DOI:
10.1039/C7RA08165K
(Paper)
RSC Adv., 2017,
7, 44639-44646
Sorption and sensing properties of coordination polymers with mixed 1,3,5-tri(1-imidazolyl)benzene and 2,6-naphthalenedicarboxylate ligands†
Received
24th July 2017
, Accepted 11th September 2017
First published on 18th September 2017
Abstract
Under hydro/solvothermal conditions, the assembly of imidazole-containing ligand 1,3,5-tri(1-imidazolyl)benzene (tib) and 2,6-naphthalenedicarboxylic acid (H2NDC) with different transition metal salts provides five new coordination polymers [M2(tib)2(NDC)(H2O)2] SO4·XH2O (1: M = Cd, X = 4; 2: M = Co, X = 4; 3: M = Ni, X = 3), [Cd2(tib)2(NDC)(NO3)2] 2.5DMF·H2O (4) (DMF = N,N-dimethylformamide) and [Zn(tib)2/3(NDC)] (5). 1–3 have the same three-fold interpenetrating 3D framework structures with SO42− as counteranion, while 4 with NO3− as terminal ligand exhibits a wave-like 2D layer structure, which is further extended into a 3D framework by hydrogen bonds. In addition, complex 5 obtained at high temperature is an interesting two-fold interpenetrating 3D structure. Sorption and photoluminescence properties of the complexes were investigated. 4 displays selective adsorption of CO2 over N2. Furthermore, 1 and 4 can be used to probe acetone molecules selectively and rapidly via fluorescence quenching.
Introduction
Crystal engineering of coordination polymers (CPs) as well as metal–organic frameworks (MOFs) has been extensively studied in the past years, however, further systematic study is required for predictable and controllable synthesis of desired MOFs with specific properties and potential applications in gas storage/separation, catalysis, magnetism, photoluminescence and so on.1 In recent years, MOFs have been developed and show perspective in molecular sensing and probes, particularly in monitoring harmful components in industrial and environmental fields as well as the active components in organisms.2
With respect to fluorescence sensing, lanthanide-MOFs have superiority benefitting from metal-based emissions.3 In addition, some transition metal ions such as cadmium(II) ion has also been used to construct MOFs with sensing properties. For example, a porous three-dimensional (3D) MOF [Cd(NDC)0.5(PCA)] Gx (H2NDC = 2,6-naphthalenedicarboxylic acid, HPCA = 4-pyridinecarboxylic acid, G = guest molecules) was synthesized by reaction of Cd(II) salt with H2NDC and HPCA ligands, and can sensitively and selectively detect TNP (2,4,6-trinitrophenol) from mixed nitro explosives.4 In 2014, another Cd-MOF CZJ-3 with formula of [CdL(H2O)] 4DMF·2H2O [H2L = (E)-4-(2-carboxyvinyl)benzoic acid] has been reported. There are one-dimensional (1D) hexagonal nanotube channels in CZJ-3, which can absorb rhodamine B molecules to accomplish luminescent Rho@CZJ-3, as a result, sensing various volatile organic molecules can be easily realized by monitoring the photoluminescence of Rho@CZJ-3.5 Furthermore, Cd-MOFs also made gratifying achievements in the aspect of ion sensing. A dual-emission 3D MOF {[Cd1.5(EDDA)](H3O)(H2O)3}n (H4EDDA = 5,5′-[ethane-1,2-diylbis(oxy)]diisophthalic acid) has been recognized as the first example of MOF-implicated sensor for Hg(II) ions in water rapidly and sensitively.6 There are some other luminescent Cd-MOFs based on carboxylate ligands that are applied to chemical sensing.7 However, examples of imidazole-containing ligand based MOFs with sensing property are quite limited. In this work, we introduced rigid tripodal imidazole-containing ligand 1,3,5-tris(1-imidazolyl)benzene (tib) and dicarboxylic acid of 2,6-naphthalenedicarboxylic acid (H2NDC) to react with different metal salts and five new MOFs, [M2(tib)2(NDC)(H2O)2] SO4·XH2O (1: M = Cd, X = 4; 2: M = Co, X = 4; 3: M = Ni, X = 3), [Cd2(tib)2(NDC)(NO3)2] 2.5DMF·H2O (4) (DMF = N,N-dimethylformamide) and [Zn(tib)2/3(NDC)] (5) were obtained. Selective gas sorption and sensing properties of the complexes were investigated and the Cd-MOFs were proved to be capable of sensing acetone molecules.
Experimental
Materials and methods
Ligand tib was prepared according to the previously published procedure,8 and other commercially available chemicals were used as received. Elemental analyses (EA) for C, H and N were performed on a Perkin–Elmer 240C elemental analyzer at the analysis center of Nanjing University. IR spectra were measured on a Bruker Vector 22 FT-IR spectrometer by the KBr method. Powder X-ray diffraction (PXRD) measurements were made on a Bruker D8 Advance instrument using Cu Kα radiation (λ = 1.5418 Å). Thermogravimetric analyses (TGA) were carried out on a Mettler-Toledo (TGA/DSC1) thermal analyzer from 30 °C to 800 °C under nitrogen flow with a heating rate of 10 °C min−1. Gas sorption tests were taken on a Belsorp-max volumetric gas sorption instrument. Photoluminescence spectra were recorded on a Perkin–Elmer LS 55 spectrofluorometer and xenon arc lamp acts as the light source.
Preparation of [Cd2(tib)2(NDC)(H2O)2] SO4·4H2O (1)
A mixture of tib (27.6 mg, 0.10 mmol), CdSO4·8/3H2O (25.6 mg, 0.10 mmol), H2NDC (21.6 mg, 0.10 mmol) and H2O (6 ml) was sealed in a Teflon-lined stainless steel container and heated at 90 °C for 3 days. After being cooled to room temperature, colorless block crystals of 1 were obtained in 57% yield. Anal. calcd for C42H42N12O14SCd2: C, 42.19; H, 3.54; N, 14.06%. Found: C, 42.11; H, 3.59; N, 14.11%. IR (KBr pellet, cm−1): 3442 (w), 3105 (m), 1622 (s), 1551 (s), 1513 (vs), 1411 (s), 1355 (m), 1264 (s), 1096 (s), 1018 (s), 935 (m), 849 (m), 806 (m), 652 (m).
