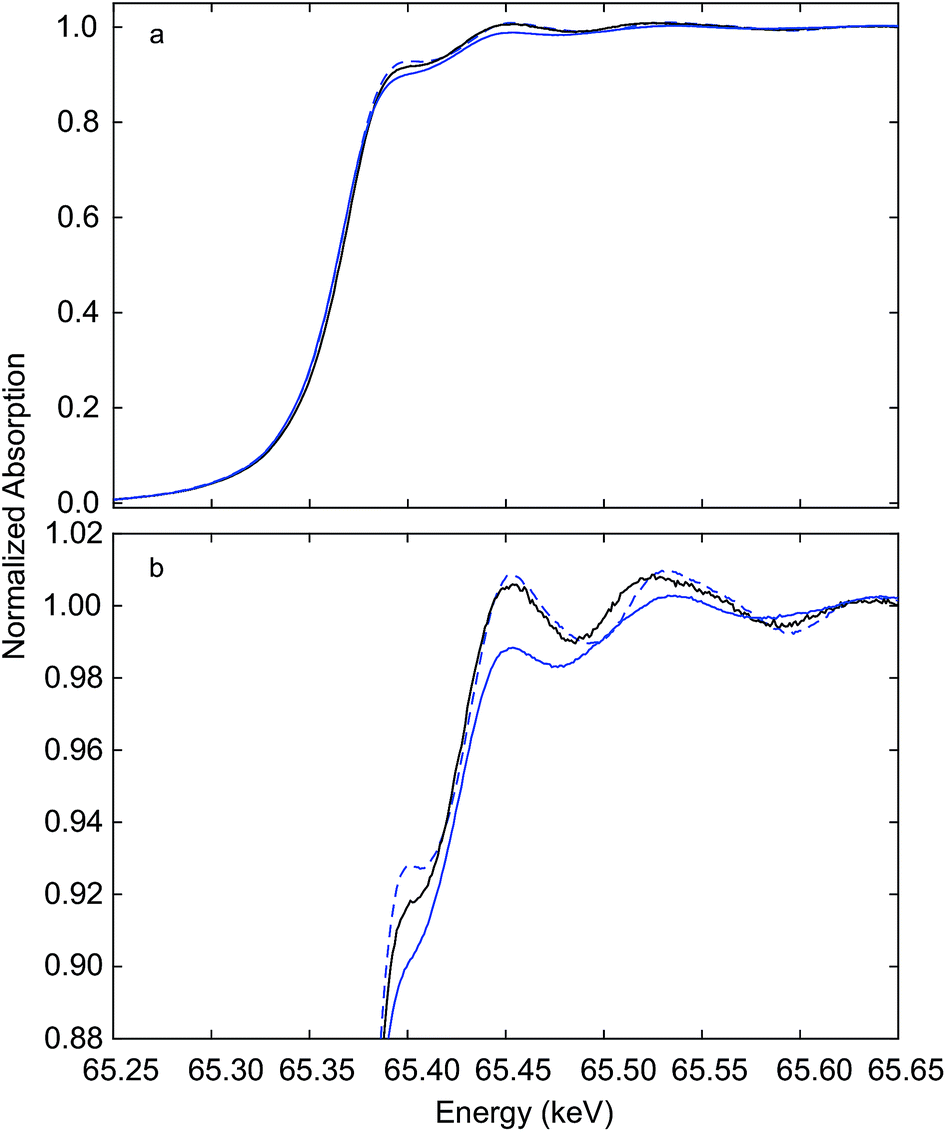 | ||
| Fig. 6 Hf K edge XANES recorded at room temperature. Hf50C (solid blue line), auto-reduced Hf50CR (dashed blue line) recorded in a flow of argon immediately after the experiment (see Fig. 4 and 5), and hafnia reference (solid black line) (b) shows a close-up of the XANES region given in (a). | ||
3.5 Hf K edge EXAFS
Fig. 7 shows the Hf K edge EXAFS signals of Hf50C and Hf20C recorded before and after thermal reduction (Fig. 4 and 5). There were significant differences in the amplitude and the signal/noise ratio of the data. The EXAFS signal of auto-reduced Hf50C had the largest amplitude and the best signal/noise ratio. In the Fourier-transform of all the spectra except that of calcined Hf50C, two main peaks were identified, the first and most intense originating from the first coordination shell of oxygen atoms and the second from the second coordination shell consisting of cerium and hafnium atoms at higher radial distance. The crystallinity of the samples is much higher in the dense ceramic body of Hf50CR compared to Hf50C, which consisted of calcined powder. In the pyrochlore ceria–zirconia structure,60 the first coordination shell of zirconium consists of six oxygen atoms at a distance of 2.097 Å.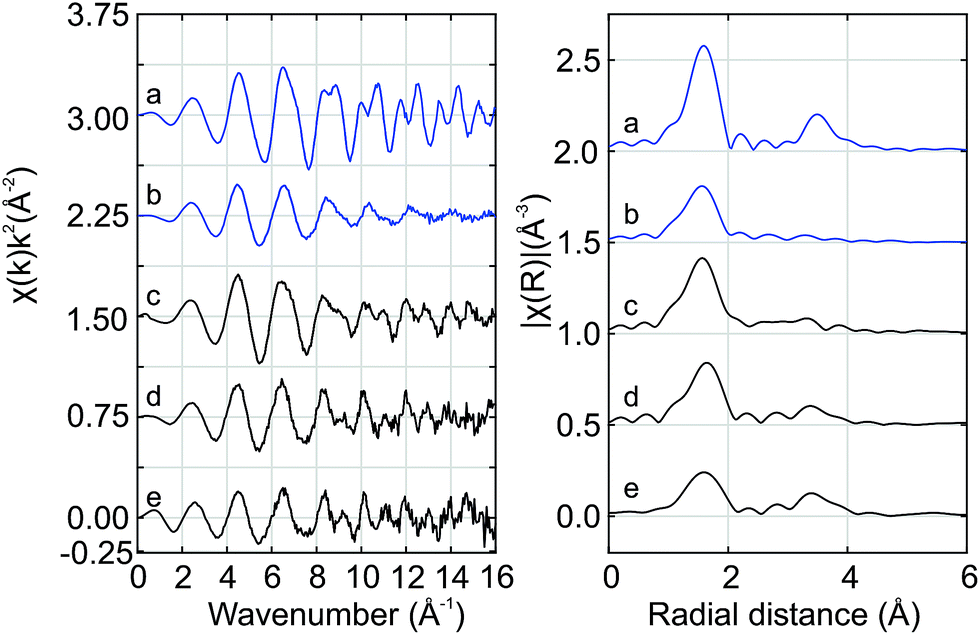 | ||
| Fig. 7 Hf K edge EXAFS recorded at room temperature. (a) auto-reduced Hf50CR, (b) calcined Hf50C, (c) HfO2 reference, (d) auto-reduced Hf20CR, and (e) calcined Hf20C. The k-range for the Fourier transform was 4.0 to 11.6 Å−1. | ||
Fig. 8 presents a fit of the Hf K edge EXAFS of Hf50CR. Table 2 presents the numerical results of coordination numbers, Debye–Waller factors, and interatomic distances. The real part and the magnitude of the fit are in excellent agreement with the data, which is reflected in the excellent goodness-of-fit parameters (reduced χ2 = 462.6 and R-factor Rf = 0.0063). Deviations at low radial distance are small and due to low frequency components in the background. Results of the fit are in very good agreement with the pyrochlore structure. The hafnium–oxygen distance is slightly shorter than that of the