DOI:
10.1039/C7RA08172C
(Paper)
RSC Adv., 2017,
7, 43030-43039
Phenol and Cr(VI) degradation with Mn ion doped ZnO under visible light photocatalysis†
Received
24th July 2017
, Accepted 23rd August 2017
First published on 5th September 2017
Abstract
Mn ion doped ZnO with different percentages of Mn content (Zn0.9Mn0.1O (1), Zn0.8Mn0.2O (2), Zn0.7Mn0.3O (3), and Zn0.6Mn0.4O (4)) was synthesized via a solution combustion method, with urea used as the fuel. The optical, morphological, and structural properties were studied using Raman, UV-DRS, SEM, TEM, XPS, and powder XRD techniques. The average crystallite sizes of Zn1−xMnxO (1, 2, 3, 4), which are around 30–60 nm, were confirmed via powder X-ray diffraction studies, whereas transmission electron microscopy studies confirmed the formation of a ZnO wurtzite crystal phase. Scanning electron microscopy indicated the spherical morphology of the samples. Raman spectroscopy studies confirmed a decrease in oxygen vacancies with increasing Mn content, whereas confirmation of the doping of Mn ions into the ZnO lattice was obtained using X-ray photoelectron spectroscopy. The band gap energies of samples were calculated using UV-DRS spectroscopy, whereas BET surface area measurements confirmed the surface area. The visible light activity of Zn1−xMnxO (1, 2, 3, 4) was identified through studies of phenol degradation and Cr(VI) reduction under visible light photocatalysis, which highlight that Zn0.8Mn0.2O (2) shows the best activity. Typical degradation profiles indicated that the simultaneous degradation of pollutants is more effective than the removal of individual pollutants.
1. Introduction
Removing highly toxic materials/pollutants from industrial wastewater and pure water supplies has received much attention, because of their highly toxic and endocrine-disrupting properties.1 Photocatalytic degradation is one of the options that scientists have been working on, as this route of degradation utilizes natural resources.2–4 Phenolic compounds, polychlorinated biphenyls and heavy metals, like Cr(VI), Hg(II), Pd(II), Pt(IV), etc., are some of the highly toxic pollutants, whose existence in water would cause serious health/environmental effects. Industries like petroleum refining, automobile manufacturing, film making, leather tanning, and wood processing, and industrial wastewater, etc. are major contributors.5,6 Among the different treatments for removing such pollutants from wastewater, the use of photocatalytic technology under visible light is the most important known technique, and it has the ability to mineralize the greatest number of toxic organic pollutants using radiant energy. In general, semiconductor photocatalysts are mainly useful materials for treating a number of different water pollutants, and they also play an important role in environmental purification because of their simplicity, the gentle reaction conditions needed, and their low energy consumption. Several metal oxides, like TiO2, ZnO, and Fe2O3, have been tested in photocatalytic degradation processes. Most of the research in this direction has been focused on the removal of individual pollutants, whereas actual effluent is highly diverse in nature. Hence, the simultaneous elimination of both organic and inorganic pollutants is of interest and is essential in the treatment of water for further use in the current climate.7–10 Dong et al. suggested a Bi2WO6/RGO nanocomposite for the removal of organic pollutants. His group observed a much lower amount of phenol degradation (39.68%) after 8 h under sunlight irradiation. In this method the irradiation time was very high.11 Wan et al. reported a Bi12GeO20/g-C3N4 photocatalyst for Cr(VI) reduction; only 50% reduction was observed after 3 h. The main disadvantage is that the percentage reduction was very low.12 Diao et al. proposed an Fe/TiO2 nanocomposite for Cr(VI) reduction with the use of UV light.13 Dingze Lu et al. reported phenol oxidation and Cr(VI) reduction simultaneously, with the use of terbium oxide nanoparticles loaded on TiO2.14 Mani et al. also reported the simultaneous degradation of such pollutants using C and N doped TiO2.15 Zhao et al. reported the simultaneous removal of phenol and Cr(VI) by using Y2O3/TiO2 in the presence of UV light within 3 h. The disadvantage of these experiments was that the removal percentage of phenol and Cr(VI) was very low (70–80%).16 In general, ZnO is a semiconductor and an excellent raw material in the glass, ceramic, textile, and cosmetic industries. Its band gap of 3.37 eV makes it active in the UV region. Yet ZnO has desirable attractiveness because of its non-toxicity and low cost. Furthermore, under UV light irradiation, ZnO shows excellent activity toward organic pollutant degradation in aqueous suspensions through photocatalysis and produces photo-generated strong oxidizing agents, like hydroxyl radicals (OH˙) and superoxide radical anions (O2−˙). To shift the absorption into the visible region, ZnO has been doped with metals and non-metals, and coupled with small conductors, like CdS, etc.17 When compared to these methods, combustion synthesis has certain advantages, like ease of operation, one step doping, etc. Several reports deal with the synthesis of Mn ion doped ZnO via different methods.18–20 For example, Yildirimcan et al. reported Mn ion doped ZnO synthesized via a facile chemical method and the photocatalytic activity was tested by degrading Orange-G dye (C16H10N2O7S2Na2).20 Barzgari et al. synthesized Mn/ZnO, which was also prepared via a precipitation method, and the photocatalytic activity was tested through the degradation of cresols21 Bordbar et al. reported Mn doped ZnO synthesized via a hydrothermal method, and the photocatalytic activity was tested via methyl orange degradation and 78% degradation was observed within 3 h.22 Achouri et al. proposed a Mn doped ZnO photocatalyst via a solvothermal method, and observed 75% orange II dye degradation within 240 min under visible light.23
To the best of our knowledge, no reports are available in the literature on the synthesis of Mn ion doped ZnO via a combustion method and its application towards the removal of pollutants (phenol and Cr(VI)), individually and simultaneously, using visible light.
The present manuscript deals with the preparation of Mn doped ZnO and photocatalytic activity estimations under visible light for the individual/simultaneous degradation of phenols and Cr(VI) in waste water. In the presence of visible light, the photocatalytic activities of the synthesized photocatalysts 1, 2, 3, and 4 were tested through phenol degradation and Cr(VI) reduction, and the results were compared with those for ZnO.
2. Experimental section
2.1. Materials
Zinc nitrate (Zn(NO3)2·6H2O) (Sigma-Aldrich), manganese nitrate (Mn(NO3)2·4H2O) (Sigma-Aldrich), urea (Sigma-Aldrich), phenol (Sigma-Aldrich), and potassium dichromate (K2Cr2O7) were used without any further purification. Millipore water was employed for all experiments.
