
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and structure–property relationships of SIS-g-PB copolymers and their application in hot-melt pressure-sensitive adhesives†
Zhongfu Zhao *a,
Peiying Liua,
Chunqing Zhang*a,
Wei Liua,
Yifu Dingb,
Yandong Zhanga,
Fanzhi Menga and
Tao Tangc
*a,
Peiying Liua,
Chunqing Zhang*a,
Wei Liua,
Yifu Dingb,
Yandong Zhanga,
Fanzhi Menga and
Tao Tangc
aState Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, No. 2 Linggong Road, Dalian 116024, China. E-mail: zfzhao@dlut.edu.cn; zhangchq@dlut.edu.cn; Tel: +86-0411-8498-6102
bDepartment of Mechanical Engineering, University of Colorado Boulder, Boulder, CO 80309-0427, USA
cState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China
First published on 12th September 2017
Abstract
A “graft onto” method was combined with an epoxidation reaction and living anionic polymerization to successfully synthesize a series of SIS-g-PB copolymers with defined branch numbers and branch lengths. These copolymers were utilized to formulate various hot-melt pressure-sensitive adhesives (HMPSAs). Their molecular structure and bulk properties were characterized by 1H-nuclear magnetic resonance (1H-NMR), gel permeation chromatography (GPC), differential scanning calorimetry (DSC) and rheometry. The adhesion performances were characterized in terms of holding power and 180° peel strength. The epoxidation reaction alone would negatively influence the rheological properties of the parent SIS copolymers, particularly for low-temperature applications. Controlled addition of the low-Tg PB blocks can significantly improve the low-temperature properties of the SIS copolymers. Both η* and G′ increased in the lower shear frequency regime (<101 rad s−1) but decreased in the higher shear frequency regime (>101 rad s−1) with branch number and branch length, in which branch length had a greater effect than the branch number. As a result, the 180° peel strength of the SIS-g-PB based HMPSAs displayed reached 0.23 kN m−1, which is more than twice the value for SIS-based HMPSAs.
1. Introduction
Styrene-b-isoprene-b-styrene (SIS) and styrene-b-butadiene-b-styrene (SBS) copolymers are a class of polymers that behave like vulcanized-rubbers at room temperature but can be processed like conventional thermoplastics at elevated temperatures. In comparison, SIS copolymers have much lower viscosity than SBS copolymers since the polyisoprene (PI) chains have much higher entanglement molecular weight than the polybutadiene (PB) chains. On the other hand, SBS copolymers display a much higher elastic modulus and better low temperature properties.1 Thus, most SIS copolymers are traditionally used in hot-melt pressure sensitive adhesives (HMPSAs) while SBS copolymers are mainly used in footwear and asphalt modification.1–9SIS-based HMPSAs are mainly formulated with SIS copolymers, tackifier resins and antioxidants.10–14 Their nonpolar feature greatly limits their applications due to their poor adhesion with polar materials or weak compatibility with hydrophilic components.15–18 To improve their polarity, addition of polar tackifiers such as rosin and its derivatives were examined. However, the partial compatibility between the additives and the styrene end-block might compromise the integrity of the HMPSAs.3,13,19 Other hydrophilic components were also introduced into the formulation of the SIS-based HMPSAs for transdermal delivery of hydrophilic drugs.20,21 Similarly, this method sacrificed the adhesive performance of the SIS-based HMPSAs due to the weak interface interaction.
Another strategy to improve the polarity of the SIS-based HMPSAs is to functionalize the SIS copolymers.22 One of such approach is to epoxidize SIS (ESIS).23 Indeed, the ESIS markedly improved the polarity of the HMPSAs. However, the compatibility between ESIS and tackifier resins is reduced, which results in a decrease of the adhesive performance of HMPSAs. To overcome this shortcoming, parent SIS resins and ESIS resins were combined in the formulation HMPSAs.24 This method could greatly improve the adhesive performance of the adhesive matrix, but again lowered the polarity of the adhesives.
In this article, we explore a new synthetic approach to improve both the polarity of SIS copolymers and the compatibility with components in the HMPSAs formulations. The main idea is to controllably graft PB blocks onto ESIS, as PB is known to have excellent compatibility with tackifier resins in HMPSAs.25 The influence of the degree of epoxidation, degree of PB grafting on the properties of the SIS-PB copolymers as well as the performance of the corresponding HMPSAs were examined systematically.
2. Experimental
2.1. Materials
SIS copolymers (YH-1209, Mn ∼ 9 × 104 g mol−1, diblock content < 1%, styrene content ≈ 29 wt%) were obtained from the Sinopec Baling Petroleum & Chemical Co., Ltd. Formic acid (AR grade, 88 vol%) and hydrogen peroxide (AR grade, 30 vol%) were used as received. Toluene, tetrahydrofuran and cyclohexane were refluxed over calcium hydride and distilled under argon atmosphere. n-BuLi (J&K Scientific Ltd.) was analysed by the double-titration method with 1,2-dibromoethane. 1,3-butadiene (Sinopec Beijing Yanshan Petrochemical Corp) was treated with minor n-BuLi to remove the residual water and inhibitor. C5 resin (C-100R, Eastman Chemical Company, USA) was used as a tackifier. A plasticizer (KN-4010) and antioxidant Irganox 1010 were purchased from Kelamayi refinery and Beijing Jiyi chemical limited company, respectively.2.2. In situ epoxidation of SIS copolymers
ESIS was prepared by an in situ epoxidation procedure. Typically, 5.0 g SIS copolymer was first dissolved into 100 g toluene in a 250 ml three-necked glass flask at room temperature. At 4 °C (ice bath), a designed amount of formic acid and hydrogen peroxide (with equivalent mole to the isoprene units in SIS) was successively added into the system, forming performic acid (an oxidizing agent). Then, the reaction occurred at a stirring speed of 300 rpm for a designed reaction time. ESIS was precipitated in ethanol, washed with distilled water for three times till pH neutral, and dried at 60 °C in a vacuum oven till constant weight. In the following discussions, the copolymers were referred to as ESISx, where x represents the epoxidation degree (ED, defined as the mole percentage of epoxidized C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds among all the CC bonds within the PI blocks of the parent SIS copolymer). Specifically, the ED is controlled by their epoxidation time and estimated by eqn (1).
C bonds among all the CC bonds within the PI blocks of the parent SIS copolymer). Specifically, the ED is controlled by their epoxidation time and estimated by eqn (1).