zirconium–oxygen bond of the initial model, which is in agreement with the slightly smaller ion radius of Hf4+ compared to Zr4+ (0.72 and 0.71 Å) in a six-fold coordination. S02 was manually adjusted such that a Hf–O coordination number of 6.0 was obtained in the fit. The Hf–Ce and Hf–Hf coordination numbers are in agreement with the expected theoretical value of 6.0. While the Hf–Ce coordination number is lower and the bond length is shorter compared to the starting value, the Hf–Hf coordination number is higher than that of Hf–Ce with longer bonds. Consistent with and very similar to the results with a slightly smaller statistical uncertainty were obtained by the fitting of the data in a larger k-range.
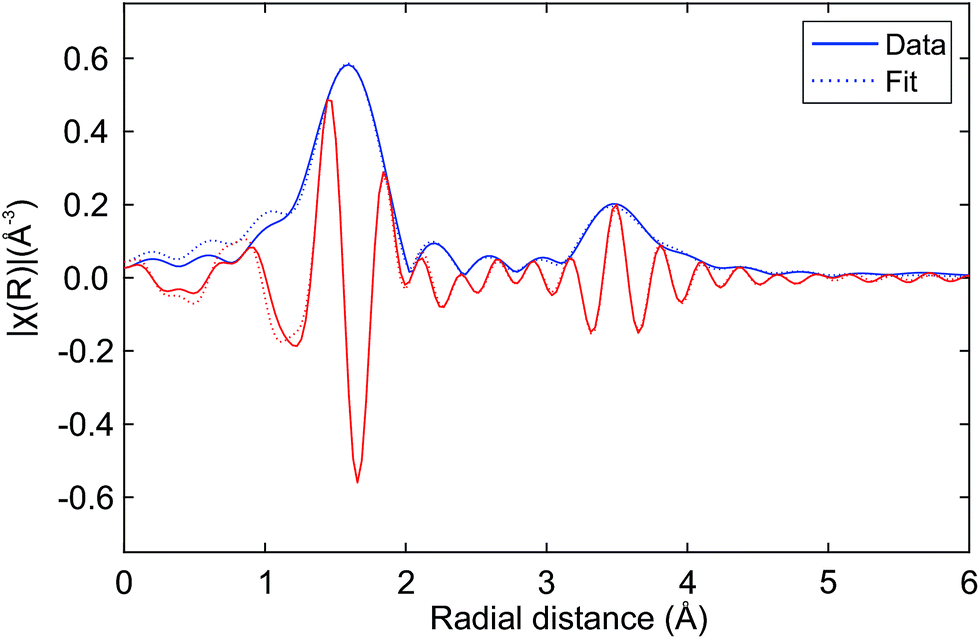 | ||
| Fig. 8 Fit of the Hf K edge EXAFS of auto-reduced Hf50CR to the pyrochlore structure. Magnitude (blue line) and real part (red line) of the data (solid lines) and the fit (dashed lines). The k-range for the Fourier transform was 4.0 to 11.6 Å−1. Table 2 gives numerical details. | ||
| Path | CN | σ2 × 103 (Å2) | R (Å) | ΔR (Å) |
|---|---|---|---|---|
| Hf–O | 6.0 ± 0.5 | 3.11 ± 0.68 | 2.09 | −0.01 ± 0.01 |
| Hf–Ce | 2.5 ± 4.3 | 2.23 ± 7.84 | 3.77 | −0.03 ± 0.05 |
| Hf–Hf | 5.2 ± 5.0 | 3.17 ± 5.29 | 3.83 | +0.03 ± 0.03 |
3.6 In situ X-ray diffraction
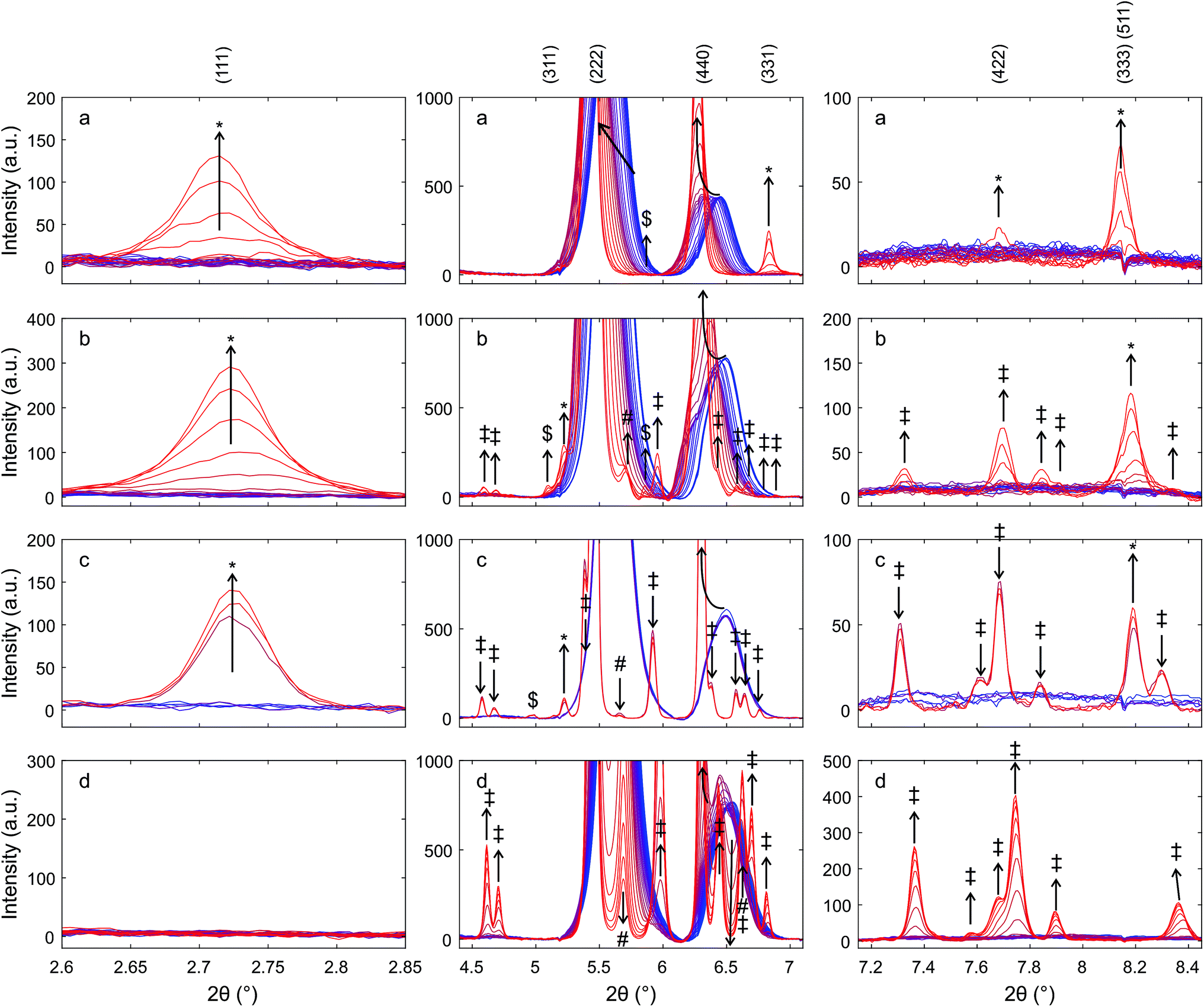 | ||
| Fig. 9 Details of XRD patterns, highlighting reflections related to the pyrochlore structure recorded during heating of (a) Zr50C in hydrogen/helium, (b) Hf50C in hydrogen/helium, (c) Hf50C in argon, and (d) Hf50C in air during heating at about 50 K min−1 from room temperature to 1623 ± 50 K in (hydrogen/helium) and to 1823 ± 50 K in argon and air. Blue lines indicate room temperature and red lines indicate high temperature. Arrows indicate directions of changes. Reflections are assigned to the pyrochlore ceria–hafnia phase (*), monoclinic hafnia (‡), and orthorhombic hafnia (#). Some peaks could not be definitely assigned ($). | ||