2.2. Synthesis
ZnO, Zn0.9Mn0.1O (1), Zn0.8Mn0.2O (2), Zn0.7Mn0.3O (3), and Zn0.6Mn0.4O (4) were synthesized via a combustion method with different weight ratios of Zn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mn ions (w/w%) (0.9:0.1, 0.8:0.2, 0.7:0.3, and 0.6:0.4). The required amounts of zinc nitrate (w%), manganese nitrate (w%) and urea (0.90 g) were mixed in 15 mL of Millipore water in a china dish. This solution was preheated for initial dehydration by using a hot plate at 120 °C. After that, the heated compound was transferred to a muffle furnace and heated at 450 °C for 10 minutes. Colorless and brown colored compounds can be obtained, which correspond to ZnO and Mn/ZnO samples; the color of the sample increases upon increasing the Mn2+ content.
:Mn ions (w/w%) (0.9:0.1, 0.8:0.2, 0.7:0.3, and 0.6:0.4). The required amounts of zinc nitrate (w%), manganese nitrate (w%) and urea (0.90 g) were mixed in 15 mL of Millipore water in a china dish. This solution was preheated for initial dehydration by using a hot plate at 120 °C. After that, the heated compound was transferred to a muffle furnace and heated at 450 °C for 10 minutes. Colorless and brown colored compounds can be obtained, which correspond to ZnO and Mn/ZnO samples; the color of the sample increases upon increasing the Mn2+ content.
2.3. Characterization
Phase identification and the characterization of the crystallite sizes of undoped ZnO and the samples Zn0.9Mn0.1O (1), Zn0.8Mn0.2O (2), Zn0.7Mn0.3O (3), and Zn0.6Mn0.4O (4) are carried out using a PANalytical X'pert Pro Powder X-ray diffractometer with Cu-Kα radiation, with a wavelength λ = 1.54 Å, and Ni is used as a filter. The applied current and accelerating voltage are maintained at 30 mA h and 40 kV respectively during the measurements. The optical properties of the prepared samples can be characterized using a UV-visible diffuse reflectance spectrophotometer (Shimadzu-3600), with BaSO4 as a standard reference. Raman spectra of undoped ZnO and all samples were collected using a Bruker Senterra dispersive Raman microscope with a 532 nm laser excitation wavelength. The surface areas of the ZnO samples were measured using N2 physio-sorption studies conducted at liquid nitrogen temperature. According to BET analysis, compound 2 shows the highest surface area (54.035 m2 g−1) compared to the other materials (1, 3, and 4). The morphologies of the prepared samples were investigated using SEM (FESEM-FEI Quanta 400, the Netherlands) at a 20 kV accelerating voltage, and the magnifications of the instrument were 1000–5000. The samples are placed on a copper grid with an applied operating voltage of 200 kV, and images are recorded using a TEM instrument (TECNAI G-2). The chemical or electronic states of each element on the surface of the Mn doped ZnO nanoparticles are characterized via X-ray photoelectron spectroscopy (XPS) analysis, carried out using a Kratos analytical spectrophotometer, with Mg Kα monochromatic excited radiation of 1253.6 eV; the residual pressure in the analysis chamber is around 10−9 mbar.
2.4. Photocatalytic experiments
The photocatalytic activities of the synthesized catalysts are tested using a specially designed reactor, and three non-halogen lamps (250 W, 24 V) are arranged inside the reactor with a light intensity of 750–800 W m−2; the intensity is tested using power meter. Individual and combined experiments for phenol degradation and Cr(VI) reduction in an aqueous medium are carried out with pure ZnO and 1, 2, 3, and 4 under visible light photocatalysis. To reach adsorption–desorption equilibrium, an aqueous solution of pollutants with a particular amount of catalyst is taken in a 100 mL beaker and placed under dark conditions for 30 minutes. No phenol degradation or Cr(VI) reduction was observed in the absence of light and without catalyst in aqueous solution. Under light exposure, 1–2 mL of sample was collected using 5 mL syringes every 30 minutes; that was centrifuged and the catalyst was finally separated through filtering with 40 μm Millipore filter paper. A UV-visible spectrophotometer was used to estimate the concentration of phenol and Cr(VI).
Before spectrophotometric analysis, phenol was converted into a brownish red antipyrine dye complex, with the addition of 1 mL of buffer (pH = 9), 1 mL of 0.05 M 4-aminoantipyrine and 1 mL of a 0.05 M aqueous solution of potassium ferricyanide, and it was examined at a wavelength at 504 nm. The purple colour of the Cr(VI) complex was recorded at 540 nm, and it was prepared via the addition of an acidic solution (10% H2SO4) of Cr(VI) and was also converted into a purple complex through the addition of 1,5-diphenyl hydrazide (1 mL) solution. The efficiency of the photocatalytic degradation of phenol and Cr(VI) aqueous solutions was calculated using the following formula:
where
C0 is the initial concentration,
C is the concentration after visible light irradiation, and
η is the efficiency of photocatalytic degradation.