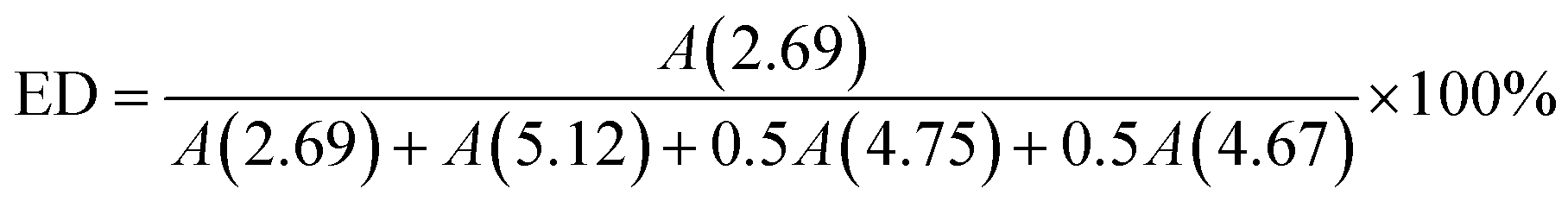 | (1) |
2.3. Synthesis of SIS-g-PB copolymers
SIS-g-PB copolymers were synthesized by grafting living PB lithium macroanions onto the ESIS copolymers. First, given amounts of butadiene and cyclohexane were transferred into a 300 ml glass reactor through stainless steel capillaries. After being initiated by a stoichiometric amount (according to a designed molecular weight of PB branches) of n-BuLi, living PB lithium macroanions was polymerized at 50 °C for 4 h. Subsequently, the reactor was injected with a 5 wt% ESIS/cyclohexane solution and stirred at 50 °C for 8 h. By design, the molar ratio of PB lithium macroanions to the epoxide groups in ESIS ranged between 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 and 1:20. The graft reaction was terminated by a small amount of degassed ethanol and the product was precipitated in an excess amount of ethanol. Finally, ethanol and tetrahydrofuran were utilized to carefully remove the unreacted PB homopolymer.26
:5 and 1:20. The graft reaction was terminated by a small amount of degassed ethanol and the product was precipitated in an excess amount of ethanol. Finally, ethanol and tetrahydrofuran were utilized to carefully remove the unreacted PB homopolymer.26
2.4. Characterization of SIS-g-PB copolymers
1H-NMR spectrum was recorded on a Varian INOVA 400 MHz spectrometer in CDCl3 at ambient temperature. The number molecular weight (Mn), molecular weight distribution (PDI) and intrinsic viscosity were measured by gel permeation chromatograph (GPC) consisting of a Viscotek TDA-302 (Viscotek Co., USA), equipped with tetra detectors [refractive index (RI), UV, viscosity (VISC), and two-angle laser light scattering]. THF was used as eluent at a flow rate of 1.0 ml min−1 at 30 °C, and polystyrene standards were used to calibrate the instrument.For all samples, 10.0 ± 0.1 mg was used in differential scanning calorimetry (DSC, NETZSCH DSC 204 instrument, Germany) measurements. They were first scanned from room temperature to 150 °C, kept for 5 min. Then, they were cooled to −120 °C with a cooling rate of 10 °C min−1 in N2 stream and kept for 5 min. Finally, they were reheated to 150 °C with a heating rate of 10 °C min−1.
A rotational rheometer (AR2000ex, TA Instrument, U.S.) was used to measure the rheological property of SIS-g-PB copolymers. SIS-g-PB copolymers (∼0.5 g) were compressed by a thermal press into discs with a diameter of ∼25 mm and a thickness of ∼1 mm at 170 °C. The rheological measurements were conducted under 170 °C and nitrogen atmosphere, with a frequency range of 0.01 Hz–100 Hz and a strain level of 1%, to ensure that all samples were measured within the linear viscoelastic response region.
2.5. Preparation and characterization of HMPSAs
In a N2 atmosphere, the copolymers were melt-blended with a tackifier, a plasticizer (at a weight ratio: 3:5:2) and an antioxidant (0.5 wt%) to prepare HMPSAs. First, all the components were slowly mixed at 120 °C in an internal mixer until the tackifier was fully melted. Then, the processing temperature was gradually elevated to 170 °C. After being stirred at a rate of 500 rpm for 30 min, the mixture was placed on PET (polyethylene terephthalate) films (backing layer) to prepare the HMPSA specimens with an average thickness of 100 μm using a TX2003-1 film applicator at 130 °C. Finally, these specimens were covered with another PET film (release liner) and were pressed with a 2 kg rubber roller for three times and stored at room temperature for 48 h prior to the adhesive performances tests. In practice, the adhesive performance mainly includes 180° peel strength and holding power. The former is the force required to remove the adhesive from the substrate surface under standard conditions. The latter is characterized by the break time of the adhesive held under a defined shear load. Their specimens were cut into strips with 25 mm × 100 mm dimensions for the measurement of 180° peel strength, tested with a BLD-S electronic all-powerful stripping machine (Labthink Instruments Co., Ltd., China) at a peeling rate of 300 mm min−1 in accordance with GB/2792-1998. The average value of 180° peel strength for the sample was determined for at least three times. Their specimens were cut into strips with 25 mm × 70 mm dimensions for the measurement of their holding power, tested with a CZY-6 holding adhesive testing instrument (Labthink Instruments Co., Ltd., China). They were tested under a load of 1 kg at room temperature for three times and the reported data was their average. Prior to the above measurements, these strips were pressed onto a clean stainless steel substrate with a 2 kg rubber roller (moved backward and forward for three times) and left to stand for over 20 min at 23 ± 2 °C.
3. Results and discussion
3.1. Fabrication of ESIS copolymers and their HMPSAs
A range of ESIS copolymers were synthesized with varying epoxidation time. Fig. 1 displays the 1H-NMR spectra of the copolymers, from which the ED of the ESIS copolymers was calculated to be 5, 11, 15 and 20 according to eqn (1). The typical signals of epoxide groups appear at 2.69 ppm in the 1H-NMR spectra and become stronger at higher ED.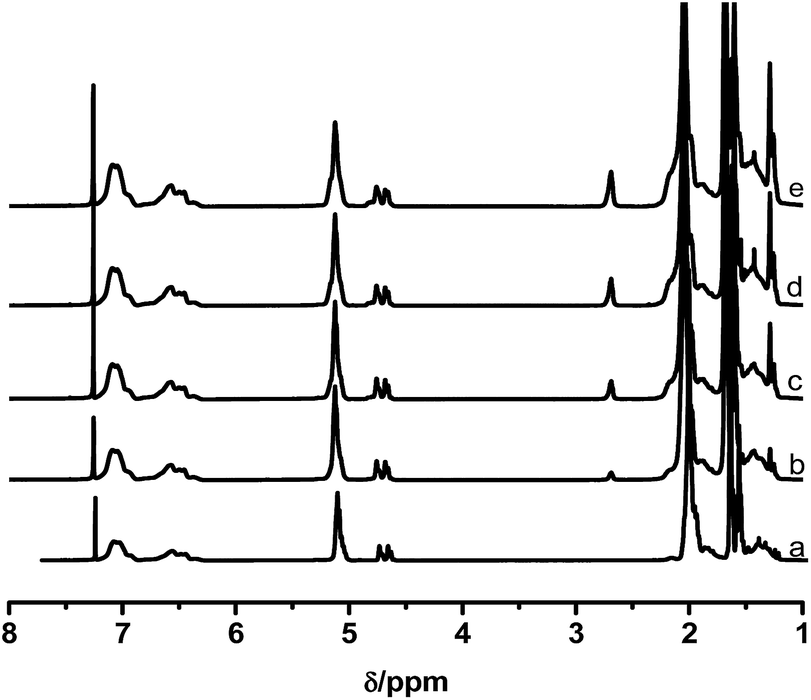 | ||
| Fig. 1 1H-NMR spectra of (a) parent SIS, (b) ESIS5, (c) ESIS11, (d) ESIS15, and (e) ESIS20. | ||
These ESISx and parent SIS copolymers were used to fabricate HMPSAs, referred to as H-ESISx and HSIS in the following discussions, respectively. The adhesion performances of all the HMPSAs are summarized in Table 1. Although the holding power remained rather constant within the range of experimental uncertainty, the 180° peel strength showed strong dependency on the epoxidation degree. As the ED exceeds 10%, the corresponding HMPSAs displayed very poor adhesion performance because the compatibility between ESIS and tackifier resins reduced significantly by the excess epoxide groups.23 Since ESIS15 copolymers had sufficient amounts of epoxide groups and correspondingly H-ESIS15 showed only slightly lower adhesion performance compared with SIS-based HMPSA, they were chosen as the main precursors to synthesize graft copolymers. These synthesized branch copolymers were then used to investigate the effect of the branch structures on the adhesion performance of the corresponding HMPSAs in the following paragraphs.