In the diffraction patterns of materials that were exposed to reducing conditions, the reflections of the cubic fluorite-type structure appeared at higher scattering angles than expected for pure ceria and contained additional very weak reflections, some of which indicated the formation of the cation-ordered pyrochlore structure. At the end of the heating ramp the peaks in the pattern of Hf50C that had been heated in a flow of hydrogen/helium were wider than the peaks in the pattern of Zr50C. During chemical reduction in a flow of hydrogen/helium, more peaks appeared in the diffraction pattern of Hf50C than in that of Zr50C. Many of these reflections were tentatively assigned to the monoclinic or the orthorhombic hafnia phases. The patterns of Hf50C heated in air and the pattern recorded after sintering in air at 1873 K (Fig. 1) were very similar and differed only in the intensity of the reflections. In both diffraction patterns, reflections of hafnia were present and reflections indicating cation ordering were absent. Most reflections of ceria–zirconia pyrochlore Ce2Zr2O7 emerged during reduction of Zr50C in a flow of hydrogen/helium at a maximum temperature of 1623 ± 50 K: the (111), (331) and (333), (511) and more reflections were identifiable. The peak of the very weak (442) reflection of the pyrochlore could not be assigned due to potential overlap with a reflection of the monoclinic zirconia phase that emerged after the temperature ramp.
In the patterns of Hf50C recorded during reduction in hydrogen/helium, there were a larger number of small peaks, many of which were assigned to monoclinic and/or orthorhombic hafnia phases. The (111) reflection of the pyrochlore phases of chemically reduced Zr50C and Hf50C in a flow of hydrogen/helium emerged at a similar temperature. The (333), (511) diffraction peaks of auto-reduced and chemically reduced Hf50C were clearly detected close to the position of the peaks of reduced Zr50C. Compared to reduced Zr50C, the intensity of the (333), (511) peaks relative to the (111) reflection were slightly weaker. Close to the position of the (311) reflection of the Zr50C pyrochlore, three very small peaks were detected in the pattern of chemically reduced and auto-reduced Hf50C, which could not be clearly assigned to the pyrochlore phase.