15
3. Results and discussion
3.1. X-ray diffraction
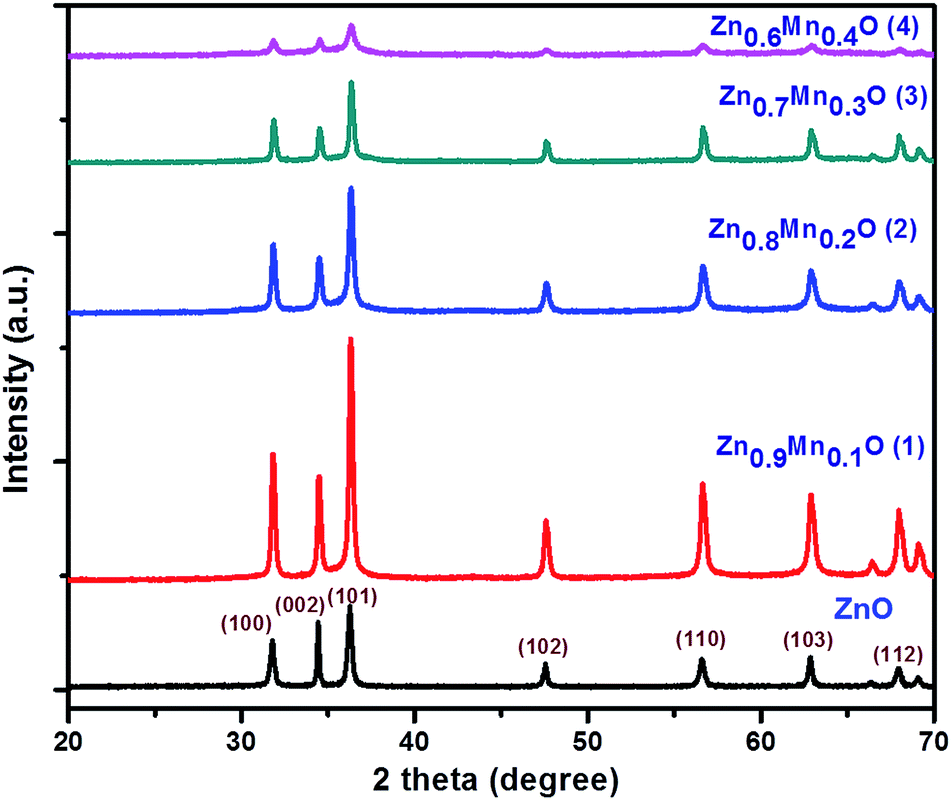
Powder XRD patterns of undoped ZnO and Mn doped ZnO nanoparticles (1–4) are shown in Fig. 1. All diffraction peaks in the graph confirm the formation of a ZnO hexagonal wurtzite structure and show the space group P63mc with the lattice parameters a = 3.249 Å and c = 5.206 Å (JCPDS No. 361451). The diffraction peaks observed at 2θ values of 31.77°, 34.42°, 36.25°, 47.53°, 56.60°, 62.86°, 66.38°, and 67.96° correspond to the (100), (002), (101), (102), (110), (103), and (112) crystalline planes, respectively. From Fig. 1, it is revealed that there is no secondary phase observed and the synthesized material has a single phase. The XRD results also explain that the Mn2+ ions replace the Zn2+ ions in the ZnO lattice, without changing the wurtzite structure of ZnO. The high intensity peak at (101) is slightly shifted to the lower angle side, as well as there being a lowering of the intensity. The unit cell parameters and d-spacing values, evaluated as a function of Mn content, are shown in Table 1.24,25 From Table 1, the d-spacing values decrease with an increase in the Mn content in the ZnO lattice. The average crystallite sizes of ZnO and the 1, 2, 3, and 4 samples were determined using the Debye Scherrer formula.
| D = Kλ/βcosθ |
where D = the size of the crystallite, K = the shape factor, λ = wavelength, θ = diffraction angle, and β = full width at half maximum. The average crystallite sizes of ZnO and the 1, 2, 3, and 4 samples are around 55, 51, 50, 34, and 33 nm, respectively, confirming a decrease in the crystallite size upon increasing the Mn concentration.
 |
| | Fig. 1 Powder XRD spectra for ZnO and Mn doped ZnO (1–4). | |
Table 1 The unit cell parameters and d-spacing values for ZnO and nanomaterials 1, 2, 3, and 4
| Sample |
a (Å) |
c (Å) |
d-Spacing (Å) |
| ZnO |
3.249 |
5.206 |
2.4756 |
| Mn0.1Zn0.9O (1) |
3.255 |
5.212 |
2.4742 |
| Mn0.2Zn0.8O (2) |
3.270 |
5.236 |
2.4732 |
| Mn0.3Zn0.7O (3) |
3.277 |
5.243 |
2.4720 |
| Mn0.4Zn0.6O (4) |
3.283 |
5.253 |
2.4713 |
3.2. Raman spectroscopy
The crystalline nature of ZnO (wurtzite) and 1, 2, 3, and 4 was further confirmed via Raman spectroscopy analysis. As seen in Fig. 2, the peaks centered at 503 cm−1, 562 cm−1, 580 cm−1 and 665 cm−1 were related to the E2, A1, E1 and E2 (high) vibration modes of ZnO, with P63mc symmetry, respectively. The peaks between 1000 cm−1 and 1200 cm−1 correspond to multiphonon processes. XRD and Raman analysis clearly indicates that ZnO and 1, 2, 3, and 4 nanoparticles were produced, with a hexagonal ZnO crystal structure and good crystal quality.26
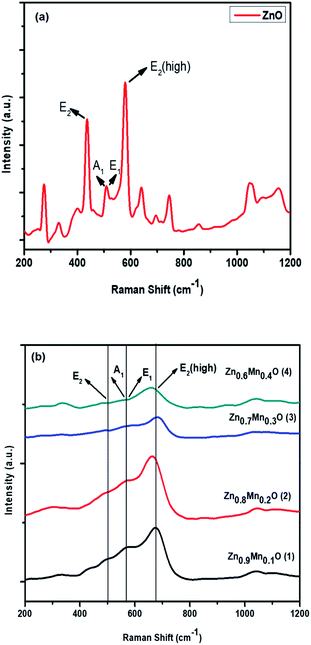 |
| | Fig. 2 Raman spectra of (a) ZnO and (b) Mn doped ZnO (1–4) nanomaterials. | |
3.3. UV-visible spectroscopy (DRS) analysis
The optical properties of ZnO and samples 1–4 are further confirmed using UV-visible diffuse reflectance spectroscopy, and the absorption peaks are presented in Fig. 3. The absorption peak corresponding to ZnO is found at 316 nm, with a band gap of 3.9 eV, and the peaks corresponding to samples 1–4 are shifted to the higher wavelength side (red shifted) upon increasing the Mn content from 0.1 to 0.4%. All peaks corresponding to the ZnO and Mn–ZnO (1–4) samples are identified in the UV region and the decrement in band gap energies is also very small. The band gap energies of ZnO and samples 1–4 are 3.9 eV, and 3.8 eV, 3.7 eV, 3.5 eV, and 3.4 eV, respectively. The slight decrease in the band gap values for 1, 2, 3 and 4, compared to ZnO, may be due to different optically active sublevels being located through the band gap.
 |
| | Fig. 3 UV-vis-DRS spectra for ZnO and materials 1–4. | |
3.4. Scanning electron microscopy analysis
The influence of different percentages of Mn doping on the morphology of ZnO (1–4 samples) was investigated via scanning electron microscopy. SEM images of sample 2 show a well-ordered morphology with spherical nanoparticles, low aggregation and better particle size distribution.27 This can be seen in Fig. 4.
