DOI:
10.1039/C7RA08409A
(Paper)
RSC Adv., 2017,
7, 43681-43688
Efficient mineralization of phenol by a temperature-responsive polyoxometalate catalyst under wet peroxide oxidation at lower temperatures†
Received
31st July 2017
, Accepted 29th August 2017
First published on 11th September 2017
Abstract
Herein, a temperature-responsive polyoxometalate (POM) catalyst [C16H33(CH3)3N]3[PO4{WO(O2)2}4]/poly(N-isopropylacrylamide) (abbreviated as (C16PW(O2)2/PNIPAM) was prepared and used in the catalytic wet peroxide oxidation (CWPO) of phenol under mild conditions. The POM catalyst C16PW(O2)2/PNIPAM showed a higher degradation efficiency and mineralization of phenol with H2O2 at room temperature or even at lower temperature (0 °C) within a short time (120 min). The high efficiency at lower temperature was attributed to its temperature-responsive property, wherein the lattice of the temperature-sensitive polymer relaxed at lower temperature and then wrinkled at higher temperature. These characteristics also permitted C16PW(O2)2/PNIPAM to be easily separated for recycling. The leaching test indicated that the POM catalyst exhibited excellent stability and little leaching and can be used as a thermosensitive catalyst for about six times. C16PW(O2)2/PNIPAM has potential application in the CWPO of phenol without the limitation of temperature and pH conditions.
1. Introduction
Agricultural processes and industrial manufacturing produce a large amount of wastewater including organic pollutants,1 which are toxic and hardly biodegradable. Therefore, a wide range of scientific studies have been conducted to develop new technologies, such as physical adsorption, photo-catalysis, and chemical oxidation, to achieve the effective purification of wastewater polluted with organic compounds such as phenol and its derivatives.2–5 Among the reported degradation processes, the CWPO system has emerged as a clean and effective alternative6–9 because it has significant oxidizing ability with harmless by-products and non-toxic substances and high degradation efficiency under mild conditions (20–60 °C, atmospheric pressure). To date, some studies have been reported on the CWPO of phenol containing the catalysts of Fenton agents, metal oxides, and polyoxometalates (Table S1†). At present, the highest efficiency was obtained with nano-metallic particles with 100% degradation of phenol under ultrasound power (500 W) for 120 min and the molar ratio of phenol to catalyst to H2O2 was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12.5:170.10 However, there are three major drawbacks for Fenton or metal oxides: the need for the removal of the homogeneous catalyst, narrow acidic range of pH (∼3.0), and low utilization of hydrogen peroxide and low degree of mineralization, which lead to the limitation of their application. POM has a broad use in catalysis such as in wet air oxidation,11,12 photodegradation,13 CWPO or as anchoring sites for single-atom metal catalysts.14,15 Our group developed the polyoxometalate [C16H33(CH3)3N]4H2SiV2W10O40 for the CWPO of phenol, and highest catalytic activity was obtained (a 91.6% degradation efficiency, 93.2% COD removal, and 85.5% TOC reduction with the phenol to catalyst to H2O2 molar ratio of 1:60:156.4 under ambient conditions for 90 min).16 A higher usage of H2O2 was required to achieve high efficiency, and it did not exhibit activity at lower temperatures. Despite the higher efficiency and higher degree of mineralization, the separation was relatively difficult due to its nanostructure. In addition, our city, Changchun, is located in the Northeast of China, where for up to six months, the temperature remains below 10 °C. Normal CWPO treating temperature does not favor the degradation of phenol in our city. Therefore, procedures with high efficiency and deep mineralization at low temperatures are of great value and highly desirable.
:12.5:170.10 However, there are three major drawbacks for Fenton or metal oxides: the need for the removal of the homogeneous catalyst, narrow acidic range of pH (∼3.0), and low utilization of hydrogen peroxide and low degree of mineralization, which lead to the limitation of their application. POM has a broad use in catalysis such as in wet air oxidation,11,12 photodegradation,13 CWPO or as anchoring sites for single-atom metal catalysts.14,15 Our group developed the polyoxometalate [C16H33(CH3)3N]4H2SiV2W10O40 for the CWPO of phenol, and highest catalytic activity was obtained (a 91.6% degradation efficiency, 93.2% COD removal, and 85.5% TOC reduction with the phenol to catalyst to H2O2 molar ratio of 1:60:156.4 under ambient conditions for 90 min).16 A higher usage of H2O2 was required to achieve high efficiency, and it did not exhibit activity at lower temperatures. Despite the higher efficiency and higher degree of mineralization, the separation was relatively difficult due to its nanostructure. In addition, our city, Changchun, is located in the Northeast of China, where for up to six months, the temperature remains below 10 °C. Normal CWPO treating temperature does not favor the degradation of phenol in our city. Therefore, procedures with high efficiency and deep mineralization at low temperatures are of great value and highly desirable.
Thermoresponsive polymers exhibit very sensitive and reversible temperature-dependant water solubility, which have attracted significant attention in terms of the catalytic loading.17,18 In our desulfurization process based on POMs, we have designed temperature-responsive POMs [C16H33N(CH3)3]3[PO4{MO(O2)2}4]/PNIPAM (M = Mo and W) using poly-N-isopropylacrylamide (PNIPAM) as the support19 in the H2O2 oxidation of organic sulfurs. It was found that the thermoresponsive hybrid [C16H33N(CH3)3]3[PO4{MO(O2)2}4]/PNIPAM exhibited a novel switchable property based on a change of temperature: it dissolved in organic solvents at higher temperatures and became gradually insoluble upon decreasing the temperature; we speculated that these thermo-responsive POM hybrids could exhibit good activity in the CWPO of phenol, especially at lower temperatures.
Herein, we used a thermo-sensitive POM C16PW(O2)2/PNIPAM hybrid in the CWPO of phenol at various temperatures. It exhibited fast adsorption and higher activity than its parent at room temperature and even at lower temperatures, showing great potential in the cold temperature region. In addition, the degradation procedure of phenol under CWPO using the abovementioned POM was studied (Scheme S1†).
2. Experimental
2.1 Materials
All reagents were of AR grade and used without further purification. A 0.72 mM phenol solution was prepared. C16PW(O2)2/PANIPAM was synthesized according to ref. 20 and characterized by IR spectroscopy.
2.2 Physical measurement
Elemental analysis was carried out using a Leeman Plasma Spec (I) ICP-ES and a P-E 2400 CHN elemental analyzer. The FT-IR spectra (4000–400 cm−1) were obtained using KBr discs via a Nicolet Magna 560 IR spectrometer. The 31P MAS NMR spectra of the samples were obtained using a Bruker AVANCE III 400 WB spectrometer equipped with a 4 mm standard bore CPMAS probe with the X channel tuned to 162 MHz. Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDAX) were performed using a XL30 ESEM FEG at 25 kV (PhilipsXL-30). Calorimetric measurements were performed using an SDT Q600 instrument from 16 °C to 80 °C at a scan rate of 0.5 °C min−1. The analysis of phenol during the reaction was performed using a closed reflux colorimetric method via a 756 CRT UV-vis spectrophotometer at a wavelength of 505 nm. Total organic carbon (TOC) was monitored using a Shimadzu TOC-VCPH total organic carbon analysis system. Chemical oxygen demand (COD) was determined using a closed reflux colorimetric method via a 756 CRT UV-vis spectrophotometer at a wavelength of 600 nm. The intermediates were analyzed via high pressure liquid chromatography (HPLC) with a UV detector using a YWG (4.6 mm × 150 mm, 10 μm) column. The mobile phase was a solution of H3PO4 (0.02% w/w) in H2O/acetonitrile (95/5 v/v) with a flow rate of 0.7 mL min−1.