Preparation of [Co2(tib)2(NDC)(H2O)2] SO4·4H2O (2)
Complex 2 was obtained by the same procedure used for preparation of 1 except that CdSO4·8/3H2O was replaced by CoSO4·7H2O (28.1 mg, 0.1 mmol). Purple block crystals of 2 were obtained in 66% yield. Anal. calcd for C42H42N12O14SCo2: C, 46.33; H, 3.89; N, 15.44%. Found: C, 46.29; H, 3.86; N, 15.41%. IR (KBr pellet, cm−1): 3392 (m), 1679 (m), 1614 (s), 1509 (vs), 1419 (s), 1357 (w), 1301 (m), 1249 (s), 1105 (vs), 1006 (w), 929 (w), 788 (m).
Preparation of [Ni2(tib)2(NDC)(H2O)2] SO4·3H2O (3)
The title complex 3 was also isolated by the same procedure used for the preparation of 1 except that NiSO4·6H2O (26.3 mg, 0.1 mmol) was used instead of CdSO4·8/3H2O. Green block crystals of 3 were obtained in 61% yield. Anal. calcd for C84H80N24O26S2Ni4: C, 47.13; H, 3.77; N, 15.70%. Found: C, 47.10; H, 3.74; N, 15.75%. IR (KBr pellet, cm−1): 3419 (vs), 1618 (s), 1548 (m), 1508 (m), 1419 (s), 1307 (w), 1257 (m), 1089 (s), 1018 (m), 790 (w).
Preparation of [Cd2(tib)2(NDC)(NO3)2] 2.5DMF·H2O (4)
A mixture of tib (27.6 mg, 0.10 mmol), Cd(NO3)2·4H2O (30.8 mg, 0.10 mmol), H2NDC (21.6 mg, 0.10 mmol) and DMF (6 ml) was sealed in a Teflon-lined stainless steel container and heated at 90 °C for 3 days. After being cooled to room temperature, colorless block crystals of 4 were obtained in 55% yield. Anal. calcd for C49.5H49.5N16.5O13.5Cd2: C, 45.17; H, 3.79; N, 17.56%. Found: C, 45.21; H, 3.75; N, 17.58%. IR (KBr pellet, cm−1): 3421 (s), 1622 (vs), 1517 (s), 1407 (w), 1322 (w), 1263 (s), 1101 (s), 1082 (s), 1016 (m), 952 (w), 847 (w), 828 (m), 763 (m).
Preparation of [Zn(tib)2/3(NDC)] (5)
A mixture of tib (27.6 mg, 0.10 mmol), ZnSO4·7H2O (28.7 mg, 0.10 mmol), H2NDC (21.6 mg, 0.10 mmol) and H2O (6 ml) was sealed in a Teflon-lined stainless steel container and heated at 180 °C for 3 days. After being cooled to room temperature, colorless block crystals of 5 were obtained in 48% yield. Anal. calcd for C22H14N4O4Zn: C, 56.98; H, 3.04; N, 12.08%. Found: C, 56.81; H, 3.02; N, 12.11%. IR (KBr pellet, cm−1): 3419 (m), 3131 (m), 1602 (s), 1554 (s), 1497 (s), 1397 (s), 1375 (s), 1328 (vs), 1235 (m), 1188 (m), 1076 (w), 1013 (m), 930 (w), 788 (s), 688 (m).
Crystal structure determination
Crystallographic data for 1–5 were collected on a Bruker Smart Apex II CCD single-crystal X-ray diffractometer with a graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) at 293(2) K. Multi-scan absorption corrections were applied using the SADABS program.9 All the structures were solved by direct methods using the SHELXS-2016 program of the SHELXTL package and refined by the full-matrix least-squares method using SHELXL-2016.10 All the non-hydrogen atoms in the structures were refined on F2 with anisotropic displacement parameters. Except those of water molecules, the hydrogen atoms were placed geometrically and refined isotropically according to the riding model. The hydrogen atoms of water molecules in 1–3 were found directly. The atom O5 in 1 is disordered in two positions, each