 |
| | Fig. 4 SEM image of Zn0.8Mn0.2O (2). | |
3.5. Transmission electron microscopy analysis
A TEM image of sample 2 (Fig. 5(a)) shows that the nanoparticles have a spherical morphology, as long as the size is below 100 nm. The grain size of sample 2 is 15–20 nm. Fig. 5(b) shows the selected area electron diffraction (SAED) pattern of sample 2, which complements the observations from XRD, and confirms the wurtzite structure of ZnO.28,29
 |
| | Fig. 5 (a) A TEM image of sample 2. (b) The selected area electron diffraction pattern for sample 2. | |
3.6. X-ray photoelectron spectroscopy analysis
The elemental composition and chemical states of sample 2 were measured via XPS analysis. The C 1s peak (284.5 eV) is used as a reference peak for calibrating the binding energies of the remaining peaks. Fig. 6(b) represents the peaks corresponding to Mn 2p for sample 2. From the peaks indicated at 642.26 eV for Mn 2p3/2 and at 654.3 eV for Mn 2p1/2, it is clearly indicated that the Mn ions in sample 2 are in a +2 oxidation state. We observe that the peaks corresponding to Mn 2p are very weak, which clearly indicates a very low concentration of manganese doping. The peak appearing at 642.26 eV for Mn 2p3/2 is associated with Mn2O3 and MnO phases. In Fig. 6(b), the binding energies are 640.7 eV and 641.8 eV for MnO and Mn2O3, respectively. The binding energies of Zn 2p3/2 and Zn 2p1/2 are 1022.13 eV and 1045.1 eV, respectively, and these are presented in Fig. 6(a). These two peaks are strong and have narrow line widths, highlighting that Zn2+ ions are predominant in the nanostructure of sample 2. A broad peak at 530 eV, which indicates the binding energy of the O 1s region, is shown in Fig. 6(c). The full scan survey spectrum does not show any other elemental peaks, which suggests that sample 2 does not have any other impurities in it; this is also explained through XRD analysis.30
 |
| | Fig. 6 (a) XPS spectrum of Zn 2p, (b) XPS spectrum of Mn 2p, and (c) XPS spectrum of O 1s for sample 2. | |
3.7. Photocatalytic activity tests
3.7.1. Photocatalytic degradation of phenol. The activities of ZnO and photocatalysts 1, 2, 3, and 4 were estimated from the degradation of phenol using visible light analysis. As shown in Fig. 7, first order kinetics were used to plot the photocatalytic oxidation of phenol under visible light, and the first order rate constants were found to be 0.00529, 0.00588, 0.00644, 0.00695, and 0.00732 S−1 respectively for ZnO, Zn0.6Mn0.4O (4), Zn0.7Mn0.3O (3), Zn0.9Mn0.1O (1), and Zn0.8Mn0.2O (2). From this, it can be concluded that the rate constant increases for materials 1–4 compared with pure ZnO. Zn0.8Mn0.2O (2) has the highest photocatalytic activity compared to other materials, such as 1, 3 and 4. From Fig. S1† it is confirmed that 79% degradation was observed within 210 min after using photocatalyst 2, and 66%, 71%, 74%, and 77% were observed after using ZnO, 4, 3 and 1 respectively. The percentage of mineralization can be calculated using TOC analysis and measured using a Shimadzu TOC analyzer. The percentages of mineralization for ZnO and the Zn1−xMnxO (1–4) samples are 72%, 76%, 83%, 79%, and 74% respectively.
 |
| | Fig. 7 The first order kinetics of nanoparticles of 1–4: irradiation time = 210 min, catalyst amount = 40 mg L−1, and phenol concentration = 20 ppm. | |