2.3 Preparation of C16PW(O2)2/PNIPAM
In a typical synthesis, N-isopropylacrylamide (5 g) and N,N-methylenebisacrylamide (MBA) (0.04 g) were dissolved in a dimethyl sulfoxide (DMSO) mixed solution (4 mL) containing 1.5 g of polyvinylpyrrolidone (PVP) and 0.02 g of Pluronic F-127 (PF127). Then, 0.03 g of the initiator ammonium persulfate (APS) was added to the abovementioned monomer solution. The mixture was stirred magnetically at room temperature for 30 min. The reaction mixture was poured into a 200 mm long test tube (inside diameter 12.5 mm). The tube was clamped to a ring standing in a chamber drying oven at 25 °C. Then, 0.1 g C16PW(O2)2 was added to the upper layer of the mixture under stirring for 30 min. The mixed solutions were cast on glass substrates, followed by gelation and drying at 60 °C. The C16PW(O2)2/PNIPAM was obtained in 80.3% yield.
2.4 Catalytic procedure
For the degradation of phenol in a water system, 0.01 g of the catalyst was suspended in a fresh aqueous phenol solution (C0 = 0.72 mM, 5 mL, pH = 1.7) in a 25 mL three-neck flask at ambient temperature. Then, 0.15 mL of hydrogen peroxide (30.0%) was added to the mixture under stirring to start the reaction. After the reaction, the catalyst was separated by centrifugation at 1600 rpm for 10 min, which resulted in two layers: the upper layer was the aqueous phase and the bottom layer was the solid phase containing the catalyst. The aqueous sample was obtained at different times and immediately tested and measured using a 4-aminoantipyrine spectrophotometric method21 via a 756 CRT UV-vis spectrophotometer at a wavelength of 505 nm, which detected the degradation of phenol. The degradation efficiency of phenol was calculated using the following formula:
| Phenol degradation (%) = (C0 − Ci)/C0 |
where C0 is the initial phenol concentration in the liquid sample and Ci is the final phenol concentration in the liquid sample.
The utility of H2O2 was detected via the titration method with Ce(SO4)2.22 The temperature-control C16PW(O2)2/PNIPAM catalyst was left in the bottom of the reactor and was obtained by decanting and washing three times with water. The obtained solid was desiccated under vacuum at 60 °C for 120 min for recycling. The leaching of POMs from the polymer was tested using ICP analysis.
2.5 Adsorption experiment
An adsorption experiment was carried out to determine the adsorption capacity of the catalyst for an aqueous phenol solution. In the adsorption experiment, 0.01 g of catalyst was suspended in a fresh aqueous phenol solution (C0 = 0.72 mM, 5 mL, pH = 1.7) in a 25 mL three-neck flask for 2 h at ambient temperature. Then, the solid phase was separated by filtration. The catalyst was dried under vacuum at 60 °C and then used in determining the adsorption effect of phenol by IR spectroscopy.
2.6 Differential scanning calorimetry (DSC)
Calorimetric measurements were carried out using an SDT Q600 instrument from 16 °C to 80 °C at a scan rate of 0.5 °C min−1. Herein, 0.0006 mg of the catalyst was used. The DSC curve was integral to obtain the latent heat of the phase change.
3. Results and discussion
3.1 Evaluation of the degradation efficiency
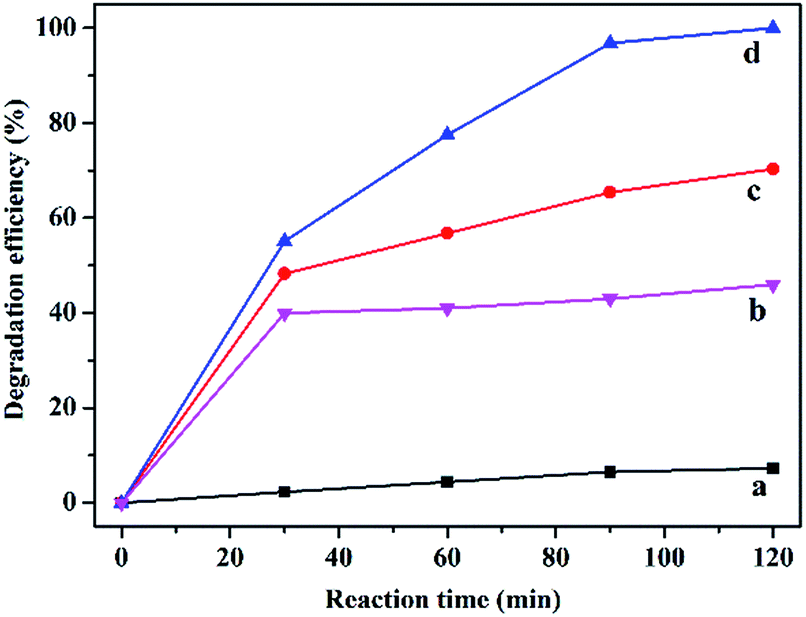
A preliminary experiment was performed at 25 °C and under atmospheric pressure in the presence of H2O2 without the catalyst and with different catalysts including C16PW(O2)2, PNIPAM and C16PW(O2)2/PNIPAM (Fig. 1). Without the catalyst, phenol was degraded negligibly (less than 8.0%); this indicated that the oxidative ability of H2O2 was weak at room temperature and pressure (Fig. 1a). In addition, the degradation of phenol by H2O2 was enhanced to a maximum of 46.0% using PNIPAM (Fig. 1b) at 120 min, whereas the phenol degradation reached 40.0% at 30 min and then hardly increased upon prolonging the reaction time. This may be attributed to the adsorption of phenol and no catalytic degradation for the support PNIPAM (Fig. S1a†). Fig. S1† displays the adsorption of phenol by PNIPAM and C16PW(O2)2/PNIPAM, which shows some difference for these two species in the amount of adsorbed phenol. However, the adsorption amount of phenol per gram of the polymer was almost the same for C16PW(O2)2/PNIPAM and PNIPAM (Fig. S1b†). This indicated that the decrease of phenol in the PNIPAM system was mainly attributed to the adsorption effect. In addition, the product distribution of the reaction system using PNIPAM was tested with only phenol; this also determined the adsorption of phenol and no oxidation occurred. These two aspects indicate that PNIPAM was the only support. Therefore, the decrease in phenol was mainly attributed to the adsorption of phenol by the polymer support, whereas the adsorption effect mainly depended on the polymer. Addition of POMs can significantly accelerate the degradation rates; C16PW(O2)2 exhibited a 70.3% degradation efficiency for about 120 min (Fig. 1c), whereas C16PW(O2)2/PNIPAM can catalyze the complete decomposition of phenol under the same reaction conditions (Fig. 1d). Therefore, the activity of the C16PW(O2)2/PNIPAM hybrid originated from C16PW(O2)2. The higher efficiency observed for C16PW(O2)2/PNIPAM was attributed to its temperature-responsive property in the water phase.