with site occupancies of 0.5, the NDC2− ligands in 1 and 5 are disordered in two positions with site occupancies of 0.786(6) and 0.214(6) for 1 and 0.814(4) and 0.186(4) for 5, the naphthalene rings of NDC2− in 2 and 3 are also disordered in two positions with site occupancies of 0.4 and 0.6 for 2, and 0.5 and 0.5 for 3. All the SO42− anions in 1–3 are passed through by two-fold axes. The highly disordered solvent molecules in 4 are difficult to resolve and their contribution of densities was modeled using the SQUEEZE routine in software PLATON.11 The definite chemical formula of 4 were obtained from the overall contribution of crystallographic, EA and TG data. The details of the crystal parameters, data collection and refinements for the complexes are summarized in Table 1, selected bond lengths and angles as well as hydrogen bonding data are listed in Tables S1 and S2,† respectively.
Table 1 Crystal data and structure refinements for 1–5
| R1 = ∑||Fo| − |Fc||/∑|Fo|. wR2 = |∑w(|Fo|2 − |Fc|2)|/∑|w(Fo)2|1/2, where w = 1/[σ2(Fo2) + (aP)2 + bP]. P = (Fo2 + 2Fc2)/3. |
| Compound |
1 |
2 |
3 |
4 |
5 |
| Formula |
C42H42N12O14SCd2 |
C84H84N24O28S2Co4 |
C84H80N24O26S2Ni4 |
C49.5H49.5N16.5O13.5Cd2 |
C22H14N4O4Zn |
| Mr |
1195.73 |
2177.59 |
2140.68 |
1316.35 |
463.74 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Triclinic |
Trigonal |
| Space group |
C2/c |
C2/c |
C2/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) |
| a (Å) |
23.6468(18) |
22.802(2) |
22.602(1) |
13.572(5) |
19.1253(9) |
| b (Å) |
13.261(1) |
13.5397(12) |
13.646(1) |
14.157(5) |
19.1253(9) |
| c (Å) |
17.991(2) |
17.2991(15) |
17.163(1) |
19.505(8) |
8.5716(8) |
| α (°) |
90 |
90 |
90 |
93.398(11) |
90 |
| β (°) |
126.758(1) |
125.990(1) |
125.812(1) |
110.063(9) |
90 |
| γ (°) |
90 |
90 |
90 |
90.003(10) |
120 |
| V (Å3) |
4519.9(8) |
4321.4(7) |
4292.7(4) |
3513(2) |
2715.2(4) |
| Z |
4 |
2 |
2 |
2 |
6 |
| Dc (g cm−3) |
1.757 |
1.674 |
1.656 |
1.244 |
1.702 |
| M (mm−1) |
1.070 |
0.903 |
1.010 |
0.667 |
1.399 |
| F (000) |
2408 |
2240 |
2208 |
1332 |
1416 |
| Reflections collected |
12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 400 |
14338 |
11888 |
20724 |
14072 |
| Unique reflections |
3984 |
3793 |
3798 |
12316 |
3463 |
| Goodness-of-fit |
1.073 |
0.991 |
1.086 |
1.031 |
1.030 |
| Final R indices [I > 2σ(I)]a,b |
R1 = 0.0212 wR2 = 0.0483 |
R1 = 0.0458 wR2 = 0.1239 |
R1 = 0.0379 wR2 = 0.1008 |
R1 = 0.1021 wR2 = 0.2452 |
R1 = 0.0357 wR2 = 0.0891 |
| R Indices (all data) |
R1 = 0.0250 wR2 = 0.0502 |
R1 = 0.0587 wR2 = 0.1353 |
R1 = 0.0426 wR2 = 0.1044 |
R1 = 0.1494 wR2 = 0.2695 |
R1 = 0.0476 wR2 = 0.0941 |
Results and discussion
Crystal structures of [M2(tib)2(NDC)(H2O)2] SO4·XH2O (1: M = Cd, X = 4; 2: M = Co, X = 4; 3: M = Ni, X = 3)
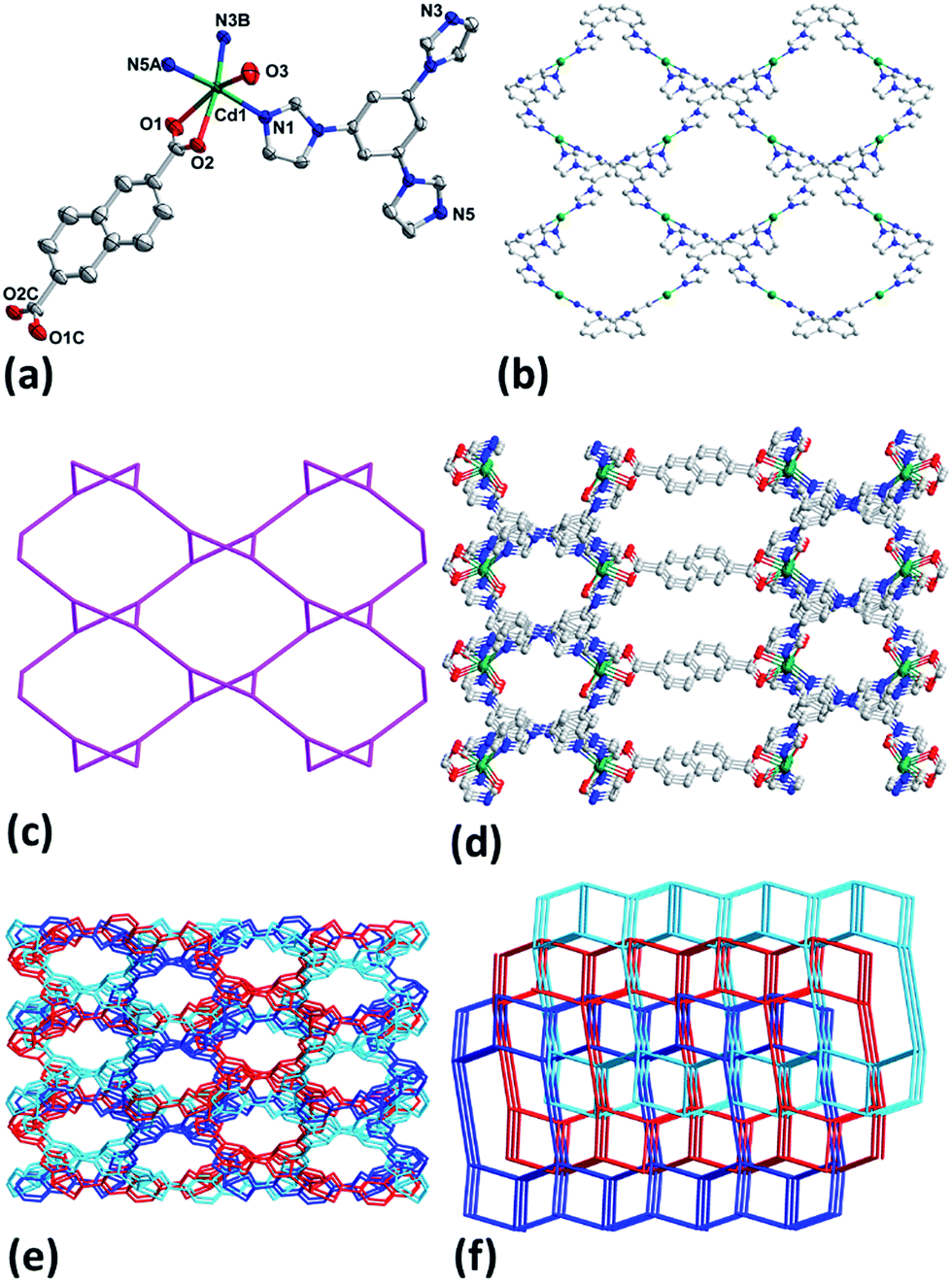