3.7.2. The effect of concentration on photocatalytic phenol oxidation. The concentration of a pollutant plays a main role in organic pollutant photodegradation. The influence of phenol on photocatalytic degradation at four different concentrations (20, 30, 40 and 50 mg L−1) was explained and kinetic plots are shown in Fig. 8. At higher concentration levels of phenol, the degree of degradation becomes low, since a greater number of molecules are adsorbed on the surface of the photocatalyst, which prevents visible light from being absorbed by the catalyst. As a result, the amount of OH˙ and O2−˙ forming on the surface of the catalyst decreases. The OH˙ generation is lower, and the number of phenol molecules reacting with OH˙ decreases, so the degradation efficiency decreases. In every case, the amount of catalyst (Zn0.8Mn0.2O), irradiation time (210 min) and intensity of light was kept constant. Photocatalytic degradation decreases upon increasing the concentration of phenol, and this can be seen in Fig. S2.†
 |
| | Fig. 8 First order kinetics plots showing the effect of the concentration of phenol for photocatalyst 2: irradiation time = 210 min, and catalyst amount = 40 mg L−1. | |
3.7.3. Effect of catalyst amount on photocatalytic phenol oxidation. The catalyst amount greatly influences organic pollutant (phenol) degradation. As expected, with an increase in the concentration of catalyst from 30 mg to 60 mg L−1, the rate constants also increase, as shown in Fig. 9. This can be explained because, as the number of photons on the surface of the catalyst increases with an increase in the catalyst amount, there is an increase in the available active sites on the catalyst surface, and finally a greater number of phenol molecules are also adsorbed on the surface. But there was no considerable increase in phenol degradation when the catalyst concentration was increased to more than 60 mg L−1. The screening effect of excess catalyst creates a shield, and consequently hinders light penetration, causing the loss of surface area for light harvesting and a reduction in catalytic activity. The optimal dosage of Mn ion doped ZnO was determined to be 60 mg L−1. The percentages of degradation also increase with an increase in the amount of catalyst 2, and results are presented in Fig. S3.†
 |
| | Fig. 9 Kinetic plots showing the effect of catalyst amount on the photocatalytic activity of ZnO:Mn (0.2%): irradiation time = 210 min, and phenol concentration = 30 ppm. | |
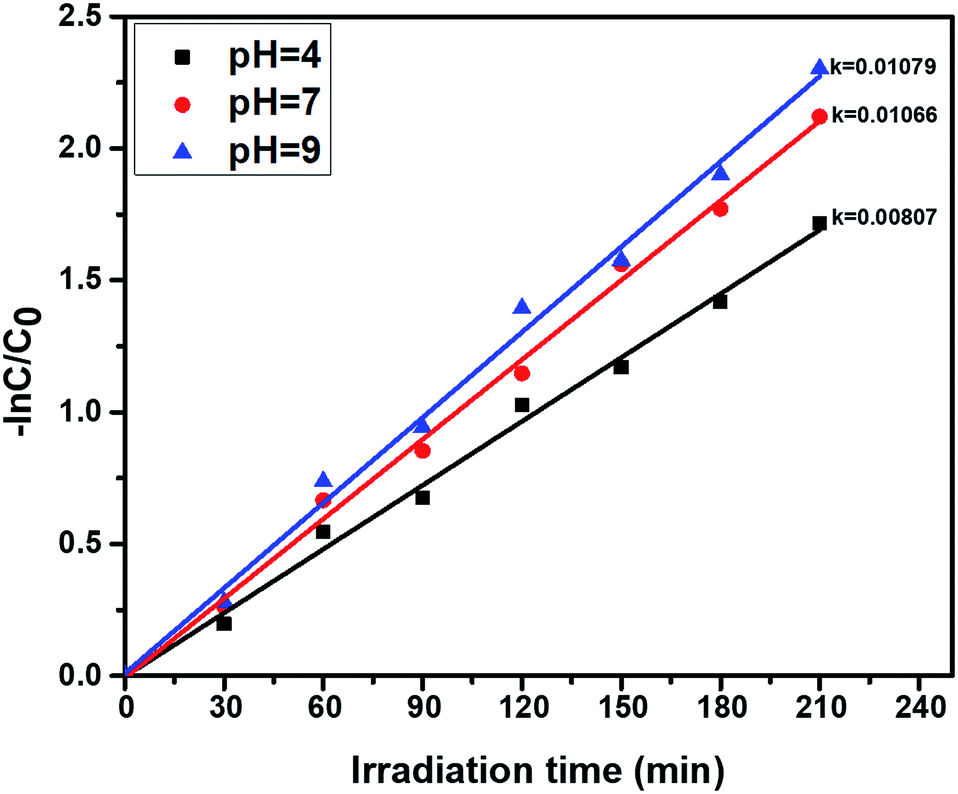
3.7.4. pH effect on photocatalytic phenol oxidation. The pH greatly influences the photocatalytic activity, due to the surface chemistry of the catalyst being changed. The effect of pH on photocatalytic phenol oxidation is shown in Fig. 10. A phenol aqueous solution with an acidic or basic pH range favours the formation of hydroxyl radicals, and the hydroxyl radical is one of the oxidants during the photocatalytic experiments. Under basic conditions (pH = 9), the OH− ion concentration will be greater, so a number of OH radicals will be formed, and the efficiency of photocatalytic degradation of phenol will be greater. In the oxidation of phenol, OH˙ radicals play a vital role during the course of degradation reactions, so, under basic conditions, better degradation efficiency was observed when compared to neutral (pH = 7) and acidic conditions (pH = 4). The percentage of degradation is also higher at pH = 9, and this is shown in Fig. S4.†
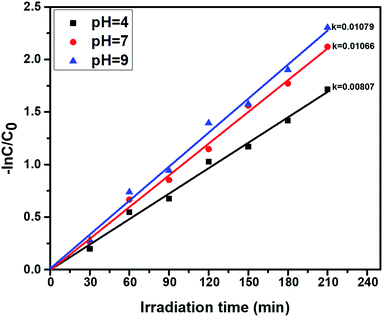 |
| | Fig. 10 First order kinetics showing the effect of the pH of phenol solutions, using the photocatalyst ZnO:Mn (0.2%): irradiation time = 210 min, catalyst amount = 40 mg L−1, and phenol concentration = 30 ppm. | |
3.7.5. Cr(VI) reduction under photocatalysis. The activities of the photocatalysts ZnO and nanomaterials 1, 2, 3, 4 were tested for Cr(VI) reduction using visible light, and the results are shown in Fig. 11, which reveals the first order reduction behavior. The first order kinetics rate constants were found to be 0.0047, 0.0050, 0.0051, 0.0059, and 0.0068 min−1 respectively for ZnO, 1, 2, 3, and 4. During the reduction process, 0.2% Mn/ZnO (2) shows higher activity compared to the other catalysts. It is confirmed from Fig. 11 that 72% degradation was achieved within 180 minutes and the percentages of degradation using pure ZnO and catalysts 1, 3 and 4 are 58%, 60%, 63%, and 66% respectively, and this is shown in Fig. S5,† under the same experimental conditions.