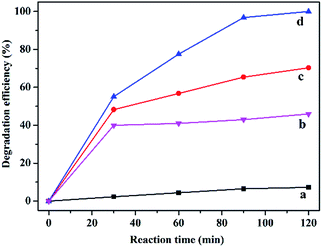 |
| | Fig. 1 The effect of different catalysts on the degradation of phenol. Blank (a), PNIPAM (b), C16PW(O2)2 (c), and C16PW(O2)2/PNIPAM (d). Reaction conditions: PNIPAM (0.0084 g), C16PW(O2)2 (0.0016 g), C16PW(O2)2/PNIPAM (0.01 g), H2O2 (0.1 mL), phenol solution (0.72 mM, 5 mL), 25 °C, 2 h. | |
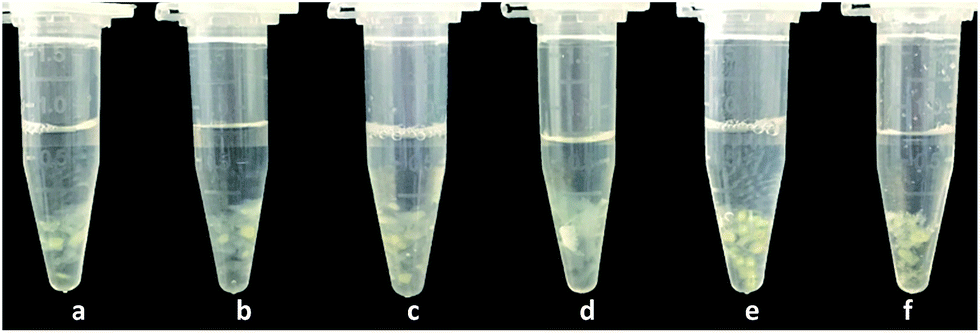
It can be seen that C16PW(O2)2/PNIPAM exhibits a temperature-dependent relaxation in water (Fig. 2): at lower temperature, relaxation occurs in water, whereas at temperatures higher than 32 °C, the catalyst wrinkles. In addition, the heat capacity of C16PW(O2)2/PNIPAM was measured using a DSC instrument. It can be seen that the peak at 32.93 °C is the temperature point of phase change with ΔH = 113.5 J g−1 (Fig. S2†). The abovementioned results indicated that C16PW(O2)2/PNIPAM could exhibit different degrees of swelling, as also indicated in the UV-visible spectra of C16PW(O2)2/PNIPAM in water upon varying the temperature (Fig. 3). It can be seen that the transmittance of C16PW(O2)2/PNIPAM increased gradually upon increasing the temperature to 32 °C and decreased at high temperature (60 °C). Therefore, the phase changes in C16PW(O2)2/PNIPAM occur upon varying the temperature; this is mainly attributed to PNIPAM.
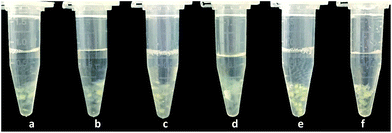 |
| | Fig. 2 Images of the phase changes observed for C16PW(O2)2/PNIPAM at (a) 0 °C, (b) 10 °C, (c) 25 °C, (d) 32 °C, (e) 45 °C, and (f) 60 °C. | |
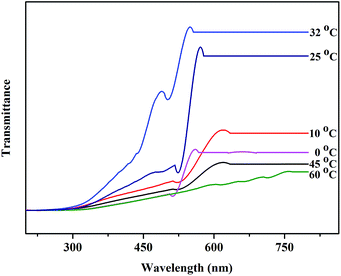 |
| | Fig. 3 The visible spectra of C16PW(O2)2/PNIPAM at different temperatures. | |
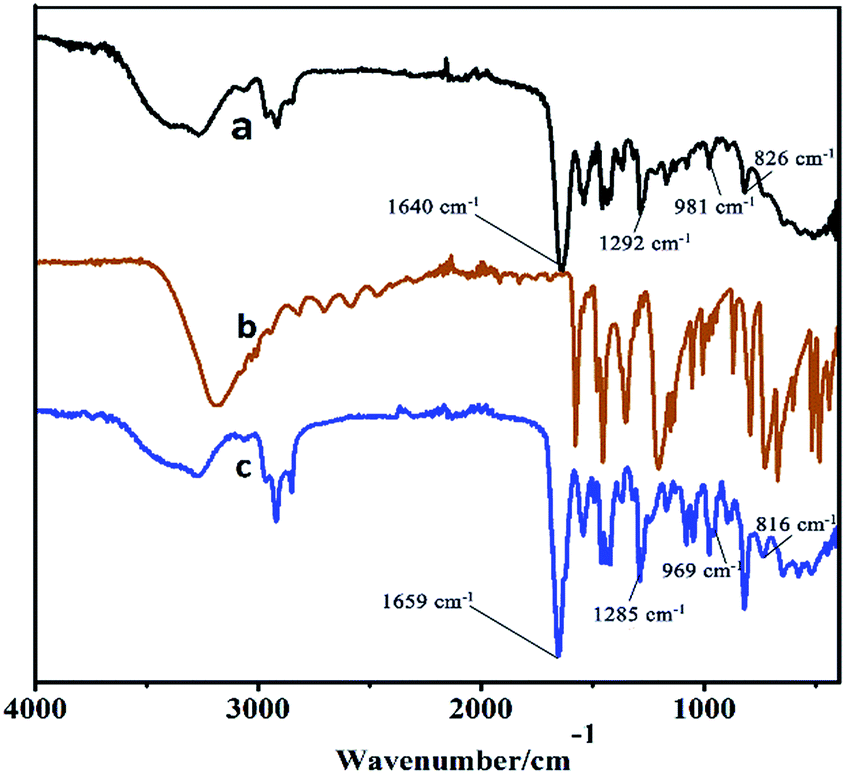
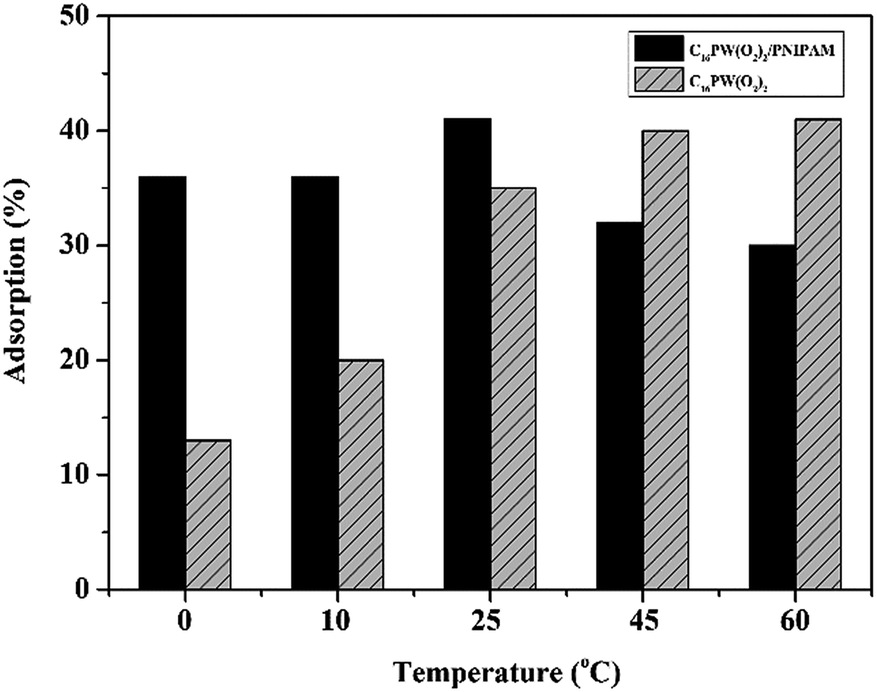
Moreover, the higher activity of C16PW(O2)2/PNIPAM was attributed to the stronger adsorption of phenol (Fig. 4) by C16PW(O2)2/PNIPAM. First, the adsorption of phenol by C16PW(O2)2/PNIPAM was determined using IR spectra at 1 h (Fig. 4). In comparison with those of C16PW(O2)2/PNIPAM, the peaks originating from phenol at 1640 cm−1 shifted to 1659 cm−1, 1292 cm−1 shifted to 1285 cm−1, 981 cm−1 shifted to 969 cm−1, and 826 cm−1 shifted to 816 cm−1; this suggested that some interaction occurred between the O atom of phenolic hydroxyl and the terminal oxygen of [PO4{WO(O2)2}4]3−. In addition, the amount of phenol absorbed was measured (Fig. S1b†). It can be seen that C16PW(O2)2/PNIPAM can rapidly adsorb phenol molecules in 30 min and then saturation is reached after 30 min. This adsorption of phenol can help them access the active sites in the POM; this promotes its oxidative degradation. Moreover, the adsorption of phenol by C16PW(O2)2 and C16PW(O2)2/PNIPAM depended on the temperature, and the adsorption capacity of C16PW(O2)2/PNIPAM was larger than that for C16PW(O2)2 below 25 °C; in addition, at higher temperatures, C16PW(O2)2 showed a higher adsorption ability than the C16PW(O2)2/PNIPAM hybrid (Fig. 5). This difference was also attributed to the temperature-responsive property of C16PW(O2)2/PNIPAM. As a result, C16PW(O2)2/PNIPAM exhibited 29.9% higher activity than C16PW(O2)2 at a lower temperature of 25 °C (Fig. 6) and a significant degradation efficiency of 89.2% at 0 °C.