| Adhesion performance | H-SIS | H-ESIS5 | H-ESIS11 | H-ESIS15 | H-ESIS20 |
|---|---|---|---|---|---|
| Holding power (day) | >7 | >7 | >7 | >7 | >7 |
| 180° peel strength (kN m−1) | 0.09 ± 0.01 | 0.11 ± 0.02 | 0.09 ± 0.01 | 0.07 ± 0.02 | 0.02 ± 0.03 |
3.2. Synthesis of SIS-g-PB copolymers
It is known that the properties of grafted polymers show significant dependency on the branching, only when the molecular weight of branched chains is well above the critical molecular weight for entanglements. In this regard, the entanglement behavior of the grafted polymers will play an important role at improving the adhesion performance of HMPSAs, since they are the matrix materials. 1.7 kg mol−1 is the critical molecular weight for entanglements of PB chains.1 Thus, PB branches with Mn of 1.9, 3.1 and 4.2 kg mol−1 were synthesized (referred to as PB1.9, PB3.1 and PB4.2) in this work. Prior to being grafted onto ESIS15, a small amount of living PB lithium macroanions were extracted from the reactor and terminated by degassed ethanol. Fig. 2 shows the GPC traces of the PB obtained. Fig. 3 displays the 1H-NMR spectra with chemical shift peaks centered at 5.57, 4.91–4.87 and 5.32–5.37 ppm, corresponding to the olefinic protons of 1,2-butadiene units (–CH), 1,2-butadiene (CH2) units and 1,4-butadiene (–CHCH–) units, respectively. By comparing the peak areas, the content of 1,4 BD and 1,2 BD can be quantified. The chemical composition of the PB branches estimated from 1H-NMR, as well as their molecular weight and PDI estimated from GPC are summarized in Table 2.
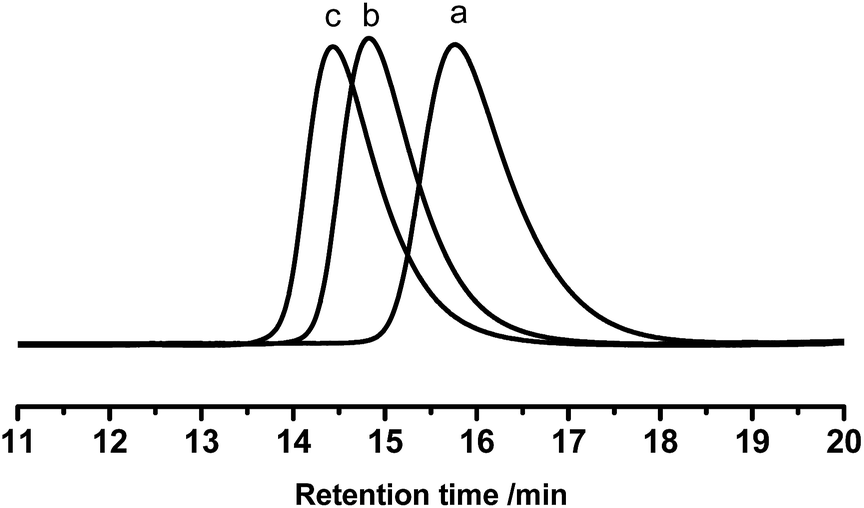 | ||
| Fig. 2 GPC traces of (a) PB1.9, (b) PB3.1, and (c) PB4.2. | ||
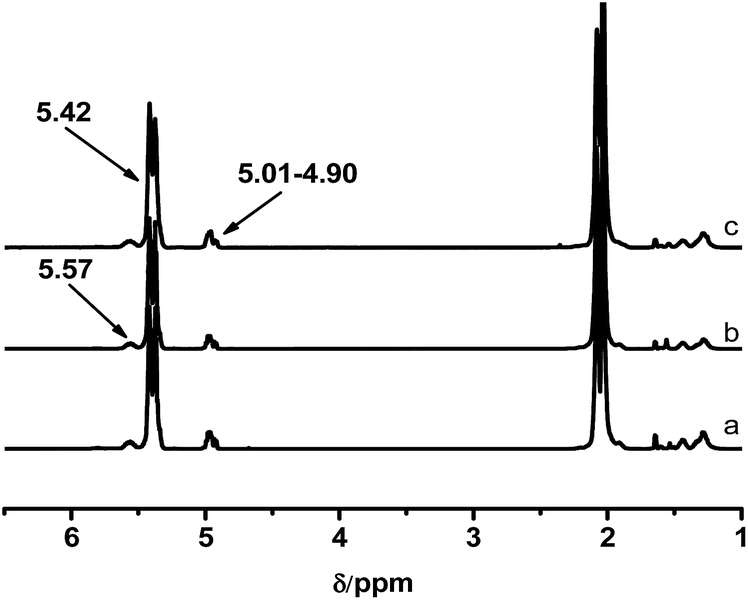 | ||
| Fig. 3 1H-NMR spectra of (a) PB1.9, (b) PB3.1, and (c) PB4.2. | ||
These PB chains were grafted onto ESIS15 to synthesize a range of SISx-yg-PBz copolymers, where x, y and z represent their ED, branch number and branch length, respectively. SIS15-10g-PB3.1 is taken for example to demonstrate the evolution of their molecular structures, measured by GPC (Fig. 4) and 1H-NMR (Fig. 5). Prior to the purification, the as-synthesized products were a mixture of free PB3.1 and SIS15-10g-PB3.1, evidenced by the two elution peaks in the GPC curve (curve b in Fig. 4). After the purification process, the elution peak corresponding to the free PB3.1 disappeared (curve c in Fig. 4). Thus, SIS15-10g-PB3.1 is successfully synthesized without linear PB3.1 chains. In the 1H-NMR spectrum (curve 3 in Fig. 5), there appear typical peaks of PB and SIS, which confirms the successful synthesis of SIS15-10g-PB3.1. Moreover, the peak of epoxide groups at 2.69 ppm is still very strong, showing that most functional groups were remained in the SIS15-10g-PB3.1.