During thermal reduction of Hf50C in a flow of argon, the (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1) reflection at 5.9° 2θ indicated the formation of monoclinic hafnia. The (222) peak of the pyrochlore corresponds to the (111) reflection of the fluorite-type structure. At higher temperature, the normalized intensities of the reflections of the monoclinic hafnia phase decreased, while the intensity of the peaks assigned to the pyrochlore increased further. In agreement with the pattern of chemically reduced Hf50C, the (333), (511) reflection of auto-reduced Hf50C was clearly detected. There were fewer reflections of other phases compared to the reduction of Zr50C and Hf50C in hydrogen/helium at a lower temperature. At the position of the notable (331) reflection of ceria–zirconia pyrochlore, there was no peak with increasing intensity related to the pyrochlore phase.
1) reflection at 5.9° 2θ indicated the formation of monoclinic hafnia. The (222) peak of the pyrochlore corresponds to the (111) reflection of the fluorite-type structure. At higher temperature, the normalized intensities of the reflections of the monoclinic hafnia phase decreased, while the intensity of the peaks assigned to the pyrochlore increased further. In agreement with the pattern of chemically reduced Hf50C, the (333), (511) reflection of auto-reduced Hf50C was clearly detected. There were fewer reflections of other phases compared to the reduction of Zr50C and Hf50C in hydrogen/helium at a lower temperature. At the position of the notable (331) reflection of ceria–zirconia pyrochlore, there was no peak with increasing intensity related to the pyrochlore phase.
The reflections at low angles, indicating cation ordering, were not observed during heating in air. In the heating ramp both the monoclinic and a small amount of the orthorhombic hafnia phase formed. The peak assigned to the (121) reflection, the most intense reflection of orthorhombic hafnia, was observed at 5.7° 2θ and decreased to below 0.2% normalized intensity after 30 min at 1823 ± 50 K (see ESI†). A peak at a very similar position as that of the (331) reflection of the pyrochlore also formed by heating in air. While the intensity of the (111), (331), and (333), (511) reflections associated with cation ordering increased in the pattern of reduced Zr50C, the corresponding intensity of the reflections of Hf50C decreased in hydrogen/helium and argon. The intensity of the (311) reflections in the patterns of reduced Zr50C and reduced Hf50C changed only slightly. In reducing atmosphere, there were only small changes in the intensity of the hafnia phases. In contrast, in air, the intensity of the reflections of monoclinic hafnia increased and the most intense peak split into two peaks, indicating the presence of substantial amounts of monoclinic hafnia and a fluorite-type phase. Upon cooling, there was no significant change in the intensity of the (121) reflection, indicating the presence of a small amount of orthorhombic hafnia.
Fig. 10 presents diffraction patterns of auto-reduced (Fig. 9), platinum-decorated Hf50 under conditions of isothermal carbon dioxide splitting. During heating in argon to 1800 K (not shown) and for 30 minutes at 1800 ± 50 K in a flow of argon, there were no significant changes in the diffraction pattern.
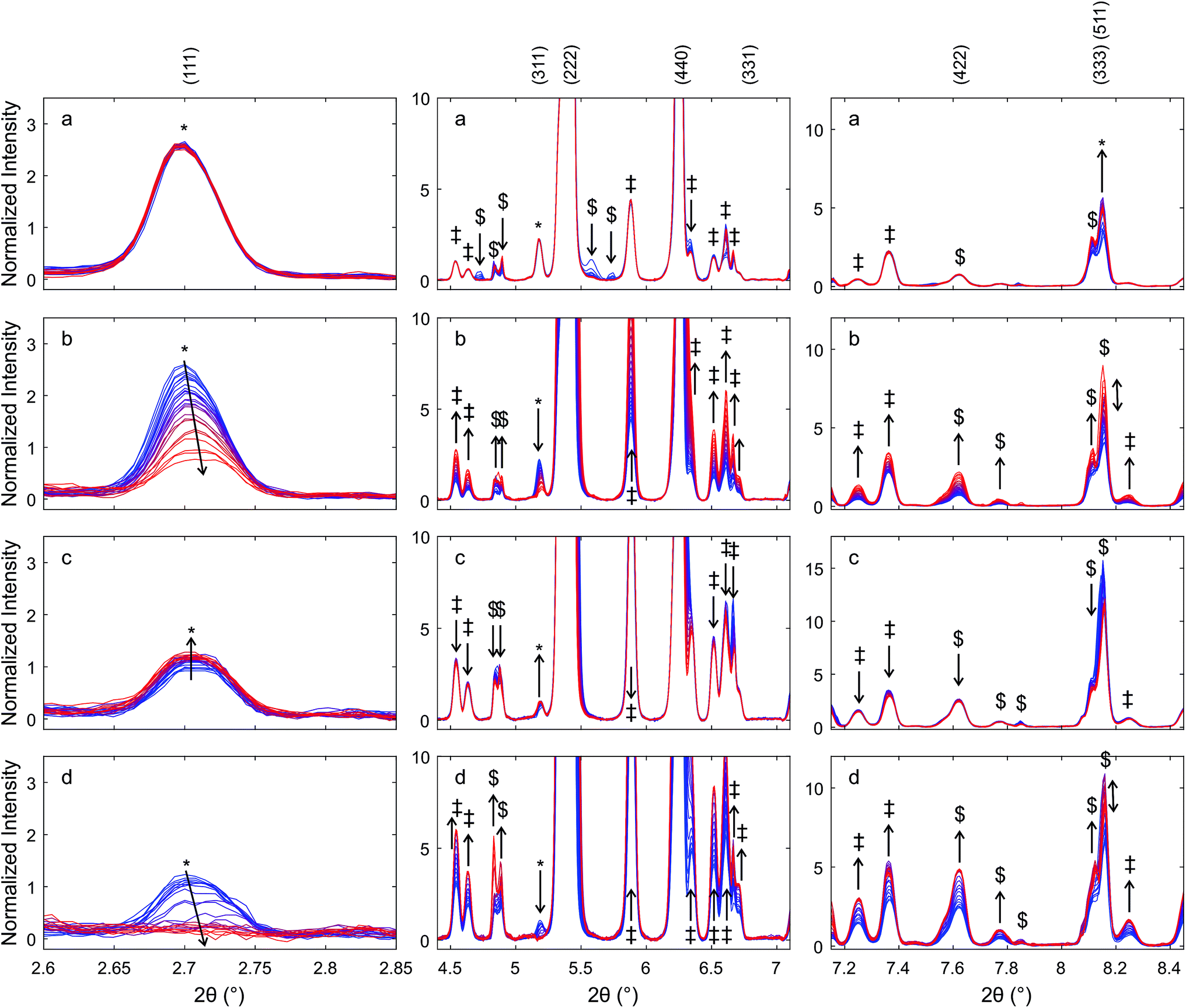 | ||
| Fig. 10 Diffraction patterns of auto-reduced Hf50C, highlighting changes in the reflections related to the pyrochlore structure under isothermal carbon dioxide splitting conditions. Normalized intensities in %. A pellet Hf50C was auto-reduced at 1823 K in a flow of argon and impregnated with platinum. Subsequently, it was heated to 1800 ± 50 K in a flow of argon and the feed gas was switched at a half period of 30 min from a flow of (a) argon to (b) carbon dioxide, (c) argon, and (d) carbon dioxide. Blue indicates the first and red the last diffraction pattern in a series. Arrows indicate changes in the intensities of the reflections assigned to the pyrochlore structure. Reflections were assigned to the pyrochlore ceria–hafnia phase (*), monoclinic hafnia (‡), orthorhombic hafnia (#). Some peaks could not be definitely assigned ($). | ||