 |
| | Fig. 11 Kinetic plots for pure ZnO and nanomaterials 1–4 for the reduction of Cr(VI) in aqueous solution: catalyst amount = 40 mg L−1, Cr(VI) concentration = 30 ppm, and irradiation time = 180 min. | |
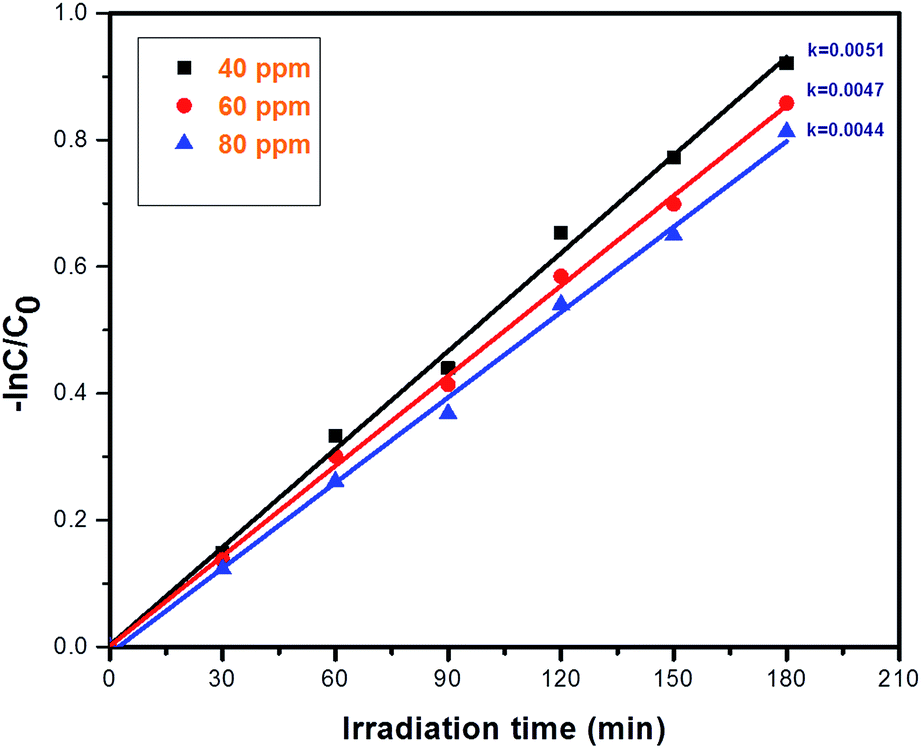
3.7.6. Concentration effect on the reduction of Cr(VI) via photocatalysis. The photo-reduction of Cr(VI) using photocatalyst 2 was investigated and, upon changing the concentration of Cr(VI) from 40 to 80 ppm, the rate constant values are increased; the results are shown in Fig. 12. If the concentration of Cr(VI) is increased, then the reduction efficiency towards Cr(VI) gradually decreases; this is because as the concentration of Cr(VI) is increased, the number of Cr(VI) molecules on the surface of photocatalyst 2 also increases, which affects the light absorption intensity and further reduces the efficiency towards Cr(VI). The kinetics of the photocatalytic reduction of Cr(VI) were investigated, which confirm first order reduction behavior. After 180 minutes, the degradation percentage drops to 80% at a concentration of 40 ppm, the percentage drops to 67% at a concentration of 60 ppm, and finally it drops to 55% at a concentration of 80 ppm, and this can be seen in Fig. S6.†
 |
| | Fig. 12 First order kinetics plots showing the effect of concentration on Cr(VI) reduction using photocatalyst 2: irradiation time = 180 min, and catalyst amount = 40 mg L−1. | |
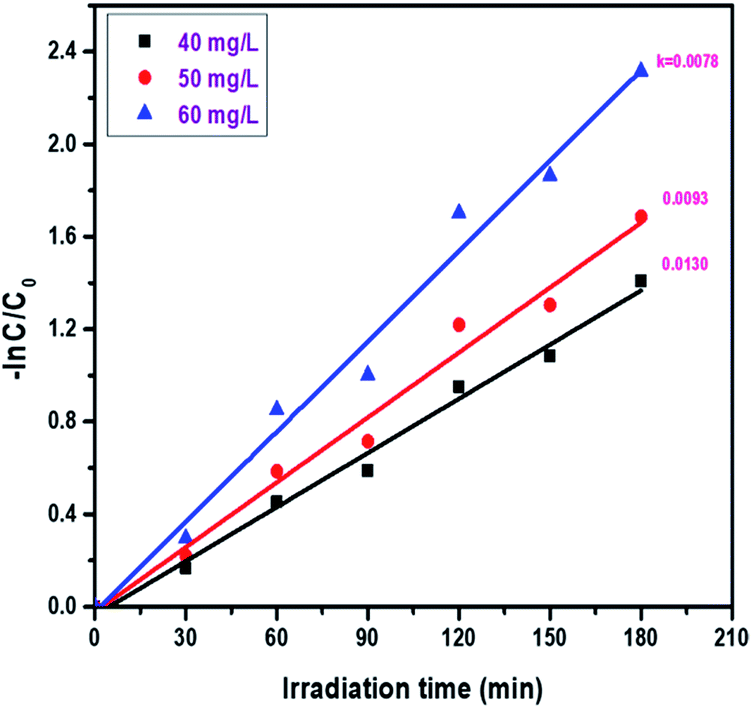
3.7.7. Catalyst effect on the reduction of Cr(VI) via photocatalysis. The effect of catalyst amount on the reduction of hexavalent chromium in aqueous solution was studied. During the present study, the catalyst amount was varied from 40 mg L−1 to 60 mg L−1. As shown in Fig. 13, the rate constant increases from 0.0078 to 0.0100 min−1 upon increasing the concentration from 40 to 60 mg L−1. The optimal dosage of Mn ion doped ZnO was determined to be 60 mg L−1. As the catalyst amount varies from 40 mg L−1 to 60 mg L−1, the percentage of reduction also increases from 75% to 90% and the results are displayed in Fig. S7.†
 |
| | Fig. 13 Kinetic plots of the effect of catalyst amount on the photocatalytic activity of ZnO:Mn (0.2%): irradiation time = 180 min, and concentration of Cr(VI) = 40 ppm. | |
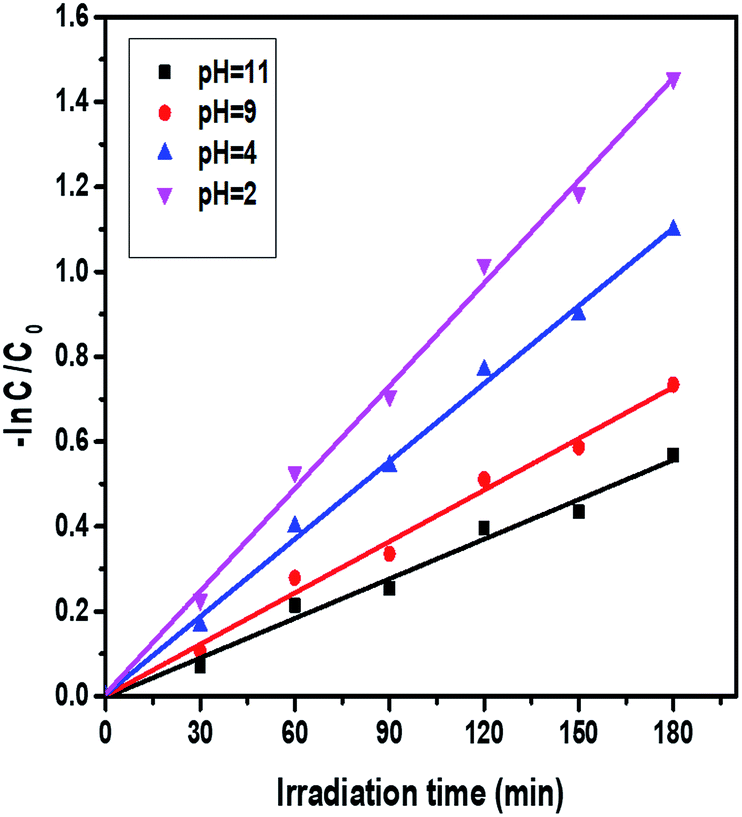
3.7.8. pH effect on the reduction of Cr(VI) via photocatalysis. The effect of pH on the photoreduction of Cr(VI) can be investigated by varying the pH range as the initial concentration (40 ppm) and catalyst amount (40 mg L−1) are kept constant. In this experiment, the photocatalytic reduction of Cr(VI) was observed at different pH values, like 2, 4, 9, and 11. The Cr(VI) reduction efficiency and rate constant values decrease upon increasing the pH from 2 to 11, and the results are shown in Fig. 14. Interestingly, the degree of reduction was greater in acidic solutions than in alkaline solutions. At pH = 2, the reduction rate was 77%, but at pH = 11, the reduction rate was only 43%, and the corresponding results are displayed in Fig. S8.† The conversion of Cr(VI) to Cr(III) consumes a greater number of protons in acidic solution and produces hydroxyls in alkaline solution. As the pH increases, the rate of the photoreduction of dichromate ions gradually decreases, since the higher pH causes a lowering of the adsorption of Cr(VI) ions onto the surface of the photocatalyst. In the presence of an alkaline pH, Cr(OH)3 covers the catalyst surface and the photocatalytic efficiency of Cr(VI) is also reduced. The kinetic data also supports this observation.