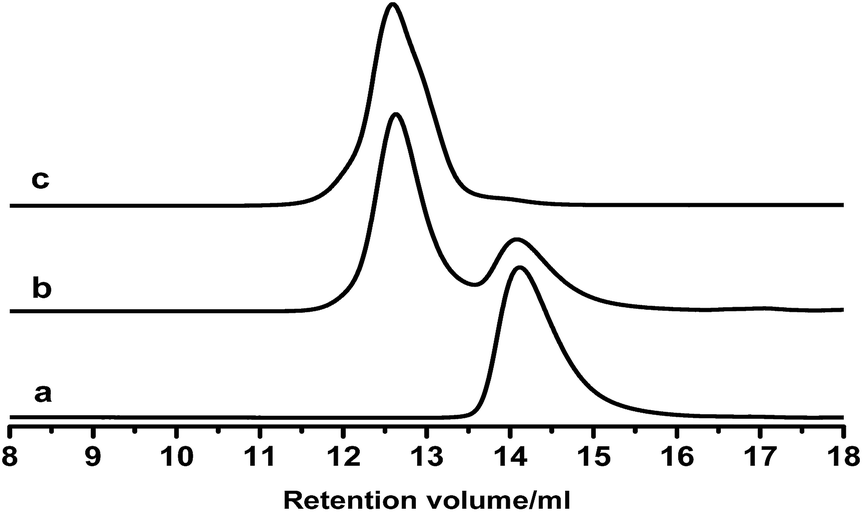 | ||
| Fig. 4 GPC traces of (a) PB3.1, (b) mixture of SIS15-10g-PB3.1 and PB3.1, and (c) SIS15-10g-PB3.1. | ||
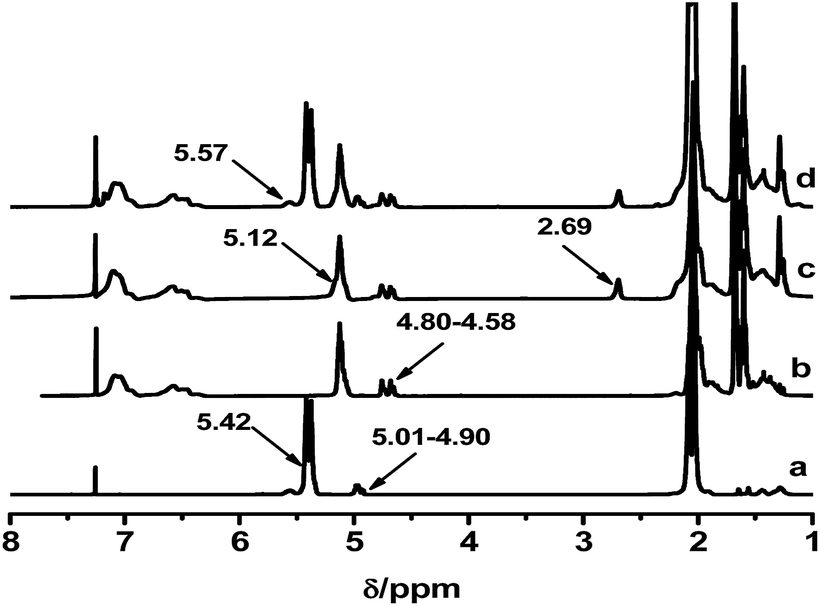 | ||
| Fig. 5 1H-NMR spectra of (a) PB3.1, (b) SIS, (c) ESIS15, and (d) SIS15-10g-PB3.1. | ||
Similarly, other SISx-yg-PBz copolymers with well-defined branch lengths and branch numbers were synthesized with various living PB lithium macroanions under designed molar ratios of PB lithium macroanions to epoxide groups. Table 3 lists their molecular structures based on the GPC results of SISx-yg-PBz, SIS, ESIS and PB branches. Among these copolymers, two series of SIS-g-PB copolymers were chosen to investigate the effects of their branch number (SIS15-8g-PB1.9, SIS15-10g-PB1.9 and SIS15-14g-PB1.9) and branch length (SIS15-10g-PB1.9, SIS15-10g-PB3.1 and SIS15-11g-PB4.2) on their thermodynamic properties and corresponding HMPSAs performances.
| Sample | Mn (kg mol−1) | St (wt%) | Bd (wt%) | Nb | PDI | [η] (dl g−1) |
|---|---|---|---|---|---|---|
| SIS | 90 | 29 | 0.0 | 0 | 1.06 | 1.170 |
| SIS15 | 92 | 31.5 | 1.190 | |||
| SIS15-14g-PB1.9 | 116 | 22.5 | 22.4 | 14 | 1.07 | 1.424 |
| SIS15-10g-PB1.9 | 109 | 23.9 | 21.1 | 10 | 1.08 | 1.411 |
| SIS15-8g-PB1.9 | 103 | 25.3 | 12.6 | 7 | 1.08 | 1.410 |
| SIS15-19g-PB3.1 | 150 | 17.4 | 40.0 | 19 | 1.07 | 1.511 |
| SIS15-12g-PB3.1 | 127 | 20.6 | 29.1 | 12 | 1.06 | 1.449 |
| SIS15-10g-PB3.1 | 121 | 21.6 | 25.6 | 10 | 1.07 | 1.425 |
| SIS15-11g-PB4.2 | 136 | 19.2 | 33.8 | 11 | 1.06 | 1.525 |
| SIS15-8g-PB4.2 | 125 | 20.9 | 28.0 | 8 | 1.07 | 1.510 |
| SIS15-5g-PB4.2 | 113 | 23.1 | 25.6 | 5 | 1.09 | 1.503 |
3.3. Physical and adhesive properties of SIS-g-PB copolymers
As SIS copolymers are epoxidized, the functional groups will increase the glass transition temperature (Tg) of PI blocks. As shown in Fig. 6, the Tg of the PI blocks increases from −57 °C to −47 °C, as the ED increases to ∼15. As a result, the working temperature window of ESIS-based HMPSAs becomes narrower (The Tg of middle PI blocks is the lower limit temperature of HMPSAs' application).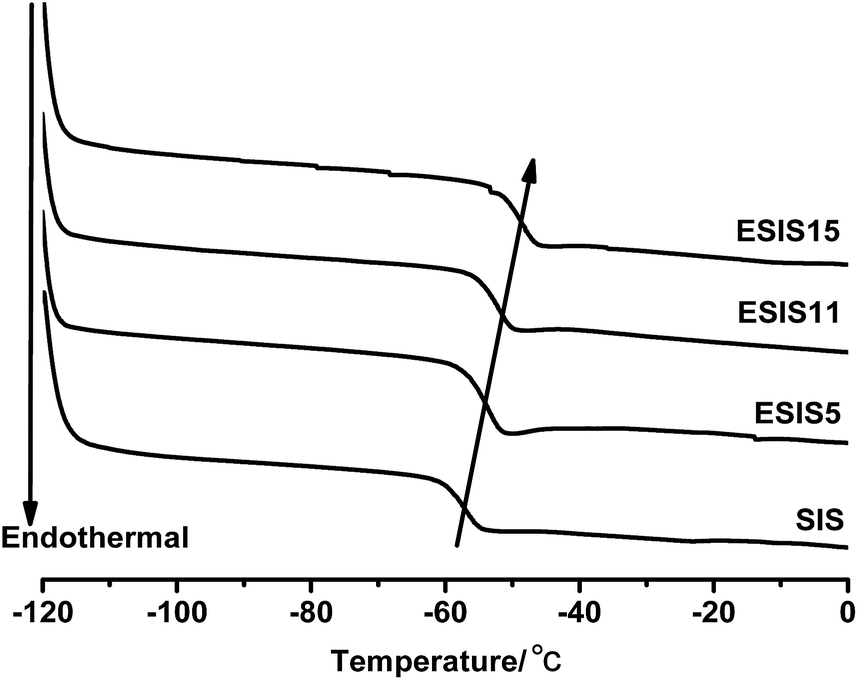 | ||
| Fig. 6 DSC curves of SIS, ESIS5, ESIS11 and ESIS15. | ||
On the other hand, once grafted onto the ESIS chains, PB blocks, which has a lower Tg than the PI blocks, could restore or even improve the low temperature property of ESIS-based HMPSAs. Fig. 7 displays the DSC curves of PB branches (PB1.9, PB3.1 and PB4.2), showing that the Tg value increases with the molecular weight from −103.4 °C for PB1.9 to −99.0 °C for PB4.2. These PB chains all have much lower Tg values than the SIS copolymers (−56 °C) and ESIS15 (−47.2 °C).