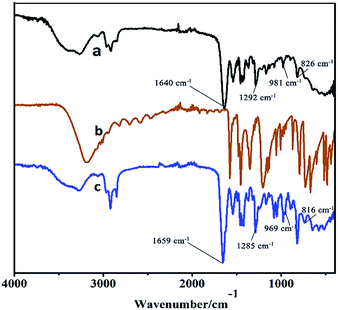 |
| | Fig. 4 The IR spectra of (a) C16PW(O2)2/PNIPAM, (b) phenol, and (c) C16PW(O2)2/PNIPAM-phenol at 25 °C for 0.5 h. | |
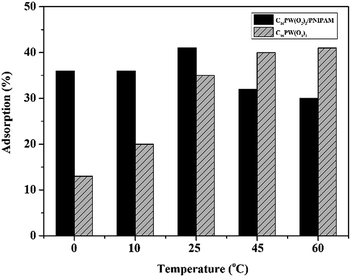 |
| | Fig. 5 Phenol adsorption on C16PW(O2)2/PNIPAM and C16PW(O2)2 at different temperatures. | |
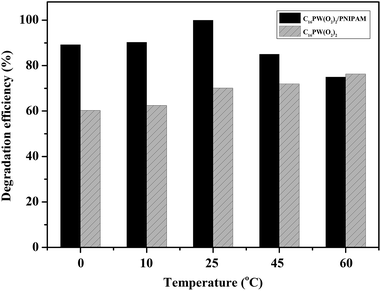 |
| | Fig. 6 The degradation efficiency between C16PW(O2)2/PNIPAM and C16PW(O2)2 at different temperatures. | |
To clarify the exact effect of C16PW(O2)2/PNIPAM on the conversion of phenol by H2O2, the COD and TOC changes were tested. It is known that the COD of water reflects the extent of contamination by a reductive substance, whereas the TOC refers to the total carbon content. At 120 min, the COD and TOC were reduced to 98.8% and ∼100%, respectively, indicating that it was the oxidative degradation of phenol and not an adsorptive effect. This indicates that the phenol molecules can be totally mineralized into simple inorganic compounds by C16PW(O2)2/PNIPAM/H2O2.
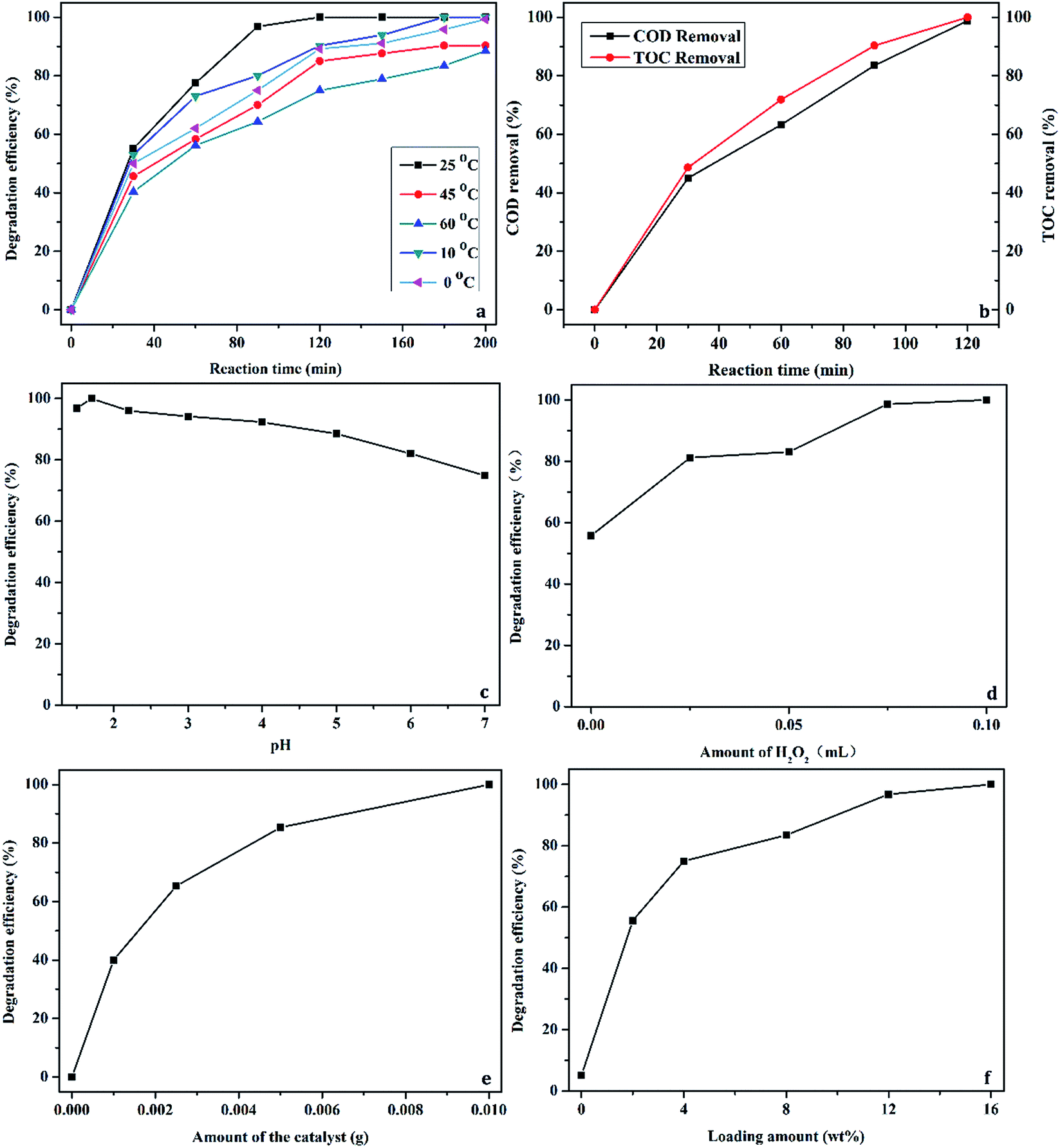
For further optimizing the degradation process with C16PW(O2)2/PNIPAM, the reaction was performed at various temperatures, reaction times, H2O2 concentrations, pH values, catalyst dosages, and loading amounts, as shown in Fig. 7. Fig. 7a shows the relationship between the degradation efficiency and reaction temperature and time when the degradation of phenol is catalyzed by C16PW(O2)2/PNIPAM. A little difference was found for various temperatures at 30 min, indicating most of the adsorption of phenol by the hybrid occurred during this period. Then, the degradation efficiency increased upon prolonging the reaction time to 120 min at all the temperatures studied herein, and the best phenol conversion was found to be ∼100% at 25 °C at 120 min. In addition, 100% degradation at 10 °C and 0 °C was obtained at 180 min and 200 min, respectively. C16PW(O2)2/PNIPAM showed excellent performance at lower temperature; this was different from the previously reported results of [C16H33(CH3)3N]4H2SiV2W10O40.16 The changes in the COD and TOC are given in Fig. 7b, showing that the removal of COD and TOC increased gradually to almost 100% at 120 min, in accordance with the phenol conversion. This result demonstrated that C16PW(O2)2/PNIPAM was more active in the CWPO treatment of phenol, whereas the intermediates formed during phenol degradation were oxidized to simple inorganic species as soon as they were formed. Further, the influence of the pH values on the degradation of phenol was investigated (Fig. 7c); it was observed that the degradation efficiency of phenol over C16PW(O2)2/PNIPAM at the pH values of 1.5, 1.7, 2.2, 3.0, 4.0, 5.0, 6.0, and 7.0 were 96.8, 100, 96.0, 94.2, 92.3, 88.5, 82.0, and 74.9%, respectively. The maximum degradation efficiency (100%) was obtained at pH 1.7. Upon increasing the pH, the degradation of phenol decreased. However, the degradation of phenol showed no significant difference in the pH range of 0–5, with only about an 8% decrease. This result shows that C16PW(O2)2/PNIPAM can perform over a wide pH range. Upon further increasing the pH to 7, the degradation is decreased to 74.9%, which is also active, and upon prolonging the reaction time, 100% conversion of phenol can be achieved. This decrease may be attributed to an increase in the pH that leads to the ionization of phenol to form Ph-O−. Because the surface charge of the POMs is negative, the phenol molecules are repelled far away from the catalytic centers; this results in a decrease in phenol adsorption by the catalyst and hence a decrease in the degradation efficiency. Fig. 7d shows the effect of H2O2 concentration when the reaction is carried out at 25 °C for 120 min using 0.01 g of catalyst. Obviously, the degradation efficiency increased markedly when the concentration of H2O2 was increased to 0.1 mL. In theory, 14 mol of H2O2 is needed to oxidize 1 mol of phenol. To achieve a high degradation of phenol, an excess of H2O2 needs to be added. The optimal hydrogen peroxide concentration was 0.19 M in our experiment, which was 19 times higher than stoichiometric concentration of 0.01 M. In addition, about 98.9% H2O2 was consumed at 120 min in the presence of C16PW(O2)2/PNIPAM. This also indicated that some organic intermediates were produced during the oxidation of phenol by H2O2. Then, further increase in the H2O2 used did not lead to a higher degradation efficiency due to the decomposition of H2O2. This usage was smaller than the previous results reported for [C16H33(CH3)3N]4H2SiV2W10O40.16
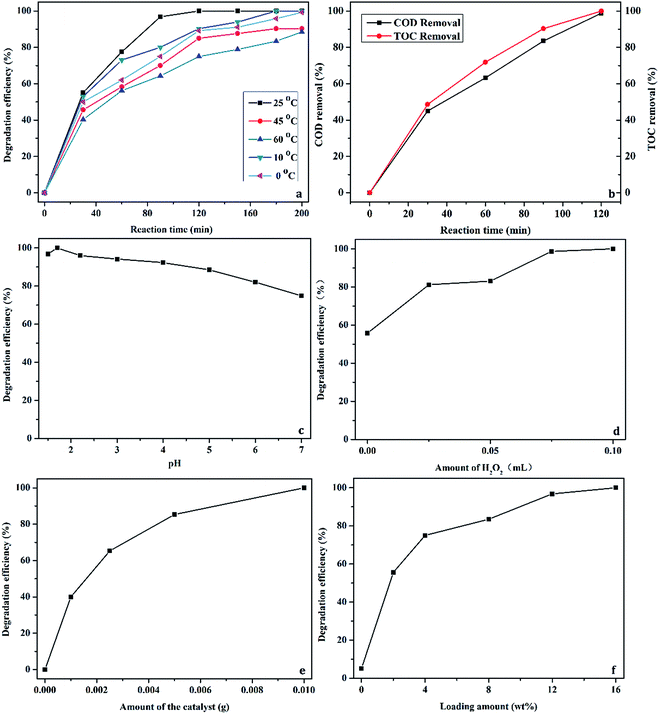 |
| | Fig. 7 The main parameters affecting the degradation of phenol: temperature (a), COD and TOC (b), pH (c), H2O2 concentration (d), catalyst dosage, (e) and loading amount (f). | |
Fig. 7e shows the effect of the catalyst dosage on the degradation of phenol. Upon increasing the catalyst dosage from 0.001 to 0.01 g, the degradation efficiency was increased from 40 and 100%, respectively. The result can be attributed to an increase in the number of catalytic active sites in the catalyst. When the amount of catalyst was increased to 0.01 g, the degradation efficiency of phenol improved slowly. Therefore, 0.01 g was selected as the most appropriate amount of catalyst with a high degradation efficiency. The loading amount of C16PW(O2)2 had a significant influence on the degradation efficiency of phenol (Fig. 7f). The degradation efficiency increased as the loading amount of POMs was increased. In addition, the degradation efficiency was 55.6, 75.0, 83.5, 96.8, and 100% at a loading amount of 2.0, 4.0, 8.0, 12.0, and 16.0 wt%, respectively. If the loading amount continued to increase, C16PW(O2)2/PNIPAM was unable to display the temperature-responsive property.