Switching from a flow of argon to a flow of carbon dioxide caused a gradual decrease in the intensity of the (111) and (311) reflections of the pyrochlore phase to less than a third of their initial intensity, while the intensity of the (333), (511) reflection increased. The normalized intensity of the peaks of monoclinic hafnia increased significantly during the oxidation step. Subsequent exposure to a flow of argon led to a slight increase in the intensity of the pyrochlore (111) and (311) reflections and a decrease in the intensity of the (333), (511) reflection. The intensity of the (333), (511) reflection increased dramatically when the gas feed was switched. After the second switch from a flow of argon to carbon dioxide, the (111) and (311) peaks decreased gradually, and after about 15 min exposure to carbon dioxide they were no longer distinguishable from noise. The intensity of the peak assigned to the (333), (511) reflection increased gradually and then decreased again. Its intensity was significantly higher in the first pattern recorded in carbon dioxide compared to the last pattern in argon before the second switch of the feed gas.
There was a weak peak at a slightly higher Bragg angle than the (440) reflection of the pyrochlore structure. Its intensity decreased slightly during heating and at 1800 K in a flow of argon. In a flow of carbon dioxide, its intensity increased and formed a shoulder in the (440) reflection. Its intensity went up during the first exposure to carbon dioxide, which led to the formation of a broad shoulder in the (440) peak. During subsequent exposure to a flow of argon, the shoulder changed to an individual peak with a higher intensity compared to that in the pattern recorded in argon after reaching isothermal conditions. During the second exposure to carbon dioxide, the intensity of the peak increased further, forming a broad shoulder of the peak assigned to the (400) reflection of the pyrochlore structure. The (400) peak of the Ce2Zr2O7.5 phase was found at a higher angle than that of the (400) reflection of the Ce2Zr2O7 pyrochlore. However, the peak may also be due to the (002) reflection of monoclinic hafnia.
4 Discussion
XRD after auto-reduction in a thermogravimetric analyzer showed the formation of ceria–hafnia pyrochlore in Hf50CT. The pyrochlore was stable for several months under ambient conditions. The release of oxygen from equimolar ceria–hafnia in the thermogravimeter indicates an average oxidation state of ceria in Hf50CT higher than +3.0, which is unusual. The reduction is probably due to a systematic error, i.e. a drift of the thermobalance in the experiment of Hf50CT, which was corrected with a linear function. On the other hand, Baidya reported51 a ceria–zirconia pyrochlore-type structure with a composition of Ce2Zr2O6.2 prepared by reduction in hydrogen/argon at 1023 K. In contrast, under the same reaction conditions, Baidya did not report the formation of ceria–hafnia pyrochlore, most likely because the mobility of hafnium cations (M = 178.5 amu) is substantially lower than that of zirconium cations (M = 91.22 amu). The Ce K edge XANES of pristine Hf50C and reduced Hf50CR indicates structural changes, but the spectral features are diffuse at 1773 K. Only after cooling to room temperature did an oscillation indicate a different cerium–oxygen coordination geometry and cerium–oxygen interatomic distances were observed. The edge shift and the drop in the intensity of the white line during thermal reduction are clear indications of changes in the electronic structure of cerium.In the acquisition of Hf K edge spectra at 65 keV, the accuracy of the movement of the monochromator is even more critical than at the Ce K edge.56 The differences in the energy and intensity of the XANES features of Hf50C and Hf50CR suggest changes in the oxygen coordination geometry. XRD of Hf50CT gave compelling evidence of cation ordering.
The ordered arrangement of cations collapsed upon exposure to a flow of carbon dioxide at 1800 K. After 30 minutes in a flow of carbon dioxide, the normalized intensity of the (111) reflection of the pyrochlore decreased by about 60% but increased by less than 10% of the initial intensity during exposure to a flow of argon. In the second cycle, the reflections of the pyrochlore structure disappeared. The reversibility of the order–disorder transformation is probably possible under more intense reducing conditions. However, preparation of pyrochlore from small particles that are not stable at high temperature lead to higher mobility of the cations compared to the oxidized bulk material that consists of at least two phases. It may be that the conditions during the reduction step of isothermal carbon dioxide splitting were not reducing enough to reverse the decomposition of the pyrochlore phase. Ceria–hafnia pyrochlore is unstable under oxidizing conditions at 1773 K, which is in agreement with the work of Andrievskaya et al.48,49 who determined the phase diagram of ceria–hafnia at 1773 K in air. It is unclear if as to whether or not a temperature range enables the complete oxidation of the cation-ordered material analogous to Ce2Zr2O8, the κ-phase of ceria–zirconia. Otobe et al. prepared Ce2Zr2O8 by oxidation of pyrochlore in a stream of oxygen at 873 K.