The results of crystallographic analysis show that 1–3 crystalize in monoclinic space group C2/c (Table 1) and have the same framework structures, thus only the structure of 1 is described in detail here. Cd1 in 1 is bounded by two chelate carboxylate oxygen atoms (O1, O2) from one NDC2−, three nitrogen ones (N1, N3B, N5A) from three different tib ligands and one coordinated water molecule (Fig. 1a). The Cd–O and Cd–N bond lengths are in the range of 2.275(2)–2.431(3) Å and 2.2854(17)–2.3017(17) Å (Table S1†), respectively. Each tib acts as a tridentate ligand and coordinates with three Cd(II) atoms, and each Cd(II) in turn links three tib to generate a rhombic-like two-dimensional (2D) network (Fig. 1b) with {4·102} topology (Fig. 1c). Ligands NDC2− join the adjoining 2D layers to attain a ladder-like 3D structure along a axis possessing two different kinds of channels with apertures of 14.1 and 9.9 Å (Fig. 1d). The large cavity allows other frameworks to interpenetrate each other to result in the final three-fold interpenetrating framework of 1 (Fig. 1e). Topologically, ligands tib and Cd1 can be simplified as 3- and 4-connected nodes and NDC2− is treated just as linker, thus 1 can be regarded as a (3, 4)-connected 2-nodal 3D net with point (Schläfli) symbol of {4·6·8}{4·62·8·102} (Fig. 1f).12
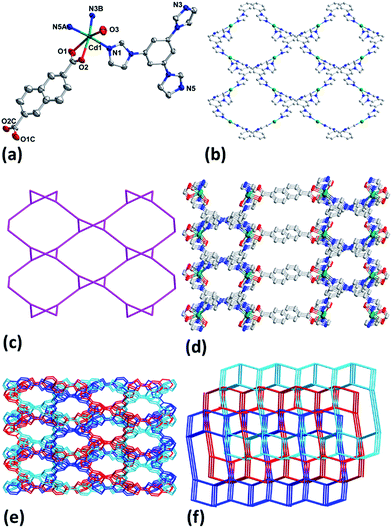 |
| | Fig. 1 (a) Coordination environment of Cd(II) in 1 with ellipsoids drawn at 30% probability level. The hydrogen atoms, free anion and water molecules are omitted for clarity. (b) 2D network of Cd-tib in 1. (c) Topology of 2D Cd-tib in 1. (d) 3D framework of 1. (e) Interpenetrating 3D framework of 1. (f) Topology of 1. | |
Crystal structure of [Cd2(tib)2(NDC)(NO3)2] 2.5DMF·H2O (4)
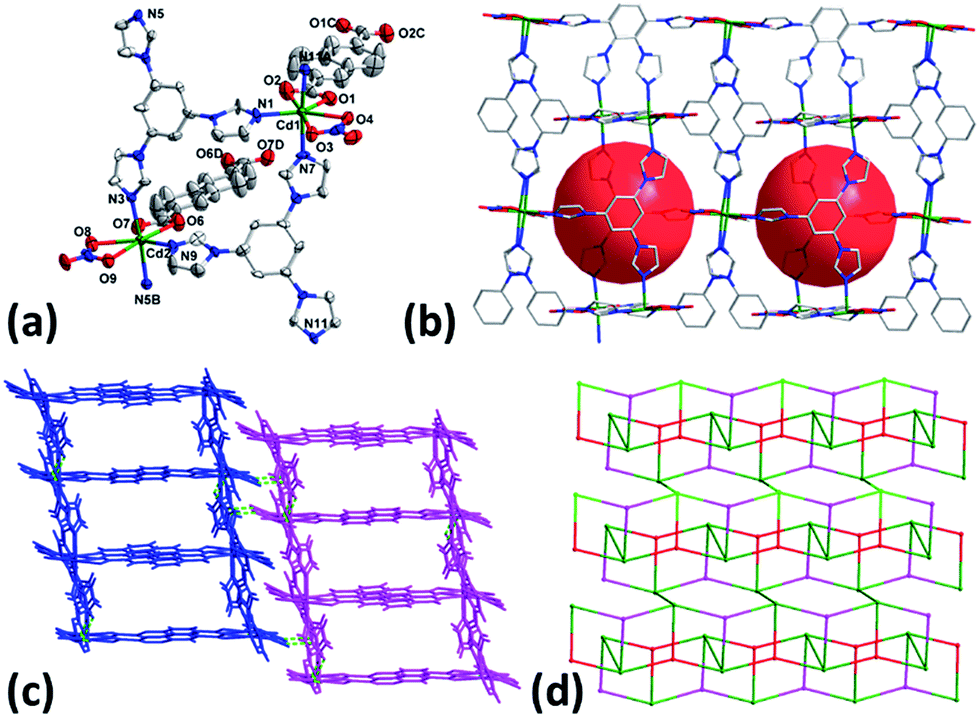
When the reaction was carried out in DMF using Cd(NO3)2·4H2O instead of CdSO4·8/3H2O, complex 4 was isolated. As illustrated in Fig. 2a, both Cd1 and Cd2 are seven- coordinated with pentagonal bipyramid coordination geometry by three nitrogen atoms from three different tib, two oxygen ones from one chelate carboxylate group of NDC2− and two other oxygen ones from coordinated NO3− anion. Each tib connects three Cd(II) to form an infinite 1D chain which is further connected by NDC2− to compose the 2D network structure of 2 with nano-sized cavities of 13.57 × 10.85 Å2 (Fig. 2b). The 2D layers finally extend to 3D supramolecular architecture through the C–H⋯O hydrogen bonds (Fig. 2c and Table S2†). It is noteworthy that in contrast to the counter anion of SO42− in 1, NO3− acts as terminal ligand in 4. The results imply that the anion plays important in determining the framework structures in this system.
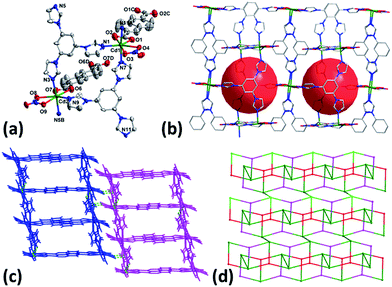 |
| | Fig. 2 (a) Coordination environment of Cd(II) in 4 with ellipsoids drawn at 30% probability level. The hydrogen atoms and free solvent molecules are omitted for clarity. (b) 2D layer of 4. (c) 3D supramolecular architecture of 4. (d) Topology of 4. | |
From topological view, Cd1, Cd2 and tib can be viewed as 4-, 4- and 3-connected nodes and ditopic carboxylate NDC2− acts as linker, leading to the formation of (3, 4)-connected 2-nodal 2D net with the point (Schläfli) symbol of {42·6}{42·63·8} (Fig. 2d).