3.2 Reusability of catalyst
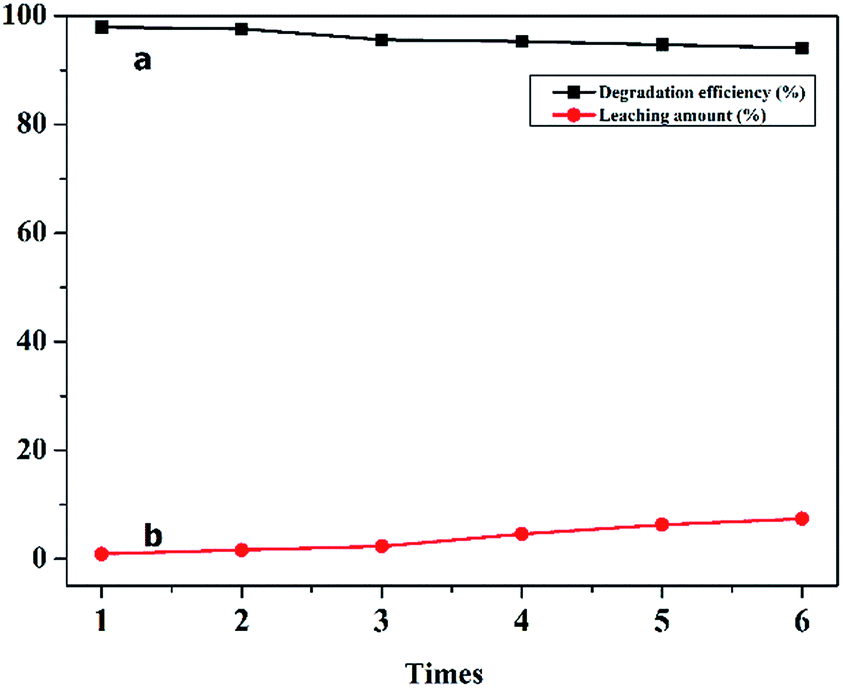
The stability of a catalyst is one of the most important factors in the heterogeneous CWPO process. The FTIR spectra of the recovered C16PW(O2)2/PNIPAM was investigated (Fig. S3†). In the IR spectrum of fresh C16PW(O2)2/PNIPAM, the peaks (Fig. S3a†) at 1166, 981, 826, and 723 cm−1 were usually assigned to ν(P–O), ν(W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), ν(O–O), and ν[W(O2)], respectively. These peaks suggest the existence of the [PO4{WO(O2)2}4]3− group in the hybrid. In addition, the peaks at 2930 and 1455 cm−1 were due to N–H, C–H, and C–N in [C16H33N(CH3)3]+. The three characteristic peaks at 3270, 1640, and 1290 cm−1 can be attributed to ν(–NH–CO), which are the intense vibrations of PNIPAM. As compared to the IR spectrum of C16PW(O2)2,23 some red shifts of the ν(WO), ν(O–O), and νasym[W(O2)] occurred; this indicated the existence of some interactions between C16PW(O2)2 and PNIPAM.24 These interactions were attributed to the hydrogen bonds formed between the terminal oxygens of C16PW(O2)2 and the NH groups in PNIPAM, which confirmed the little leaching of POMs from PNIPAM. The IR spectrum of the recovered C16PW(O2)2/PNIPAM showed no changes in the characteristic peaks for [C16H33N(CH3)3]+, [PO4{WO(O2)2}4]3−, and PNIPAM; this indicated the stability of C16PW(O2)2/PNIPAM during the oxidative reaction. The DR-UV-vis spectra of C16PW(O2)2/PNIPAM (Fig. S4a†) had a significant characteristic peak at 240 nm. In addition, C16PW(O2)2/PNIPAM after the degradation of phenol (Fig. S4b†) was the same as the original. The stability of the catalyst was also demonstrated by 31P magic-angle spinning (MAS) NMR spectroscopy (Fig. S5†). The 31P MAS NMR spectrum of C16PW(O2)2/PNIPAM after the reaction further determined its stability during the oxidation reaction. The morphology of C16PW(O2)2/PNIPAM before and after the oxidative reaction was examined by SEM (Fig. S6a†), showing that no changes occurred during the reaction. Therefore, the C16PW(O2)2/PNIPAM, as characterized by IR spectra, DR-UV-vis spectra, 31P MAS NMR spectra, and SEM before and after the degradation of phenol, kept its original structure and morphology. The results showed that the catalyst remained perfectly organized after the reaction. Moreover, the catalyst retained its high efficiency in the process of phenol degradation after six repeated experiments. Therefore, the leaching test revealed that C16PW(O2)2/PNIPAM had fantabulous stability and could be reused six times as a rapid thermosensitive catalyst via a simple treatment. Moreover, the total amount of C16PW(O2)2/PNIPAM leaching through the six runs of the reaction reached only 7.4% of the starting amount, as ascertained by ICP (Table S2†) measurements (Fig. 8).
O), ν(O–O), and ν[W(O2)], respectively. These peaks suggest the existence of the [PO4{WO(O2)2}4]3− group in the hybrid. In addition, the peaks at 2930 and 1455 cm−1 were due to N–H, C–H, and C–N in [C16H33N(CH3)3]+. The three characteristic peaks at 3270, 1640, and 1290 cm−1 can be attributed to ν(–NH–CO), which are the intense vibrations of PNIPAM. As compared to the IR spectrum of C16PW(O2)2,23 some red shifts of the ν(WO), ν(O–O), and νasym[W(O2)] occurred; this indicated the existence of some interactions between C16PW(O2)2 and PNIPAM.24 These interactions were attributed to the hydrogen bonds formed between the terminal oxygens of C16PW(O2)2 and the NH groups in PNIPAM, which confirmed the little leaching of POMs from PNIPAM. The IR spectrum of the recovered C16PW(O2)2/PNIPAM