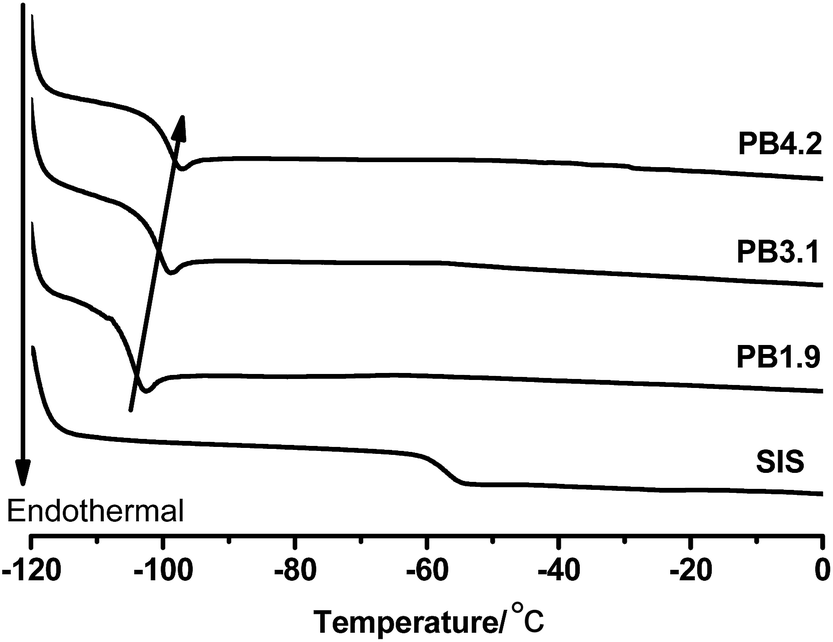 | ||
| Fig. 7 DSC curves of SIS, PB1.9, PB3.1 and PB4.2. | ||
Fig. 8 displays the DSC curves of a series of SIS15-yg-PB1.9 copolymers, showing that there is no distinctive glass transition of PB blocks. Instead, all three branched copolymers showed a single glass transition, with Tg values lower than the ESIS15. As the branch numbers increases from 8 to 14, the Tg value drops monotonically from −53.4 °C for SI5-8g-PB1.9 to −60.3 °C for SIS15-14g-PB1.9, which is even lower than that (−56 °C) of the parent SIS copolymers. Fig. 9 shows the DSC measurements of the copolymer series with constant branch number, but varying branch length. In this case, as the branch length increased from 1.9, 3.1 to 4.2 kg mol−1, the Tg value decreases from −55.0 for SIS15-10g-PB1.9, −70.7 for SIS15-10g-PB3.1 to −71.7 °C for SIS15-11g-PB4.2, which is significantly lower than that (−56 °C) of the parent SIS copolymers. Above experiments suggest that PB blocks are compatible with the PI blocks in the grafted copolymers. As a result, the mixing of PB blocks with the PI blocks effectively reduces the overall Tg values of the copolymers. With increasing number and lengths of the PB branches, the effective concentration of the PB increases, which leads to more reduction in Tg values. Such a property can be utilized to modify the low temperature property of graft copolymers and their corresponding HMPSA.
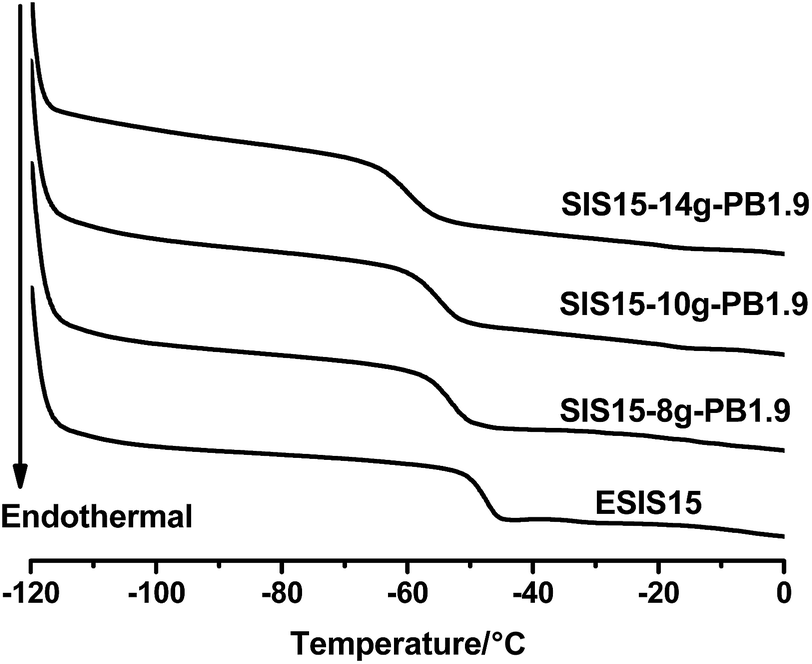 | ||
| Fig. 8 DSC curves of ESIS15, SI5-8g-PB1.9, SIS15-10g-PB1.9 and SIS15-14g-PB1.9. | ||
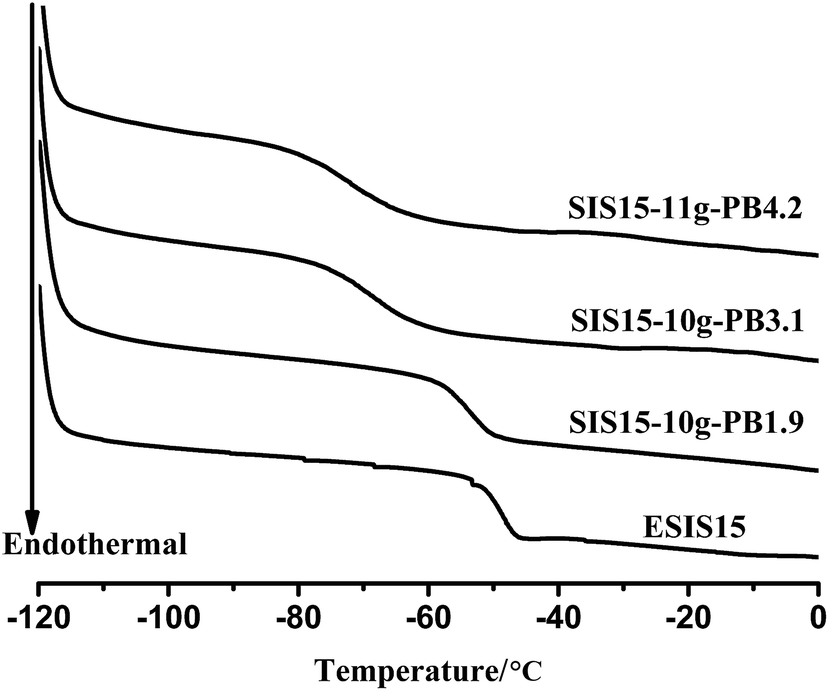 | ||
| Fig. 9 DSC curves of ESIS15, SIS15-10g-PB1.9, SIS15-10g-PB3.1 and SIS15-11g-PB4.2. | ||
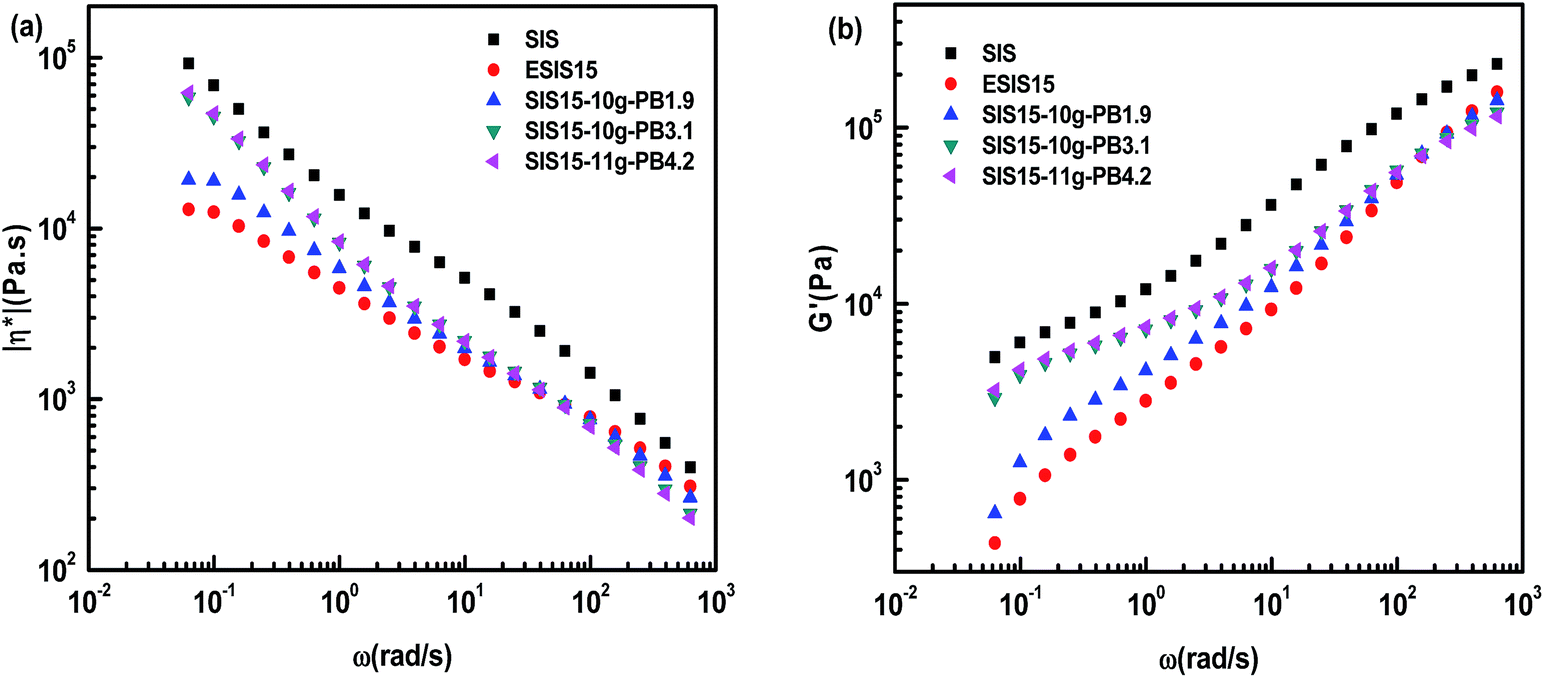
We further investigated the influences of branch numbers and branch lengths on the rheological properties of the copolymers. Fig. 10 and 11 show the complex viscosity (η*), defined in eqn (2) and storage modulus (G′) versus angular frequency (ω) at 170 °C for SIS, ESIS and various SIS-g-PB copolymers.