Crystal structure of [Zn(tib)2/3(NDC)] (5)
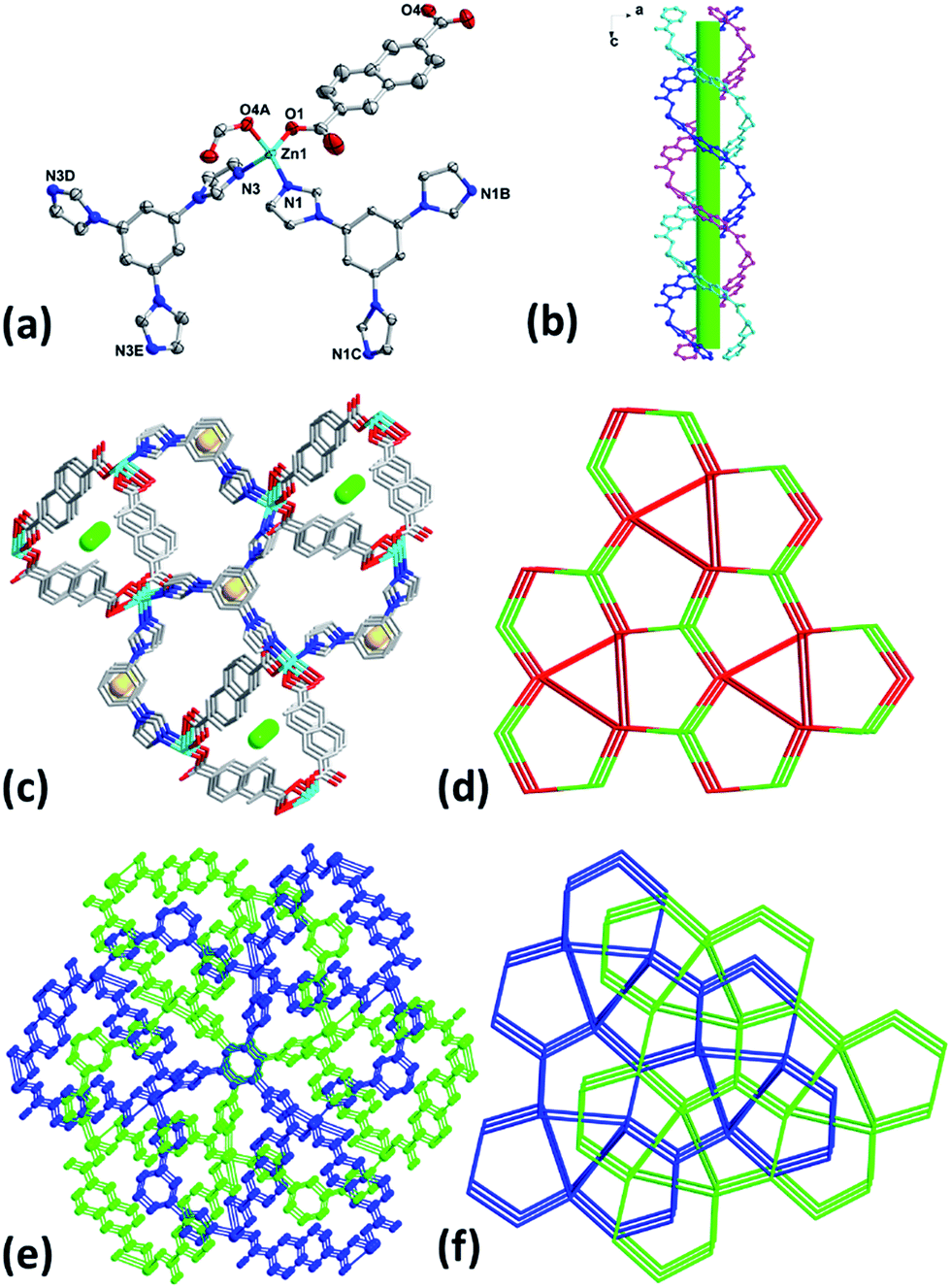
When the reaction temperature was raised to 180 °C, complex 5 was obtained. Crystal structural analysis revealed that 5 crystalizes in trigonal space group P. The asymmetric unit of 5 contains one Zn(II), two thirds of tib and one NDC2− ligand. It is noticeable that no coordinated or lattice solvent molecules were found in 5 thanks to the high reaction temperature.13 Each Zn(II) in 5 is four-coordinated with distorted tetrahedral coordination geometry by two mono-dentate carboxylate oxygen atoms (O1, O4A) from two independent NDC2− and two nitrogen ones (N1, N3) from two different tib ligands (Fig. 3a). It is worth noting that Zn(II) atoms link NDC2− to present double helical chains along b axis (Fig. 3b) and ligands tib further connect helical chains to generate the 3D framework structure of 5 (Fig. 3c). The presence of large cavity in the framework leads to the formation of two-fold interpenetrating 3D net of 5 (Fig. 3e). By simplifying the tib and Zn(II) as 3- and 4-connected nodes, 5 also belongs to a (3,4)-connected 2-nodal 3D net with Schläfli symbol of {103}2{106}3 (Fig. 3d and f).
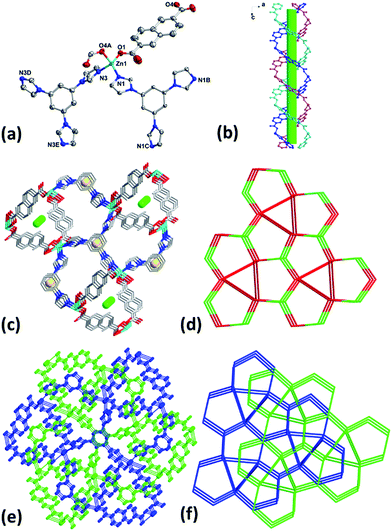 |
| | Fig. 3 (a) Coordination environment of Zn(II) in 5 with ellipsoids drawn at 30% probability level. The hydrogen atoms are omitted for clarity. (b) 1D double helical chains along b axis. (c) 3D framework with helical chains in 5. (d) Topology of 5. (e) Interpenetrating 3D framework of 5. (f) Interpenetrating topology of 5. | |
Powder X-ray diffraction (PXRD) and thermogravimetric analyses (TGA)
To ensure the phase purity of the bulk samples of 1–5 in the solid state, PXRD experiments were performed and the experimental patterns matched well with the simulated ones generated from the single crystal data (Fig. S1†), implying the pure phase of 1–5. In addition, TGA were carried out to assess the thermal stability of 1–5 and the results are exhibited in Fig. S2 and S3.† Complex 1 shows weight loss of 8.71% from room temperature to about 160 °C, which corresponds to the release of lattice and coordinated water molecules (calcd 9.03%). The decomposition of the framework occurred at 320 °C. For 2, the weight loss of 9.64% (calcd 9.92%) between 30–210 °C is due to the loss of free and coordinated water molecules, and then the framework gradually decomposed above 300 °C. 3 loses 8.84% of its weight in the temperature range of 30–240 °C, which is attributed to the departure of three lattice and two coordinated water molecules (calcd 8.41%), and the residue is stable up to about 380 °C. For 4, the weight loss of 16.65% (calcd 15.25%) between 30–235 °C corresponds to the loss of lattice water and DMF molecules, and then the framework gradually decomposed above 350 °C. 5 did not show obvious weight losses before the decomposition of the framework at about 300 °C, which is in agreement with the result of the crystal structural analysis.
Adsorption properties of 1 and 4
The porous structures of 1 and 4 discussed above suggested the possibility in the sorption aspect. Thus, the gas sorption properties of 1 and 4 were investigated. Acetone was used to exchange the guest molecules inside the frameworks for 3 days and then the activation was carried out by heating the sample at 100 °C for 10 h under vacuum, the solvent molecules were completely removed without destroying the frameworks as confirmed by the PXRD and TG data (Fig. S1 and S2†).