showed no changes in the characteristic peaks for [C16H33N(CH3)3]+, [PO4{WO(O2)2}4]3−, and PNIPAM; this indicated the stability of C16PW(O2)2/PNIPAM during the oxidative reaction. The DR-UV-vis spectra of C16PW(O2)2/PNIPAM (Fig. S4a†) had a significant characteristic peak at 240 nm. In addition, C16PW(O2)2/PNIPAM after the degradation of phenol (Fig. S4b†) was the same as the original. The stability of the catalyst was also demonstrated by 31P magic-angle spinning (MAS) NMR spectroscopy (Fig. S5†). The 31P MAS NMR spectrum of C16PW(O2)2/PNIPAM after the reaction further determined its stability during the oxidation reaction. The morphology of C16PW(O2)2/PNIPAM before and after the oxidative reaction was examined by SEM (Fig. S6a†), showing that no changes occurred during the reaction. Therefore, the C16PW(O2)2/PNIPAM, as characterized by IR spectra, DR-UV-vis spectra, 31P MAS NMR spectra, and SEM before and after the degradation of phenol, kept its original structure and morphology. The results showed that the catalyst remained perfectly organized after the reaction. Moreover, the catalyst retained its high efficiency in the process of phenol degradation after six repeated experiments. Therefore, the leaching test revealed that C16PW(O2)2/PNIPAM had fantabulous stability and could be reused six times as a rapid thermosensitive catalyst via a simple treatment. Moreover, the total amount of C16PW(O2)2/PNIPAM leaching through the six runs of the reaction reached only 7.4% of the starting amount, as ascertained by ICP (Table S2†) measurements (Fig. 8).
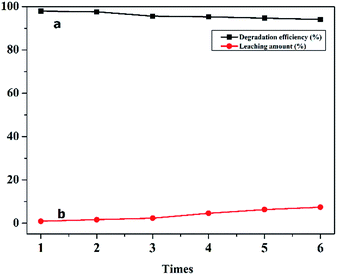 |
| | Fig. 8 The reuse of C16PMo(O2)2/PNIPAM (a) and the amount of C16PMo(O2)2 leached from the polymer (b). | |
3.3 Catalytic reaction mechanisms of the CWPO process
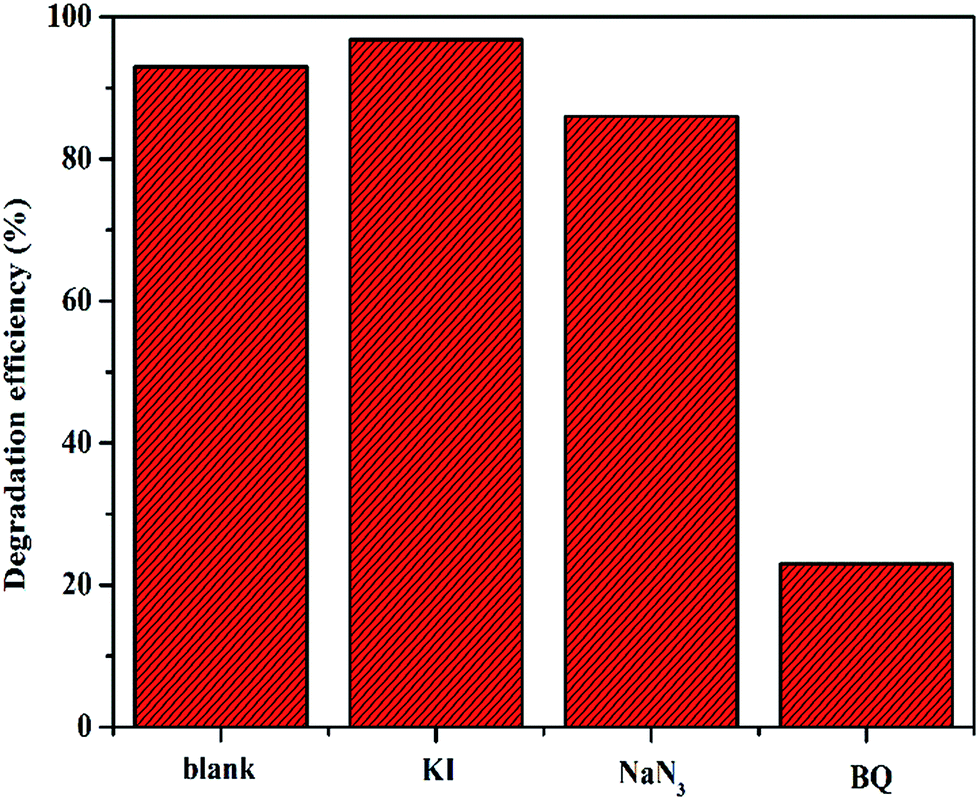
Generally, it has been shown that the oxidation process of organic molecules by POMs occurs via a free radical mechanism.25,26 Superoxide radicals (O2˙−) are extremely high-potential oxidizing agents with short lifetimes and can oxidize organic substrates and generate other free radicals.27 To confirm the main active radicals responsible for the degradation of phenol catalyzed by C16PW(O2)2/PNIPAM, a comparative experiment was carried out by adding 10 wt% of the catalyst quenchers into the initial solution under the same conditions (Fig. 9). The possible oxidative intermediate radicals, such as singlet oxygen (1O2), superoxide (O2˙−), hydroperoxy (HO2˙) or (OH˙), were demonstrated indirectly through the use of appropriate quenchers for these radicals based on otherwise identical conditions. Certain quenchers include KI (a quencher of OH˙ radicals on the catalyst surface28), sodium azide (NaN3, a singlet oxygen quencher,29 but may also interact with OH˙ radicals30), and p-benzoquinone (C6H4O2, BQ, a quencher of the superoxide radical31). The addition of p-benzoquinone had an influence on the process of phenol degradation; this suggested that the active oxidative species in this system was a combination of O2˙− and singlet oxygen (1O2) free radicals. The effect of p-benzoquinone was much higher than that of KI and higher than that of NaN3; this indicated that O2˙− was formed as a possible oxidative intermediate species during the reaction. However, KI promotes the degradation of phenol. It might be I− enhancing the transmission of electrons and accordingly improving the transmission of the peroxy-chain. The abovementioned results indicate that the reaction of phenol oxidation underwent a superoxide radical and single oxygen mechanism.