The formation of the cation-ordered phase demonstrates the influence of the preparation method on material performance in thermochemical looping. Le Gal et al.27,64 found substantial differences in the carbon monoxide yield obtained from commercial materials (Rhodia, Solvay group) and materials prepared by co-precipitation and synthesis by the polymerized complex (Pechini-type) method. Ceria–hafnia pyrochlore might also form in thermochemical cycles in inhomogeneous samples with a lower than equimolar hafnia content. Therefore, it is important to prepare materials with a homogeneous distribution of the heterocation, for example by means of the polymerized complex.18,19,22,64 Inhomogeneities and small domains with high concentrations of heterocations with cation-ordered structures might explain why the synthesis route and the thermochemical history of a sample matter. Bonk et al.19 compared materials prepared by the ceramic method and a polymerized complex method and eliminated the potential influence of the preparation method: sintering pressed pellets at 1873 K led to qualitatively equal materials in thermochemical carbon dioxide splitting.
Kuhn et al.65 attributed the difficulty in describing the thermodynamic properties of ceria–zirconia by a simple model after exposure to reducing conditions at elevated temperature to ordering phenomena. Oxidation at high temperature reversed these effects. This is in agreement with the collapse of the ceria–hafnia pyrochlore structure under oxidizing conditions at 1800 K (Fig. 10). The pyrochlore Ce2Hf2O7 is an important phase in the ceria–hafnia system, because it represents an extreme case of the density of oxygen vacancies. Cation-ordering facilitates the complete reduction of cerium from Ce4+ to Ce3+. The local structure of oxygen vacancies in fluorite-type ceria–hafnia solid solutions is closely related to the unoccupied oxygen sites in the pyrochlore. Filling these oxygen vacancies may lead to the formation of a κ Ce2Hf2O8 phase, the analogue of the ceria–zirconia κ phase. Thus, the determination of short-range and long-range ordering in ceria-based oxides containing tetravalent heterocations such as Zr4+, Hf4+ or Sn4+ under relevant reaction conditions is very important. A significant improvement in the measurement and description of ordering phenomena by quantitative determination of structural properties during oxygen uptake of pyrochlores might enable us to pinpoint the elusive oxygen vacancies and lead to a better understanding of defect association. This would lead to a significant improvement in the accuracy of thermodynamic models such as that used by Kuhn et al.65
Pyrochlore-type structural features can lead to a higher fuel yield and must be considered in the engineering of oxygen vacancies. A high fuel yield, however, does not necessarily imply an improvement in the efficiency of solar-to fuel energy conversion of a solar thermochemical reactor system.66–70 Marxer et al.71 hold the current world-record in the energy-to-fuel conversion efficiency by using pure ceria. The thermodynamic properties of cation-ordered ceria-based oxides for solar-to-fuel energy conversion efficiency of two-step thermochemical water or carbon dioxide splitting have not yet been determined.
Ruan et al.72 recently proposed a two-step solar thermochemical water splitting based on CeO2/SnO2 and the pyrochlore Ce2Sn2O7. In seven consecutive cycles, CeO2/0.15SnO2 was thermally reduced at 1673 K in a flow of argon and subsequently reacted with steam at 1073 K. Reduction led to the formation of a pyrochlore phase and a hydrogen yield 3.8 times higher than that of pure ceria.
5 Conclusions
The ceria–hafnia pyrochlore Ce2Hf2O7 with high purity can be prepared via auto-reduction of ceria–hafnia powders synthesized via a polymerized complex method. The combination of in situ XAS and XRD measurements provides insight into the dynamics of structural changes under relevant solar reactor conditions. The pyrochlore structure Ce2Hf2O7 may form during the reduction step of solar thermochemical looping, but it is converted to a mixture of fluorite-type or tetragonal ceria, monoclinic hafnia, and orthorhombic hafnia in the oxidation step at 1800 K. The ordered arrangement of cations might persist during oxidation at lower temperature. Very little is known about the thermodynamics and the structural stability of the pyrochlore and κ-phases of the ceria–hafnia and ceria–zirconia systems. The properties of pyrochlore-type materials may be further tuned for applications that require the storage, transport, and activation of oxygen by adding heterocations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Erich de Boni, Lukas Oberer, and Markus Mähr (PSI), Jan Kovacovic and Joel Jenni (ETH) for technical support; Urs Hartfelder, Flavien Morel, Kim Meyer, and Sotiria Mostrou for assistance during XAS beam time, Jerick Imbao, Petr Sot, Jacinta Moraa Xto, and Amy Jenelle Knorpp for assistance during XAS/XRD beam time, Fangjian Lin and Alwin Frei for helpful discussions and TGA experiments, Marco Taddei for help with XRD measurements, and Maarten Nachtegaal for help with the interpretation of EXAFS data. Funding by the Competence Center for Energy and Mobility (CCEM, proposal #701) is gratefully acknowledged.References
- S. Abanades and G. Flamant, Solar Energy, 2006, 80, 1611–1623 CrossRef CAS.
- W. C. Chueh and S. M. Haile, ChemSusChem, 2009, 2, 735–739 CrossRef CAS PubMed.
- W. C. Chueh and S. M. Haile, Philos. Trans. R. Soc., A, 2010, 368, 3269–3294 CrossRef CAS PubMed.
- W. C. Chueh, C. Falter, M. Abbott, D. Scipio, P. Furler, S. M. Haile and A. Steinfeld, Science, 2010, 330, 1797–1801 CrossRef CAS PubMed.