As shown in Fig. 4, the sorption isotherm of 1 exhibits that only 4.80 cm3 g−1 is adsorbed for N2 at 77 K and 0.98 atm, while the CO2 adsorption reaches 30.45 cm3 g−1 (59.81 mg g−1) at 195 K and 0.99 atm, corresponding to 1.5 CO2 molecules per formula unit of 1. The obvious hysteresis during desorption process of CO2 in 1 can be attributed to the interaction of CO2 molecules with framework of 1. On the other hand, the CO2 adsorption isotherm for 4 represents a typical type-I adsorption isotherm at 195 K as illustrated in Fig. 5.14 The CO2 uptake capacity of 4 is 73.19 cm3 g−1 (143.77 mg g−1) at 0.98 atm, amounting to 3.6 CO2 molecules per formula unit of 4. Similar to 1, there is almost no adsorption of N2 for 4 at 77 K. The selective sorption of CO2 over N2 for 1 and 4 implies the promising crystalline materials for separation of CO2.15
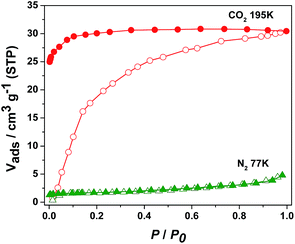 |
| | Fig. 4 N2 (77 K) and CO2 (195 K) adsorption isotherms of 1. | |
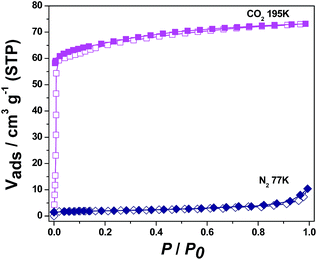 |
| | Fig. 5 N2 (77 K) and CO2 (195 K) adsorption isotherms of 4. | |
Photoluminescence properties of 1, 4 and 5
CPs containing transition-metal ions without unpaired electrons, especially d10 metal ions, are advantageous candidates for luminescent materials.16 Accordingly, the luminescent properties of free tib, 1, 4 and 5 were examined in the solid state at room temperature. Intense emission band was observed at 415 nm for tib ligand with excitation at 360 nm, while emissions at 400 nm (λex = 360 nm) for 1, 429 nm (λex = 365 nm) for 4 and 400 nm (λex = 360 nm) for 5 were observed (Fig. 6). The blue shift of 1 as well as 5 can be assigned to the coordination of organic ligands with metal ions and interpenetrated 3D framework structures, while the red shift of emission observed in 4 may be attributed to the hydrogen bonding linked supramolecular structure. It has been reported that the crystal packing of the complex in the solid state has impact on their luminescence.17
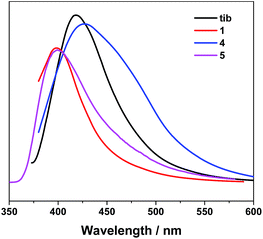 |
| | Fig. 6 Emission spectra of 1, 4, 5 and ligand tib in the solid state. | |
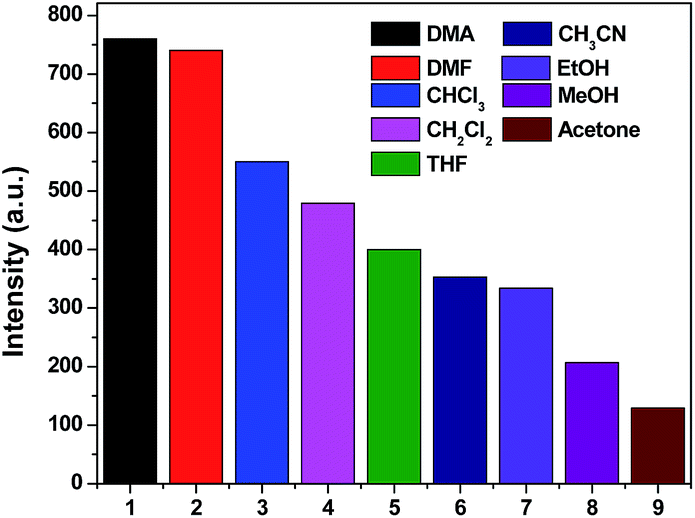
It has been well established that a considerable amount of fluorescent MOF materials are sensitive to the presence of volatile organic molecules.18 To examine whether 1, 4 and 5 have abilities to detect small solvent molecules, each 5 mg fresh sample of 1, 4 and 5 was immersed in 4 ml different organic solvent and it was found that only 1 and 4 are sensitive to the solvent molecules as evidenced by fluorescence intensity. The photoluminescence intensity of 1 and 4 depends on the identity of the solvent molecules with sequence of DMA > DMF > CHCl3 > CH2Cl2 > THF > CH3CN > EtOH > MeOH > acetone for 1 and DMF > DMA > CH3CN > CHCl3 > CH2Cl2 > THF > EtOH > MeOH > acetone for 4 (Fig. 7 and 8). It means that both 1 and 4 have significant quenching effect in the solvent of acetone, which can be attributed to the interactions between the framework and acetone molecule.19