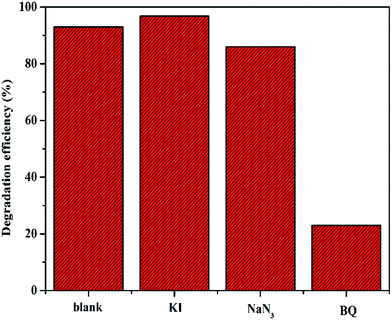 |
| | Fig. 9 The effects of scavenging agents including KI, NaN3, and 1,4-benzoquinone (BQ) on the degradation of phenol. Reaction conditions: C16PW(O2)2/PNIPAM (0.01 g), scavenging agents: 10 wt% of the catalyst, H2O2 (0.1 mL), phenol solution (0.72 mM, 5 mL), 25 °C, 2 h. | |
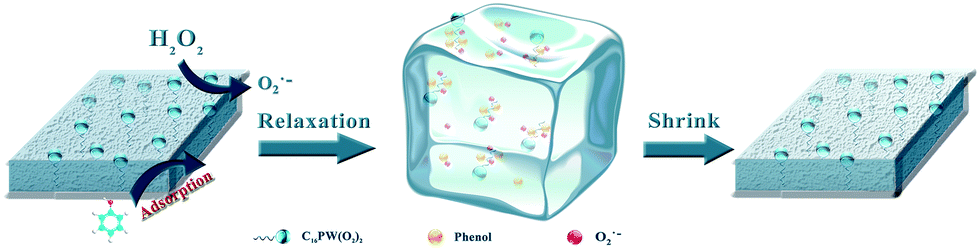
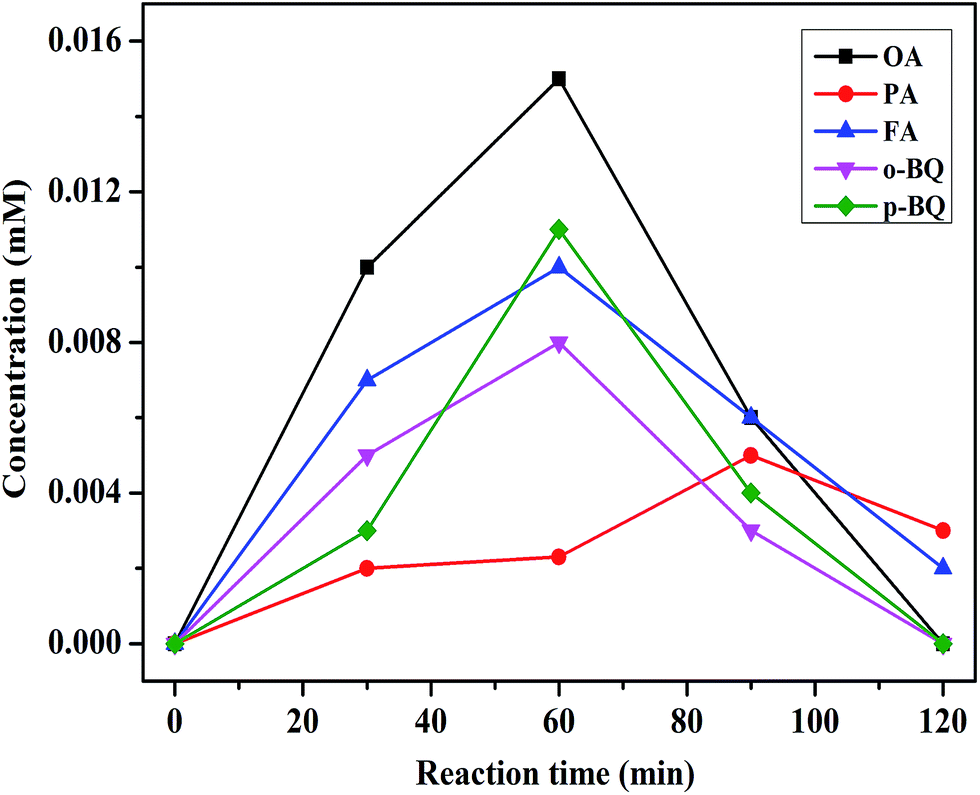
Therefore, the mechanism of CWPO degradation of phenol over C16PW(O2)2/PNIPAM catalyst was proposed as follows: (1) the reactants (phenol and H2O2) were adsorbed on the expansive membrane by the transmission peroxide chain via the interaction between hydrogen peroxide and the O–O chain in C16PW(O2)2. Then, the phenol molecules were accessible to the active sites anchored within micellar particles of C16PW(O2)2 in the lattice of PNIPAM (Scheme 1). This adsorption reached an equilibrium after the suspension was stirred for 30 min; (2) the POMred was formed. Then, O2˙− can be formed via an O–O chain auto-oxidation process in the presence of H2O2, such that the organic molecules can be degraded by the radical to accomplish the degradation of phenol. Phenol was oxidized first into catalysts and the peroxo-group was transferred to phenol molecules, while R·and reduced form of p-benzoquinone (abbreviated as p-BQ), and seldom o-benzoquinone (abbreviated as o-BQ) and then into oxalic acid (abbreviated as OA), formic acid (abbreviated as FA) and propionic acid (abbreviated as PA) measured by HPLC (Fig. 10). In the beginning of the reaction, the phenol molecules are mostly decomposed to OA and a small amount of FA and PA. Then, some of the oxalic acid molecules degraded to FA, which was tested during the degradation of oxalic acid under the same reaction conditions (Fig. S8†). Oxalic acid was rapidly degraded at the end of the reaction. In addition, they decomposed gradually and were totally mineralized into CO2 and water. (3) Finally, the catalytic cycle was completed by the reoxidation of POMred to its POMox form using H2O2.32
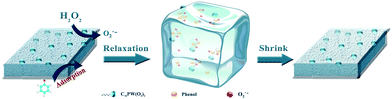 |
| | Scheme 1 A schematic of the orientation of the reactants in a membrane POM catalyst and phenol being oxidized by H2O2 to CO2. | |
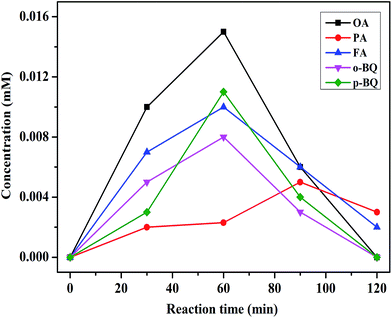 |
| | Fig. 10 The concentration of the intermediate products during the reaction. Reaction conditions: C16PW(O2)2/PNIPAM (0.01 g), phenol solution (0.72 mM, 5 mL), 25 °C, 2 h. | |
4. Conclusions
In this study, a POM thermosensitive catalyst C16PW(O2)2/PNIPAM was used in the CWPO of phenol under ambient conditions and even at lower temperatures (0 °C). This thermosensitive POM catalyst exhibited high efficiency in active H2O2 to degrade phenol at 25 °C, 10 °C, and even 0 °C within a short time (100% of degradation efficiency) due to the phase changes that occurred upon varying the temperature. The high efficiency of the catalyst was due to the catalytic center of C16PW(O2)2 and the synergistic effects of the polymer support, which enhanced the concentration of phenol molecules around the active sites. Moreover, the temperature-responsive property and high stability are beneficial in the recovery of the catalyst, which exhibited consistent activity for six reaction cycles. Therefore, CWPO based on the POM C16PW(O2)2/PNIPAM catalyst is of great value for green chemistry and C16PW(O2)2/PNIPAM has potential for the CWPO of phenol especially in low temperature regions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China [51578119].