- M. Roeb, M. Neises, N. Monnerie, F. Call, H. Simon, C. Sattler, M. Schmücker and R. Pitz-Paal, Materials, 2012, 5, 2015–2054 CrossRef CAS.
- M. Roeb and C. Sattler, Science, 2013, 341, 470–471 CrossRef CAS PubMed.
- D. Marxer, P. Furler, J. Scheffe, H. Geerlings, C. Falter, V. Batteiger, A. Sizmann and A. Steinfeld, Energy Fuels, 2015, 29, 3241–3250 CrossRef CAS.
- F. Lin, M. Rothensteiner, I. Alxneit, J. A. van Bokhoven and A. Wokaun, Energy Environ. Sci., 2016, 9, 2400–2409 CAS.
- H. Kaneko, T. Miura, H. Ishihara, S. Taku, T. Yokoyama, H. Nakajima and Y. Tamaura, Energy, 2007, 32, 656–663 CrossRef CAS.
- Q.-L. Meng, C.-I. Lee, H. Kaneko and Y. Tamaura, Thermochim. Acta, 2012, 532, 134–138 CrossRef CAS.
- C.-I. Lee, Q.-L. Meng, H. Kaneko and Y. Tamaura, J. Sol. Energy Eng., 2012, 135, 011002 CrossRef.
- S. Abanades and A. Le Gal, Fuel, 2012, 2–8 Search PubMed.
- A. Pappacena, M. Boaro, O. Šolcová and A. Trovarelli, Adv. Sci. Technol., 2014, 93, 76–81 CrossRef.
- Q. Jiang, G. Zhou, Z. Jiang and C. Li, Solar Energy, 2014, 99, 55–66 CrossRef CAS.
- E. V. Ramos-Fernandez, N. R. Shiju and G. Rothenberg, RSC Adv., 2014, 4, 16456 RSC.
- S. Lorentzou, G. Karagiannakis, D. Dimitrakis, C. Pagkoura, A. Zygogianni and A. G. Konstandopoulos, Energy Procedia, 2015, 69, 1800–1809 CrossRef CAS.
- F. Lin, I. Alxneit and A. Wokaun, CrystEngComm, 2015, 17, 1646–1653 RSC.
- J. R. Scheffe, R. Jacot, G. R. Patzke and A. Steinfeld, J. Phys. Chem. C, 2013, 117, 24104–24114 CAS.
- A. Bonk, A. C. Maier, M. V. Schlupp, D. Burnat, A. Remhof, R. Delmelle, A. Steinfeld and U. F. Vogt, J. Power Sources, 2015, 300, 261–271 CrossRef CAS.
- A. Bonk, A. Remhof, A. C. Maier, M. Trottmann, M. V. F. Schlupp, C. Battaglia and U. F. Vogt, J. Phys. Chem. C, 2016, 120, 118–125 CAS.
- A. Le Gal, S. Abanades and G. Flamant, Energy Fuels, 2011, 25, 4836–4845 CrossRef CAS.
- H. Kaneko, S. Taku and Y. Tamaura, Solar Energy, 2011, 85, 2321–2330 CrossRef CAS.
- Q.-L. Meng, C.-I. Lee, T. Ishihara, H. Kaneko and Y. Tamaura, Int. J. Hydrogen Energy, 2011, 36, 13435–13441 CrossRef CAS.
- F. Call, M. Roeb, M. Schmücker, H. Bru, D. Curulla-Ferre, C. Sattler and R. Pitz-Paal, Am. J. Anal. Chem., 2013, 04, 37–45 CrossRef.
- N. D. Petkovich, S. G. Rudisill, L. J. Venstrom, D. B. Boman, J. H. Davidson and A. Stein, J. Phys. Chem. C, 2011, 115, 21022–21033 CAS.
- A. Le Gal and S. Abanades, J. Phys. Chem. C, 2012, 116, 13516–13523 CAS.
- A. Le Gal, S. Abanades, N. Bion, T. Le Mercier and V. Harlé, Energy Fuels, 2013, 27, 6068–6078 CrossRef CAS.
- M. Kang, J. Zhang, C. Wang, F. Wang, N. Zhao, F. Xiao, W. Wei and Y. Sun, RSC Adv., 2013, 3, 18878 RSC.
- Handbook of Chemistry and Physics, ed. W. Haynes, CRC Press/Taylor and Francis, Boca Raton, FL, 92nd edn, 2012 Search PubMed.
- R. Ruh, H. J. Garrett, R. F. Domagala and N. M. Tallan, J. Am. Ceram. Soc., 1968, 51, 23–28 CrossRef CAS.
- R. Ruh and W. R. Corfiels, J. Am. Ceram. Soc., 1970, 53, 126 CrossRef CAS.
- M. Yashima, Catal. Today, 2015, 253, 3–19 CrossRef CAS.
- P. Furler, J. Scheffe, M. Gorbar, L. Moes, U. Vogt and A. Steinfeld, Energy Fuels, 2012, 26, 121024073702001 CrossRef.
- P. Furler, J. Scheffe, D. Marxer, M. Gorbar, A. Bonk, U. Vogt and A. Steinfeld, Phys. Chem. Chem. Phys., 2014, 16, 10503–10511 RSC.
- S. Phapale, R. Shukla, R. Mishra and A. K. Tyagi, J. Alloys Compd., 2014, 615, 792–794 CrossRef CAS.
- S. Otsuka-Yao-Matsuo, T. Omata, N. Izu and H. Kishimoto, J. Solid State Chem., 1998, 138, 47–54 CrossRef CAS.
- M. Subramanian, G. Aravamudan and G. Subba Rao, Prog. Solid State Chem., 1983, 15, 55–143 CrossRef CAS.
- S. N. Achary, S. K. Sali, N. K. Kulkarni, P. S. R. Krishna, A. B. Shinde and A. K. Tyagi, Chem. Mater., 2009, 21, 5848–5859 CrossRef CAS.
- T. Sasaki, Y. Ukyo, A. Suda, M. Sugiura, K. Kuroda, S. Arai and H. Saka, J. Ceram. Soc. Jpn., 2003, 111, 382–385 CrossRef CAS.