 |
| | Fig. 14 Kinetic plots of the effect of the pH of Cr(VI) solutions on the photocatalytic activity of ZnO:Mn (0.2%): irradiation time = 180 minutes, catalyst amount = 40 mg L−1, and concentration of Cr(VI) = 40 ppm. | |
3.7.9. Photocatalytic removal of Cr(VI) and phenol simultaneously. As seen in the above studies, photocatalyst 2 shows better activity compared to others (1, 3, and 4), and it was used for the reduction and oxidation of Cr(VI) and phenol. In Fig. 15, the kinetic plots can be observed. 30 ppm phenol and 20 ppm Cr(VI) are taken as a combined pollutant. From the values of the rate constants, we can observe that the individual reactions were less advantageous compared to simultaneous reactions. For this, the amount of catalyst (20 mg L−1) and irradiation time (180 min) are kept constant. The rate constants for combined pollutant removal are 0.0106 and 0.00945 S−1, whereas they are 0.00749 and 0.00603 S−1 for 30 ppm phenol and 20 ppm Cr(VI), respectively. The percentages of degradation of the combined pollutants are higher compared to individual pollutants, and the results are shown in Fig. S9.†
 |
| | Fig. 15 A kinetic plot comparison for the individual and simultaneous oxidation and reduction of both pollutants with photocatalyst 2: irradiation time = 180 minutes, and catalyst amount = 20 mg L−1. | |
3.8. The mechanism for the removal of individual pollutants under photocatalysis
The photocatalytic mechanism for phenol oxidation and chromium(VI) reduction is explained below. Under visible light irradiation, the photocatalyst is excited and produces photocatalytic excitons (e− and h+), which means that electrons are generated in the conduction band (CB) and holes are produced in the valence band (VB). The holes in the VB react with water molecules adsorbed from the atmosphere and produce hydroxyl radicals (OH˙), and the hydroxyl radical is a powerful oxidant next to fluorine.31 The electrons present in the CB reduce oxygen molecules adsorbed on the catalyst surface and convert them into O2−˙; this reacts with water to form hydroxyl radicals. The hydroxyl radicals react with phenol and convert it into nontoxic products such as CO2 and H2O.32
Mn ion doped ZnO + hν → e− (CB) + h+ (VB)
| h+ (VB) + H2O(ads) → H+ + OH˙ |
| H2O(ads) ↔ H+(ads) + OH−(ads) |
| O2−˙ + 2H2O → 2OH˙ + 2OH− + O2 |
| OH˙ + phenol → degraded products. |
Alternatively, Cr(VI) reduction is explained below. Under visible light irradiation, photocatalytic excitons (e− and h+) are produced and these excitons react with Cr(VI) and water, producing Cr(III) and protons. This is explained in the following reactions:
| Cr(VI)e− → Cr(V)e− → Cr(IV)e− → Cr(III) |
3.9. The mechanism for the photocatalytic removal of Cr(VI) and phenol simultaneously
3.9.1. Mechanism for phenol oxidation in the presence of Cr(VI). According to the above mechanism, hydroxyl radicals are produced. The produced hydroxyl radicals are highly reactive, with a high oxidation potential (2.9 eV). During phenol oxidation, the hydroxyl radical acts as an electrophile and attacks the aromatic rings of phenol molecules, producing cyclohexadienyl radicals.33,34
| C6H5OH + OH˙ → ˙C6H5(OH)2 |
The cyclohexadienyl radical undergoes disproportion to form dihydroxy benzene and phenol; in the absence of exterior oxidants like oxygen and hydrogen peroxide, this can be shown as follows:33
| C6H5(OH)2 → C6H4(OH)2 + C6H5OH |
The stoichiometric reaction of phenol with OH˙ produces dihydroxy benzene, as shown below:
| C6H5OH + 2OH˙ → C6H4(OH)2 + H2O |
From the above equation we can observe that for the oxidation of each phenol molecule we require two hydroxyl radicals.
The reaction of Cr(VI) with C6H5(OH)2˙ produces the dihydroxy phenyl cation and Cr(V); this is due to Cr(VI) acting as a good oxidizing agent, and can be shown as:35–37
| C6H5(OH)2˙ + Cr(VI) → +C6H5(OH)2 + Cr(V) |
| +C6H5(OH)2 → C6H4(OH)2 + H+ |
In the absence of a photocatalyst we cannot observe any phenol oxidation, which suggests that the reaction of phenol with Cr(VI) is insignificant.
During the photocatalysis process, we can observe the recombination of H˙ and OH˙ present in solution to form water; this can be explained as follows:38
The hydrogen radical can rapidly be oxidized by Cr(VI), as shown below:39
For phenol oxidation a greater number of OH˙ is needed, but hydrogen radicals are consuming OH˙ and suppressing the back reaction. The effect of Cr(VI) on phenol degradation is that it decreases the consumption of hydroxyl radicals and Cr(VI) consumes the hydrogen radicals. This Cr(VI) acts as a H˙ consumer and leaves more OH˙ for phenol oxidation. Finally, Cr(VI) acts as a scavenger and traps H˙ and photoelectrons present in the conduction band.
3.9.2. Mechanism for the reduction of Cr(VI) in the presence of phenol under photocatalysis. The reduction of Cr(VI) to Cr(III) is attributed to the production of hydrogen radicals, which can reduce Cr(V), followed by further reduction to Cr(III)40,41
| Cr(V) + Cr(V) → Cr(IV) + Cr(VI) |
| Cr(V) + Cr(IV) → Cr(III) + Cr(VI) |
The stoichiometric reduction of Cr(VI) to Cr(III) by H˙ can be shown as below:
| 3H˙ + Cr(VI) → Cr(III) + 3H+ |
In the same manner, there is the possibility of the formation of Cr(IV) or Cr(VI) by OH radicals, from the reduction of Cr(III) or Cr(V), respectively. This can be shown as below:
| OH˙ + Cr(III) → Cr(IV) + OH− |
| Cr(V) + OH˙ → Cr(VI) + OH− |
The presence of OH radicals causes the reduction of Cr(III) and Cr(V), which can be overcome by using phenol as a scavenger to suppress the OH radical reaction. So that which leads to the availability of more H radicals undergoes immense participation in the degradation of Cr(VI) to Cr(III).
4. Conclusions
Efficient photocatalysts based on modified Zn1−xMnxO (0.1%, 0.2%, 0.3%, and 0.4%) were synthesized via a solution combustion method and characterized using different techniques. Under visible light irradiation, photocatalyst 2 shows the highest activity for phenol oxidation and Cr(VI) reduction. It is very clear that this method is more advantageous than individual treatment, since simultaneous degradation occurs in the same reaction. The mechanism suggests that OH radicals are selectively trapped by phenol molecules, preventing the reoxidation of Cr(III). In the other direction, the presence of Cr(VI) enhances the degradation of phenol by trapping H radicals. A final summarization explains that Cr(VI) and phenol simultaneously help each other to provide better degradation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank MHRD-India (CE/2014-15/020/MHRD/KVLS/0130) for financial assistance. AS Rao would like to acknowledge DST-SERB, with project number YSS/2015/001222.
Notes and references
- Z. Guo, R. Ma and G. Li, Chem. Eng. J., 2006, 119, 55 CrossRef CAS.