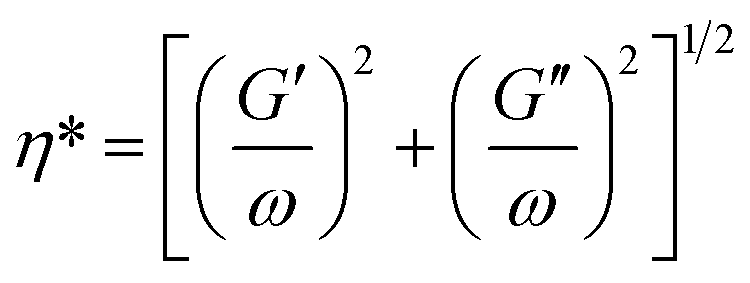 | (2) |
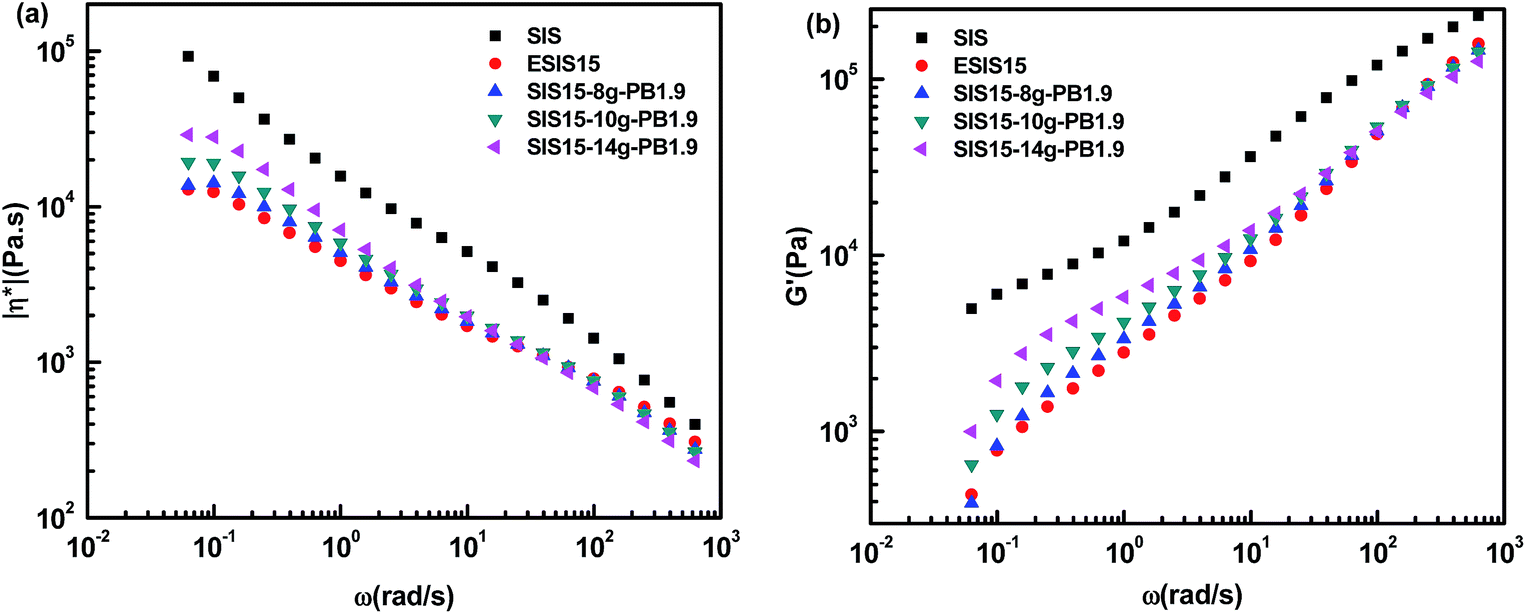 | ||
| Fig. 10 (a) η* vs. ω and (b) G′ vs. ω for SIS, ESIS15, SIS5-8g-PB1.9, SIS15-10g-PB1.9 and SIS15-14g-PB1.9 at 170 °C. | ||
 | ||
| Fig. 11 (a) η* vs. ω and (b) G′ vs. ω for SIS, ESIS15, SIS15-10g-PB1.9, SIS15-10g-PB3.1 and SIS15-11g-PB4.2 at 170 °C. | ||
Even though ESIS15 copolymer has similar molecular structures as the SIS copolymer, they display significantly different rheological behavior. In comparison, the former has much lower η* and G′ than the latter throughout the experimental shear frequency range. In the low terminal shear frequency regime, ESIS15 copolymers have a platform region of η* independent of the shear frequency and accompanying even lower G′. As PB branches are grafted onto ESIS backbones, the platform region of η* still exists for SI5-8g-PB1.9, SIS15-10g-PB1.9 and SIS15-14g-PB1.9. Besides, their η* and G′ increase in the lower shear frequency regime (<101 rad s−1) but decrease in the higher shear frequency regime (>101 rad s−1) with the branches numbers of PB for SI5-8g-PB1.9, SIS15-10g-PB1.9 and SIS15-14g-PB1.9. But, they are still lower than those of parent SIS copolymers throughout the experimental frequency range.