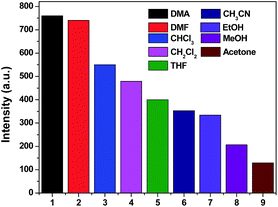 |
| | Fig. 7 Photoluminescence intensities of 1 in varied solvent when excited at 360 nm for 1. | |
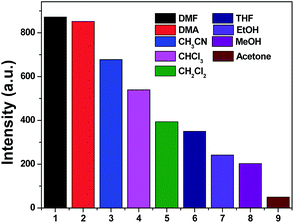 |
| | Fig. 8 Photoluminescence intensities of 4 in varied solvent when excited at 365 nm for 4. | |
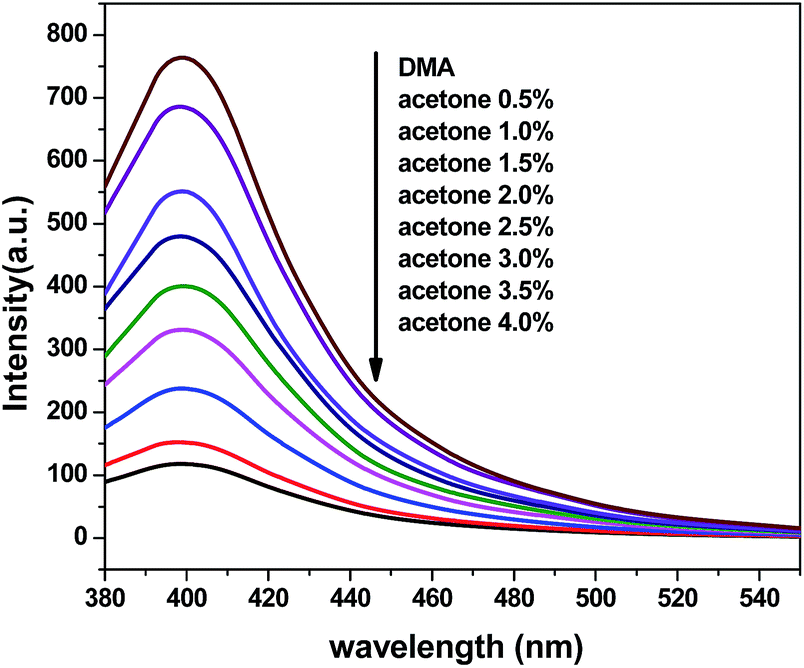
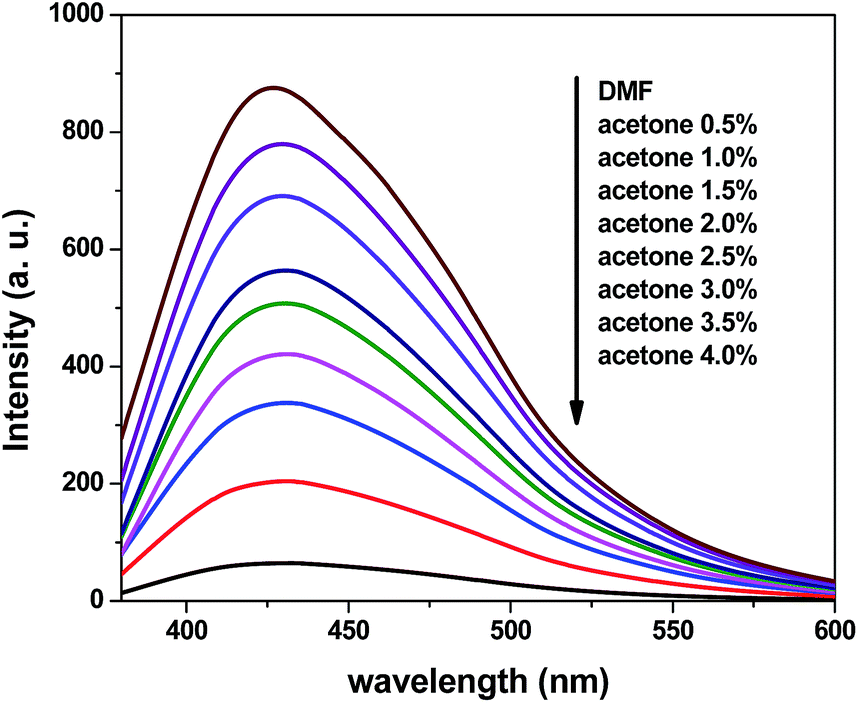
To further explore the sensing sensitivity toward acetone, a collection of powder 1 and 4 immersed in DMA and DMF solution, respectively, quantitative acetone was gradually added to the solution. The invariable skeleton structures of 1 and 4 were ensured by the PXRD of 1 and 4 immersed in DMA/DMF/acetone (Fig. S1†). By monitoring the emission response, the fluorescence intensity decrease was observed with addition of acetone to the DMA and DMF emulsion of 1 and 4 (Fig. 9 and 11). The decreasing trend of the fluorescence intensity at 400 nm for 1 and 430 nm for 4 vs. the concentrations of acetone fitted well with a first-order exponential decay (Fig. 10 and 12), suggesting that fluorescence quenching of 1 and 4 by acetone are diffusion-controlled.20 As a consequence, 1 and 4 can be potent candidates for acetone molecule sensing.
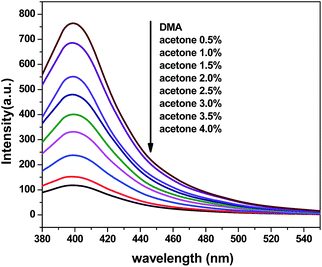 |
| | Fig. 9 Photoluminescence spectra of the dispersed 1 in DMA in the presence of different contents of the acetone solvent (excited at 360 nm). | |
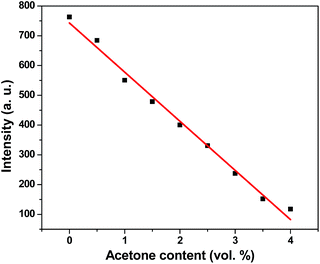 |
| | Fig. 10 Photoluminescence intensities of 1 in DMA as a function of acetone content. | |
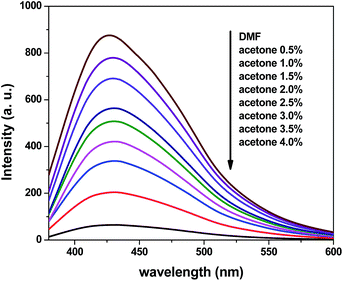 |
| | Fig. 11 Photoluminescence spectra of the dispersed 4 in DMF in the presence of different contents of the acetone solvent (excited at 365 nm). | |
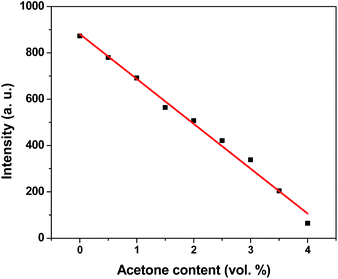 |
| | Fig. 12 Photoluminescence intensities of 4 in DMF as a function of acetone content. | |
The results show that there are differences of the adsorption and luminescence properties of 1 and 4, which may originate from their different framework structures. The large hysteresis and low adsorption capacity of CO2 observed in 1 is caused by the presence of SO42− counteranions, since SO42− can interact with CO2 but reduce the porosity of the framework. In addition, the varied hydrogen bonds within the solvent molecules and skeleton of 4 may result in the different luminescence behavior from 1.