References
- J. Guo and M. Al-Dahhan, Ind. Eng. Chem. Res., 2003, 42, 2450–2460 CrossRef CAS.
- L. Han, L. B. Qian, J. C. Yan and M. F. Chen, Chemosphere, 2016, 156, 262–271 CrossRef CAS PubMed.
- O. Sacco, V. Vaiano, C. Han, D. Sannino and D. D. Dionysiou, Appl. Catal., B, 2015, 164, 462–474 CrossRef CAS.
- J. C. Yan, L. Han, W. G. Gao, S. Xie and M. F. Chen, Bioresour. Technol., 2015, 175, 269–274 CrossRef CAS PubMed.
- K. M. Valkaj, A. Katovic and S. Zrncevic, Ind. Eng. Chem. Res., 2011, 50, 4390–4397 CrossRef CAS.
- N. S. Inchaurrondo, P. Massa, R. Fenoglio, J. Font and P. Haure, Chem. Eng. J., 2012, 198–199, 426–434 CrossRef CAS.
- D. F. Bishop, G. Stern, M. Fleischman and L. S. Marshall, Ind. Eng. Chem. Process Des. Dev., 1968, 7, 110–117 CAS.
- M. N. Timofeeva, S. T. Khankhasaeva, E. P. Talsi, V. N. Panchenko, A. V. Golovin, E. T. Dashinamzhilova and S. V. Tsybulya, Appl. Catal., B, 2009, 90, 618–627 CrossRef CAS.
- R. J. G. Lope, M. L. N. Perdigoto and R. M. Quinta-Ferreira, Appl. Catal., B, 2012, 117–118, 292–301 CrossRef.
- J. W. Singh, J. K. Yang and Y. Y. Chang, J. Environ. Manage., 2016, 175, 60–66 CrossRef CAS PubMed.
- J. Xu, S. Q. Yan, J. X. Li, S. T. Wang, X. H. Wang, M. X. Huo and Z. J. Jiang, RSC Adv., 2014, 4, 25404–25409 RSC.
- S. Zhao, C. L. Sun, L. Wang, X. B. Xu and X. H. Wang, Dalton Trans., 2010, 39, 5087–5090 RSC.
- C. Jaramillo-Páez, J. A. Navío, M. C. Hidalgo, A. Bouzianib and M. E. Azzouzi, J. Photochem. Photobiol., A, 2017, 332, 521–533 CrossRef.
- B. Zhang, H. Asakura and N. Yan, Ind. Eng. Chem. Res., 2017, 56, 3578–3587 CrossRef CAS.
- B. Zhang, H. Asakura, J. Zhang, J. G. Zhang, S. De and N. Yan, Angew. Chem., Int. Ed., 2016, 55, 8319–8323 CrossRef CAS PubMed.
- H. C. Li, X. Yu, H. W. Zheng, Y. M. Li, X. H. Wang and M. X. Huo, RSC Adv., 2014, 4, 7266–7274 RSC.
- J. G. Zhang, Y. Yuan, K. J. Kilpin, Y. Kou, P. J. Dyson and N. Yan, J. Mol. Catal. A: Chem., 2013, 371, 29–35 CrossRef CAS.
- N. Yan, J. G. Zhang, Y. Yuan, G. T. Chen, P. J. Dyson, Z. C. Li and Y. Kou, Chem. Commun., 2010, 46, 1631–1633 RSC.
- S. C. Sun, X. Yu, Y. N. Guo, L. Chen, X. H. Wang and Z. J. Jiang, Catal. Surv. Asia, 2016, 20, 98–108 CrossRef CAS.
- Q. Feng, L. Z. Du, Q. Z. Yan and C. C. Ge, Adv. Mater. Res., 2011, 295–297, 1193–1197 CrossRef CAS.
- Y. F. Cai and C. Y. Wu, Environmental Science Survey, 2012, 31, 85–87 Search PubMed.
- Y.
Y. Gu and D. Li, J. Hunan Inst. Sci. Technol., Nat. Sci., 2005, 18, 55–58 CAS.
- C. Venturello and R. D'Aloisio, J. Org. Chem., 1988, 53, 1553–1557 CrossRef CAS.
- D. F. Li, Y. H. Guo, C. W. Hu, L. Mao and E. B. Wang, Appl. Catal., A, 2002, 235, 11–20 CrossRef CAS.
- S. Alexander, G. Pierre and S. L. Kachkarova-Sorokina, Fr. Pat., 2842200, 2004.
- S. L. Kachkarova-Sorokina, P. Gallezot and A. B. Sorokin, Chem. Commun., 2004, 24, 2844–2845 RSC.
- I. A. Alaton and J. L. Ferry, Dyes Pigm., 2002, 54, 25–36 CrossRef.
- C. Karunakaran and R. Dhanalakshmi, Sol. Energy Mater. Sol. Cells, 2008, 92, 1315–1321 CrossRef CAS.
- F. Wu, N. S. Deng and H. L. Hua, Chemosphere, 2000, 41, 1233–1238 CrossRef CAS.
- S. H. Yoon and J. H. Lee, Environ. Sci. Technol., 2005, 39, 9695–9701 CrossRef CAS PubMed.
- C. Schweitzer and R. Schmidt, Chem. Rev., 2003, 103, 1685–1757 CrossRef CAS PubMed.
- S. Zhao, X. H. Wang and M. X. Huo, Appl. Catal., B, 2010, 97, 127–134 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08409a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  * and
Shengtian Wang*
* and
Shengtian Wang*