Fig. 11 illustrates the influence of the branches length on the η* and G′ of SIS-g-PB copolymers. For SIS15-10g-PB1.9, SIS15-10g-PB3.1 and SIS15-11g-PB4.2, they also have lower η* and G′ than the parent SIS copolymers throughout the experimental shear frequency range. A similar trend takes place. Their η* and G′ increase with the branches length in a low shear frequency regime (<101 rad s−1) but decrease with the branches length in the high shear frequency regime (>101 rad s−1). Their outstanding difference from SIS15-8g-PB1.9, SIS15-10g-PB1.9 and SIS15-14g-PB1.9 lies in the low terminal shear frequency regime. Clearly, the platform region in the low terminal shear frequency regime disappears for SIS15-10g-PB3.1 and SIS15-11g-PB4.2. Both their η* and G′ have a definite improvement, close to those of parent SIS copolymers. Their complex viscosity curves become higher leading to more prominent shear-thinning phenomenon. Considering the fact that their physical crosslinking sites of PS phase disappear at 170 °C, their rheological behavior could be interpreted as the typical character of polymers with long chain branching.27 For SIS15-8g-PB1.9, SIS15-10g-PB1.9 and SIS15-14g-PB1.9, their branches length is very close to the critical molecular weight for entanglements of PB chains so that they have no the typical character of polymers with long chain branching. The results also show that the branches length has greater effects on the rheology of SIS-g-PB copolymers than the branches number in the experimental range.
In Fig. 12, these SIS-g-PB copolymers are analyzed by their van Gurp–Palmen (vGP) plots, in which the loss angle, δ (tanδ = G′′/G′), is plotted as a function of the absolute value of complex modulus (|G*|). The vGP plot is a useful tool for getting insight into the molecular structure of long chain branched polymers.27 It has been pointed out that materials are almost completely viscous when the δ terminal value is close to 90° and almost completely elastic when the δ terminal value is close to 0°. The δ terminal value of parent SIS copolymers is very low (<30°). By comparison, ESIS15 copolymers have much higher δ terminal value (close to 60°), demonstrating that the elasticity of parent SIS copolymers is significantly reduced due to the introduction of epoxide groups. SIS15-8g-PB1.9, SIS15-10g-PB1.9 and SIS15-14g-PB1.9 have similar δ terminal value with ESIS15, demonstrating that PB1.9 is not long enough to dramatically influence the rheological properties and the effect of branch number can be ignored in these copolymers. As their branch length increases in SIS15-10g-PB3.1 and SIS15-11g-PB4.2, their similar δ terminal value dramatically decreases, approaching that of SIS copolymers. Obviously, the elasticity of SIS-g-PB copolymers can be restored only if the branch length is longer enough than the critical molecular weight for entanglements of PB chains.
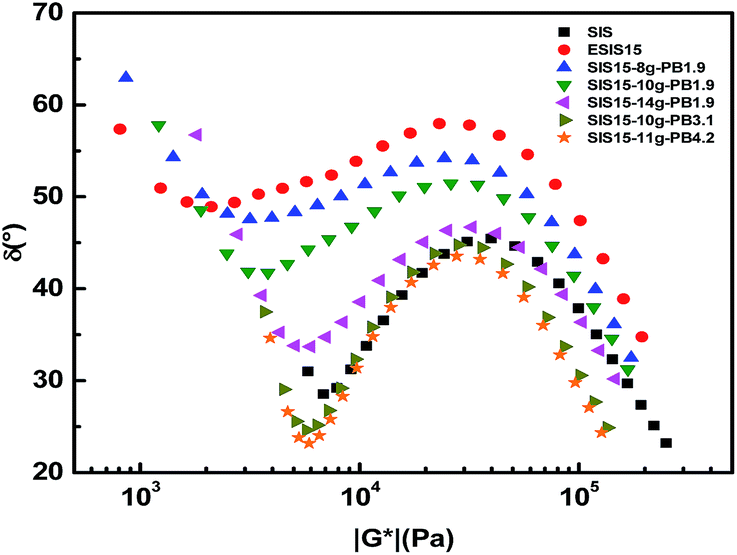 | ||
| Fig. 12 Van Gurp–Palmen plots of SIS, ESIS15 and various SIS-g-PB copolymers at 170 °C. | ||
Two series of SIS-g-PB copolymers were chosen to fabricate various HMPSAs in order to investigate the effects of the branch number (SIS15-8g-PB1.9, SIS15-10g-PB1.9 and SIS15-14g-PB1.9) and branch length (SIS15-10g-PB1.9, SIS15-10g-PB3.1 and SIS15-11g-PB4.2) on the HMPSAs performances. These HMPSAs are named as H-SISx-yg-PBz, where SISx-yg-PBz represents the corresponding SIS-g-PB copolymers. The adhesion performances were summarized in Table 4, mainly including holding power and 180° peel strength measurement results. The holding power of the grafted copolymers was similar with that of H-SIS and H-ESIS15, and showed no obvious change with the topological structures of SIS-g-PB copolymers in the experimental range (7 days). However, their 180° peel strength is prominently influenced by the branch number and branch length of SIS-g-PB copolymers.
| Adhesion performance | H-SIS15-8g-PB1.9 | H-SIS15-10g-PB1.9 | H-SIS15-14g-PB1.9 | H-SIS15-10g-PB3.1 | H-SIS15-11g-PB4.2 |
|---|---|---|---|---|---|
| Holding power (day) | >7 | >7 | >7 | >7 | >7 |
| 180° peel strength (kN m−1) | 0.15 ± 0.02 | 0.21 ± 0.01 | 0.23 ± 0.02 | 0.20 ± 0.01 | 0.18 ± 0.03 |
In comparison, their 180° peel strength is much higher than the SIS-based HMPSAs in practical applications (Table 1). To elaborate the mechanism underlying the enhanced adhesion performance, SIS15-10g-PB1.9 is taken as an example for analysis. From DSC measurements (Fig. 13), C5 resin is completely compatible with the PI blocks of SIS copolymers so that the glass transition of PI markedly shifts to higher temperature. In the case of ESIS15/C5 blends, the Tg of PI slightly shifts to higher temperature. It is clear that the compatibility between SIS and C5 resin is negatively impacted by the introduction of epoxide groups, which results in the lower 180° peel strength of H-SIS15. As discussed in Fig. 8, SIS15-10g-PB1.9 has a low Tg value, due to the PB branches, which notably shifts to much higher temperature in the blend of SIS15-10g-PB1.9 and C5 resin. To some degree, the long PB branches restore the compatibility between the rubber phase of copolymers and the C5 resin. As a result, H-SIS15-10g-PB1.9 displayed higher 180° peel strength than H-SIS.