- J. Wang, X. Gao, F. Fu, L. Zhang and Y. Wu, J. Residuals Sci. Technol., 2012, 9, 101 Search PubMed.
- Y. H. Lin, C. H. Weng, J. H. Tzeng and Y. T. Lin, Int. J. Photoenergy, 2016, 9, 1 CrossRef.
- S. Martha, P. C. Sahoo and K. M. Parida, RSC Adv., 2015, 5, 61535 RSC.
- World Health Organization, Phenol: Environmental Health Criteria 161, 1994 Search PubMed.
- World Health Organization, Cresols: Environmental Health Criteria 168, 1995 Search PubMed.
- H. Liu, T. Liu, Z. Zhang, X. Dong, Y. Liu and Z. Zhu, J. Mol. Catal. A: Chem., 2015, 410, 41 CrossRef CAS.
- Z. Yang, B. Wang, J. Zhang, H. Cui, Y. Pan, H. An and J. Zhai, Phys. Chem. Chem. Phys., 2015, 17, 18670 RSC.
- F. Liu, J. Yu, G. Tu, L. Qu, J. Xiao, Y. Liu and J. Zhang, Appl. Catal., B, 2017, 201, 1 CrossRef CAS.
- J. C. Wang, J. Ren, H. C. Yao, L. Zhang, J. S. Wang, S. Q. Zang and Z. J. Li, J. Hazard. Mater., 2016, 311, 11 CrossRef CAS PubMed.
- S. Dong, X. Ding, T. Guo, X. Yue, X. Han and J. Sun, Chem. Eng. J., 2017, 316, 778 CrossRef CAS.
- Z. Wan, G. Zhang, X. Wu and S. Yin, Appl. Catal., B, 2017, 207, 17 CrossRef CAS.
- Z. H. Diao, X. R. Xu, D. Jiang, J. J. Liu, L. J. Kong, G. Li, L. Z. Zuo and Q. H. Wu, Chem. Eng. J., 2017, 315, 167 CrossRef CAS.
- D. Lu, M. Yang, P. Fang, C. Li and L. Jiang, Appl. Surf. Sci., 2017, 399, 167 CrossRef CAS.
- A. D. Mani, P. M. K. Reddy, M. Srinivas, P. Ghosal, N. Xanthopoulos and C. Subrahmanyam, Mater. Res. Bull., 2015, 61, 391 CrossRef CAS.
- X. Zhao, P. Wu, M. Liu, D. Lu, J. Ming, C. Li, J. Ding, Q. Yan and P. Fang, Appl. Surf. Sci., 2017, 410, 134 CrossRef CAS.
- K. Umar, A. Aris, T. Parveen, J. Jaafar, Z. A. Majid, A. V. B. Reddy and J. Talib, Appl. Catal., A, 2015, 505, 507 CrossRef CAS.
- B. Meenatchi, K. R. Nandhine Deve, A. Manikandan, V. Renuga and V. Sathiyalakshmi, Adv. Sci., Eng. Med., 2016, 8, 653 CrossRef.
- V. D. Mote, J. S. Dargad and B. N. Dole, Nanosci. Nanoeng., 2013, 1, 116 Search PubMed.
- S. Yildirimcan, K. Ocakoglu, S. Erat, F. M. Emen, S. Repp and E. Erdem, RSC Adv., 2016, 6, 39511 RSC.
- Z. Barzgari, A. Ghazizadeh and S. Z. Askari, Res. Chem. Intermed., 2016, 42, 4203 CrossRef.
- M. Bordbar, S. Jafari, A. Yeganeh-Faal and B. Khodadadi, J. Iran. Chem. Soc., 2017, 14, 897 CrossRef CAS.
- F. Achouri, S. Corbel, L. Balan, K. Mozet, E. Girot, G. Medjahdi, M. B. Said, A. Ghrabi and R. Schneider, Mater. Des., 2016, 101, 309 CrossRef CAS.
- Y. Abdollahi, A. H. Abdullah, Z. Zainal and N. A. Yusof, Int. J. Basic Appl. Sci., 2011, 11, 62 Search PubMed.
- Y. M. Hao, S. Y. Lou, S. M. Zhou, R. J. Yuan, G. Y. Zhu and N. Li, Nanoscale Res. Lett., 2012, 7, 100 CrossRef PubMed.
- F. Achouri, S. Corbel, L. Balan, K. Mozet, E. Girot, G. Medjahdi and R. Schneider, Mater. Des., 2016, 101, 309 CrossRef CAS.
- J. Jayabharathi, C. Karunakaran, A. Arunpandiyan and P. Vinayagamoorthy, RSC Adv., 2014, 4, 59908 RSC.
- S. M. Yakout and A. M. El-Sayed, J. Supercond. Novel Magn., 2016, 29, 1593 CrossRef CAS.
- B. Roy, B. Karmakar, P. M. G. Nambissan and M. Pal, Nano, 2011, 6, 173 CrossRef CAS.
- M. Shatnawi, A. M. Alsmadi, I. Bsoul, B. Salameh, M. Mathai, G. Alnawashi and M. S. Bawa'aneh, Results Phys., 2016, 6, 1064 CrossRef.
- P. R. Gogate and A. B. Pandit, Adv. Environ. Res., 2004, 8, 501 CrossRef CAS.
- X. Zhu, J. Zhang and F. Chen, Appl. Catal., B, 2011, 102, 316 CrossRef CAS.
- H. Kušić, N. Koprivanac, A. L. Božić and I. Selanec, J. Hazard. Mater., 2006, 136, 632 CrossRef PubMed.
- Y. Liu, J. Hazard. Mater., 2009, 168, 992 CrossRef CAS PubMed.
- C. Walling, Acc. Chem. Res., 1998, 31, 155 CrossRef CAS.
- G. V. Buxton, J. R. Langan and J. R. L. Smith, J. Phys. Chem., 1986, 90, 6309 CrossRef CAS.
- A. K. Pikaev, L. I. Kartasheva, T. K. Yurik, V. N. Chulkov, O. A. Didenko, D. K. Kim and B. Han, Mendeleev Commun., 1997, 7, 52 CrossRef.
- M. K. Eberhardt, J. Phys. Chem., 1975, 79, 1913 CrossRef CAS.
- A. Hickling and M. D. Ingram, Trans. Faraday Soc., 1964, 60, 783 RSC.
- M. Al-Sheikhly and W. L. McLaughlin, Int. J. Radiat. Appl. Instrum., Part C, 1991, 38, 203 CAS.
- L. Chandana, B. Lakshminarayana and C. Subrahmanyam, J. Environ. Chem. Eng., 2015, 3, 2760 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08172c |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a