- T. Sasaki, Y. Ukyo, K. Kuroda, S. Arai, S. Muto and H. Saka, J. Ceram. Soc. Jpn., 2004, 112, 440–444 CrossRef CAS.
- H. Otobe, A. Nakamura, T. Yamashita and K. Minato, J. Phys. Chem. Solids, 2005, 66, 329–334 CrossRef CAS.
- T. Yamamoto, A. Suzuki, Y. Nagai, T. Tanabe, F. Dong, Y. Inada, M. Nomura, M. Tada and Y. Iwasawa, Angew. Chem., 2007, 46, 9253–9256 CrossRef CAS PubMed.
- H. F. Wang, Y. L. Guo, G. Z. Lu and P. Hu, Angew. Chem., Int. Ed., 2009, 48, 8289–8292 CrossRef CAS PubMed.
- M. Yashima, T. Sekikawa, D. Sato, H. Nakano and K. Omoto, Cryst. Growth Des., 2013, 13, 829–837 CAS.
- T. Wakita and M. Yashima, Appl. Phys. Lett., 2008, 92, 101921 CrossRef.
- F. Zhang, C.-H. Chen, J. C. Hanson, R. D. Robinson, I. P. Herman and S.-W. Chan, J. Am. Ceram. Soc., 2006, 89, 1028–1036 CrossRef CAS.
- V. V. Kharton, E. N. Naumovich and A. A. Vecher, J. Solid State Electrochem., 1999, 3, 61–81 CrossRef CAS.
- E. R. Andrievskaya, V. P. Redko and L. M. Lopato, Powder Metall. Met. Ceram., 2001, 40, 405–413 CrossRef CAS.
- E. R. Andrievskaya, G. I. Gerasimyuk, O. A. Kornienko, A. V. Samelyuk, L. M. Lopato and V. P. Redko, Powder Metall. Met. Ceram., 2006, 45, 448–456 CrossRef CAS.
- C. R. Stanek and R. W. Grimes, J. Am. Ceram. Soc., 2002, 85, 2139–2141 CrossRef CAS.
- T. Baidya, M. S. Hegde and J. Gopalakrishnan, J. Phys. Chem. B, 2007, 111, 5149–5154 CrossRef CAS PubMed.
- G. Zhou and R. J. Gorte, J. Phys. Chem. B, 2008, 112, 9869–9875 CrossRef CAS PubMed.
- J. M. Raitano, S. Khalid, N. Marinkovic and S.-W. Chan, J. Alloys Compd., 2015, 644, 996–1002 CrossRef CAS.
- A. Sharma, M. Varshney, H.-J. Shin, Y. J. Park, M.-G. Kim, T.-K. Ha, K. H. Chae and S. Gautam, Phys. Chem. Chem. Phys., 2014, 16, 19909 RSC.
- H. Fujimori, M. Yashima, S. Sasaki, M. Kakihana, T. Mori, M. Tanaka and M. Yoshimura, Phys. Rev. B, 2001, 64, 1–5 CrossRef.
- M. Rothensteiner, S. Sala, A. Bonk, U. Vogt, H. Emerich and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2015, 17, 26988–26996 RSC.
- M. Rothensteiner, J. Jenni, A. Bonk, U. F. Vogt, H. Emerich and J. A. van Bokhoven, Rev. Sci. Instrum., 2017, 1, 1 Search PubMed.
- W. van Beek, O. V. Safonova, G. Wiker and H. Emerich, Phase Transitions, 2011, 84, 726–732 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- P. E. Raison, C. C. Pavel, R. Jardin, E. Suard, R. G. Haire and K. Popa, Phys. Chem. Miner., 2010, 37, 555–559 CrossRef CAS.
- G. Ashiotis, A. Deschildre, Z. Nawaz, J. P. Wright, D. Karkoulis, F. E. Picca and J. Kieffer, J. Appl. Crystallogr., 2015, 48, 510–519 CrossRef CAS PubMed.
- X. Ning, I. W. Selesnick and L. Duval, Chemom. Intell. Lab. Syst., 2014, 139, 156–167 CrossRef CAS.
- C. Paun, O. V. Safonova, J. Szlachetko, P. M. Abdala, M. Nachtegaal, J. Sa, E. Kleymenov, A. Cervellino, F. Krumeich and J. A. van Bokhoven, J. Phys. Chem. C, 2012, 116, 7312–7317 CAS.
- A. Le Gal, S. Abanades and G. Flamant, Energy Fuels, 2011, 25, 4836–4845 CrossRef CAS.
- M. Kuhn, S. R. Bishop, J. L. M. Rupp and H. L. Tuller, Acta Mater., 2013, 61, 4277–4288 CrossRef CAS.
- J. R. Scheffe and A. Steinfeld, Mater. Today, 2014, 17, 341–348 CrossRef CAS.
- L. J. Venstrom, R. M. De Smith, Y. Hao, S. M. Haile and J. H. Davidson, Energy Fuels, 2014, 28, 2732–2742 CrossRef CAS.
- Y. Hao, C. Yang and S. M. Haile, Chem. Mater., 2014, 26(20), 6073–6082 CrossRef CAS.
- M. Takacs, J. R. Scheffe and A. Steinfeld, Phys. Chem. Chem. Phys., 2015, 17, 7813–7822 RSC.
- B. Bulfin, M. Lange, L. de Oliveira, M. Roeb and C. Sattler, Int. J. Hydrogen Energy, 2016, 49, 5–13 Search PubMed.
- D. Marxer, P. Furler, M. Takacs and A. Steinfeld, Energy Environ. Sci., 2017, 10, 1142–1149 CAS.
- C. Ruan, Y. Tan, L. Li, J. Wang, X. Liu and X. Wang, AIChE J., 2017, 1–13 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 6 figures. See DOI: 10.1039/c7ra09261j |
| This journal is © The Royal Society of Chemistry 2017 |