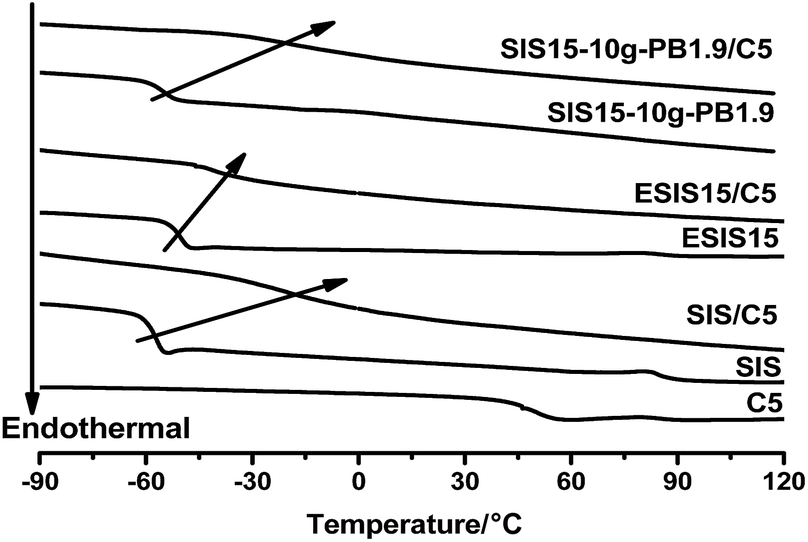 | ||
| Fig. 13 DSC curves of SIS, ESIS15, SIS, SIS15-10g-PB1.9, and their blends with C5 resin (1:1). | ||
In Table 4, the 180° peel strength increases with the branch number of copolymers for H-SIS15-8g-PB1.9, H-SIS15-10g-PB1.9 and H-SIS15-14g-PB1.9, but slightly changes with the branch length of copolymers for H-SIS15-10g-PB1.9, H-SIS15-10g-PB3.1 and H-SIS15-11g-PB4.2. These copolymers all have excellent compatibility with C5 resin just like the blend of SIS15-10g-PB1.9 and C5 resin (see ESI Fig. S1 and 2†). Thus, the SIS-g-PB/C5 compatibility cannot be directly used to explain the different adhesive performance of these HMPSAs, different from H-SIS and H-ESIS15. It is well-known that the branch chains usually enhance the bulk properties of branched copolymers by increasing the entanglement of the chains. This does not agree with the trend of 180° peel strength for H-SIS15-10g-PB1.9, H-SIS15-10g-PB3.1 and H-SIS15-11g-PB4.2. As described in the experimental section, these HMPSAs contain about 70 wt% of a tackifier and a plasticizer. These SIS-g-PB copolymers can “dissolve” into the “solid solution” of the tackifier and the plasticizer through the excellent compatibility between the rubber phases and C5 resins, in which the entanglement of PB branches can be ignored even at the longer branch length. These SIS-g-PB copolymers still work as skeletons in their HMPSAs through the entanglement of polystyrene blocks. As a result, their adhesive performances increase with the branch number because the epoxide groups can be covered by the corresponding branches. As the increase of their branch length has no effect on the epoxide groups, the 180° peel strength has nearly no change for H-SIS15-10g-PB1.9, H-SIS15-10g-PB3.1 and H-SIS15-11g-PB4.2.
4. Conclusions
A series of SIS-g-PB copolymers with well-defined branch number and branch lengths were successfully synthesized by the “graft onto” method. Living anionic polymerization was used to control their branch lengths and their branch number is controlled by the molar ratios of living PB lithium macroanions to epoxide groups. These copolymers were utilized to formulate HMPSAs. Systematical experiments were performed to investigate the relationships between their molecular structures, bulk properties, and the adhesive performances of the corresponding HMPSAs.The results demonstrated that SIS-g-PB copolymer based HMPSAs displayed much higher adhesive performances than SIS and ESIS based HMPSAs. The enhancement was attributed to the improved combability between the ESIS and the matrix resin of the HMPSAs. Furthermore, for SIS-g-PB copolymers with constant PB concentration, the branch numbers appeared to have a greater impact than the branch lengths on the performance of the HMPSAs. The method reported here offers a new approach to improve both the polarity and adhesive performance of the SIS-based HMPSAs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support from the National Natural Science Foundation of China for the projects (51073029, 51233005 and 51673034) and the financial support of Open Research Fund of State Key Laboratory of Polymer Physics and Chemistry (Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, No. 110000R088)References
- I. Benedek and M. M. Feldstein, Technology of pressure-sensitive adhesives and products, CRC Press, Boca Raton, FL, 2009 Search PubMed.
- S. A. da Silva, C. L. Marques and N. S. M. Cardozo, J. Adhes., 2012, 88, 187–199 CrossRef.
- D. J. Kim, H. J. Kim and G. H. Yoon, J. Appl. Polym. Sci., 2006, 100, 825–831 CrossRef CAS.
- C. Y. Wu, G. Z. Wu and C. F. Wu, J. Appl. Polym. Sci., 2006, 102, 4157–4164 CrossRef CAS.
- M. N. Cazenave, C. Derail, F. Leonardi, G. Marin and N. Kappes, J. Adhes., 2005, 81, 623–643 CrossRef CAS.
- C. Derail, M. N. Cazenave, F. X. Gibert, G. Marin, N. Kappes and J. Lechat, J. Adhes., 2004, 80, 1131–1151 CrossRef CAS.
- F. X. Gibert, G. Marin, C. Derail, A. Allal and J. Lechat, J. Adhes., 2003, 79, 825–852 CrossRef CAS.
- S. Akiyama, Y. Kobori, A. Sugisaki, T. Koyama and I. Akiba, Polymer, 2000, 41, 4021–4027 CrossRef CAS.
- N. A. Lynd, F. T. Oyerokun, D. L. O'Donoghue, D. L. Handlin and G. H. Fredrickson, Macromolecules, 2010, 43, 3479–3486 CrossRef CAS.
- D. J. Kim, H. J. Kim and G. H. Yoon, J. Adhes. Sci. Technol., 2004, 18, 1783–1797 CrossRef CAS.
- D. J. Kim, H. J. Kim and G. H. Yoon, Int. J. Adhes. Adhes., 2005, 25, 288–295 CrossRef CAS.
- D. J. Kim, H. J. Kim and G. H. Yoon, J. Adhes. Sci. Technol., 2006, 20, 1367–1381 CrossRef CAS.
- D. H. Lim, H. S. Do and H. J. Kim, J. Appl. Polym. Sci., 2006, 102, 2839–2846 CrossRef CAS.
- M. Sasaki, Y. Nakamura, K. Fujita, Y. Kinugawa, T. Iida and Y. Urahama, J. Adhes. Sci. Technol., 2005, 19, 1445–1457 CrossRef CAS.
- J. F. Ma, C. X. Wang, H. F. Luo, Z. Z. Zhu, Y. B. Wu and H. Wang, J. Pharm. Sci., 2013, 102, 2221–2234 CrossRef CAS PubMed.
- C. X. Wang, W. Han, X. Z. Tang and H. Zhang, AAPS PharmSciTech, 2012, 13, 556–567 CrossRef CAS PubMed.
- C. X. Wang, R. Liu, X. Z. Tang and W. Han, AAPS PharmSciTech, 2012, 13, 1179–1189 CrossRef CAS PubMed.
- C. X. Wang, J. F. Ma, R. Liu, W. Han and X. Z. Tang, Drug Dev. Ind. Pharm., 2014, 40, 211–221 CrossRef CAS PubMed.
- E. P. O'Brien, L. T. Germinario, G. R. Robe, T. Williams, D. G. Atkins, D. A. Moroney and M. A. Peters, J. Adhes. Sci. Technol., 2007, 21, 637–661 CrossRef.
- L. L. Hua, Y. Li, Q. Wang, Y. N. Hu and Z. F. Zhao, J. Adhes. Sci. Technol., 2012, 26, 1109–1122 CAS.
- Z. F. Zhao, Y. S. Zhou, C. Q. Zhang and Z. S. Li, Int. J. Adhes. Adhes., 2016, 68, 256–262 CrossRef CAS.
- Z. F. Zhao, R. J. Zhang, C. Q. Zhang and Q. Wang, Int. J. Adhes. Adhes., 2017, 74, 86–91 CrossRef CAS.
- Z. F. Zhao, B. Fang, X. H. Li and Q. Wang, J. Adhes. Sci. Technol., 2013, 27, 143–153 CrossRef CAS.
- Q. Wang, Y. Z. Wang, Z. F. Zhao and B. Fang, Int. J. Adhes. Adhes., 2012, 34, 62–67 CrossRef.
- X. H. Deng, J. Adhes., 2016, 12, 49–57 Search PubMed.
- D. M. Knauss and T. Z. Huang, Macromolecules, 2003, 36, 6036–6042 CrossRef CAS.
- L. Wang, D. Wan, Z. J. Zhang, F. Liu, H. P. Xing, Y. H. Wang and T. Tang, Macromolecules, 2011, 44, 4167–4179 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08180d |
| This journal is © The Royal Society of Chemistry 2017 |