Conclusions
Five coordination polymers with mixed organic ligands were synthesized under solvent thermal conditions. The results show that anions and reaction temperature play important role in the construction of frameworks. Adsorption experiments reveal that 1 and 4 can selectively adsorb CO2 over N2, in addition, both 1 and 4 can selectively and efficiently detect acetone molecules by fluorescence quenching via the interactions between the frameworks and acetone molecules, implying the application of CPs/MOFs in molecular sensing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grant no. 21331002 and 21573106). The authors extend their appreciation to the International Scientific Partnership Program ISPP at King Saud University for funding this research work through ISPP#0090. This work was also supported by a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Notes and references
-
(a) D. J. Tranchemontagne, J. L. Mendoza-Cortes, M. O'Keeffe and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1257 RSC;
(b) J. Y. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. B. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC;
(c) O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. Ö. Yazaydın and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016 CrossRef CAS PubMed;
(d) Y.-J. Cui, Y.-F. Yue, G.-D. Qian and B.-L. Chen, Chem. Rev., 2012, 112, 1126 CrossRef CAS PubMed;
(e) P. Ramaswamy, N. E. Wong and G. K. H. Shimizu, Chem. Soc. Rev., 2014, 43, 5913 RSC;
(f) Y.-Q. Huang, Y. Wan, H.-Y. Chen, Y. Wang, Y. Zhao and X.-F. Xiao, New J. Chem., 2016, 40, 7587 RSC;
(g) Y.-Q. Huang, H.-D. Cheng, H.-Y. Chen, Y. Wan, C.-L. Liu, Y. Zhao, X.-F. Xiao and L.-H. Chen, CrystEngComm, 2015, 17, 5690 RSC;
(h) Z.-G. Gu, C.-H. Zhan, J. Zhang and X.-H. Bu, Chem. Soc. Rev., 2016, 45, 3122 RSC;
(i) H.-R. Fu, Z.-X. Xu and J. Zhang, Chem. Mater., 2015, 27, 205 CrossRef CAS.
-
(a) X. Du, R.-Q. Fan, X.-M. Wang, G.-Z. Yu, L.-S. Qiang, P. Wang, S. Gao and Y.-L. Yang, Cryst. Growth Des., 2016, 16, 1737 CrossRef CAS;
(b) Y.-L. Hou, H. Xu, R.-R. Cheng and B. Zhao, Chem. Commun., 2015, 51, 6769 RSC;
(c) Z.-C. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815 RSC;
(d) G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040 CrossRef PubMed;
(e) X.-J. Zhang, W.-J. Wang, Z.-J. Hu, G.-N. Wang and K. Uvdal, Coord. Chem. Rev., 2015, 284, 206 CrossRef CAS.
-
(a) J. Sahoo, R. Arunachalam, P. S. Subramanian, E. Suresh, A. Valkonen, K. Rissanen and M. Albrecht, Angew. Chem., Int. Ed., 2016, 55, 9625 CrossRef CAS PubMed;
(b) W. Liu, T. Jiao, Y. Li, Q. Liu, M. Tan, H. Wang and L. Wang, J. Am. Chem. Soc., 2004, 126, 2280 CrossRef CAS PubMed;
(c) B. Chen, Y. Yang, F. Zapata, G. Lin, G. Qian and E. B. Lobkovsky, Adv. Mater., 2007, 19, 1693 CrossRef CAS;
(d) C. Zhan, S. Ou, C. Zou, M. Zhao and C.-D. Wu, Anal. Chem., 2014, 86, 6648 CrossRef CAS PubMed;
(e) J.-X. Ma, X.-F. Huang, X.-Q. Song and W.-S. Liu, Chem.–Eur. J., 2013, 19, 3590 CrossRef CAS PubMed.
- S. S. Nagarkar, B. Joarder, A. K. Chaudhari, S. Mukherjee and S. K. Ghosh, Angew. Chem., Int.
Ed., 2013, 52, 2881 CrossRef CAS PubMed.
- M. J. Dong, M. Zhao, S. Ou, C. Zou and C.-D. Wu, Angew. Chem., Int. Ed., 2014, 53, 1575 CrossRef CAS PubMed.
- P.-Y. Wu, Y.-H. Liu, Y. Liu, J.-R. Wang, Y. Li, W. Liu and J. Wang, Inorg. Chem., 2015, 54, 11046 CrossRef CAS PubMed.
-
(a) Y. Rachuri, B. Parmar, K. K. Bishta and E. Suresh, Dalton Trans., 2016, 45, 7881 RSC;
(b) L. Li, C.-X. Li, Y.-L. Ren, M. Song, Y. Ma and R.-D. Huang, CrystEngComm, 2016, 18, 7787 RSC;
(c) M. M. Wanderley, C. Wang, C.-D. Wu and W. Lin, J. Am. Chem. Soc., 2012, 134, 9050 CrossRef CAS PubMed;
(d) Q.-K. Liu, J.-P. Ma and Y.-B. Dong, Chem. Commun., 2011, 47, 7185 RSC.
- J. Fan, W.-Y. Sun, T. Okamura, W.-X. Tang and N. Ueyama, Inorg. Chem., 2003, 42, 3168 CrossRef CAS PubMed.
- G. M. Sheldrick, SADABS, University of Göttingen, Göttingen, Germany, 2003 Search PubMed.
-
(a) G. M. Sheldrick, SHELXS-2016, Program for the crystal structure solution, University of Göttingen, Göttingen, Germany, 2016 Search PubMed;
(b) G. M. Sheldrick, SHELXL-2016, Program for the crystal structure solution, University of Göttingen, Göttingen, Germany, 2016 Search PubMed.
-
(a) A. L. Spek, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 194 CrossRef;
(b) A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- V. A. Blatov, TOPOS, A Multipurpose Crystallochemical Analysis with the Program Package, Samara State University, Russia, 2009 Search PubMed.
- Y. Deng, P. Wang, Y. Zhao, Y.-S. Kang and W.-Y. Sun, Microporous Mesoporous Mater., 2016, 227, 39 CrossRef CAS.
- Y.-L. Li, D. Zhao, Y. Zhao, P. Wang, H.-W. Wang and W.-Y. Sun, Dalton Trans., 2016, 45, 8816 RSC.
- H.-Y. Zhang, R.-L. Guo, J.-P. Hou, Z. Wei and X.-Q. Li, ACS Appl. Mater. Interfaces, 2016, 8, 29044 CAS.
-
(a) F. Gandara, A. Andres, B. Gomez-Lor, E. Gutierrez-Puebla, M. Iglesias, M. A Monge, D. M. Proserpio and N. Snejko, Cryst. Growth Des., 2008, 8, 378 CrossRef CAS;
(b) Y. Zhou and B. Yan, Nanoscale, 2015, 7, 4063 RSC;
(c) Y. Lu and B. Yan, Chem. Commun., 2014, 50, 15443 RSC.
-
(a) R.-B. Fu, S.-C. Xiang, S.-M. Hu, L.-S. Wang, Y.-M. Li, X.-H. Huang and X.-T. Wu, Chem. Commun., 2005, 5292 RSC;
(b) Y.-Q. Huang, B. Ding, H.-B. Song, B. Zhao, P. Ren, P. Cheng, H.-G. Wang, D.-Z. Liao and S.-P. Yan, Chem. Commun., 2006, 4906 RSC;
(c) G.-Z. Liu, S.-H. Li, X.-L. Li, L.-Y. Xin and L.-Y. Wang, CrystEngComm, 2013, 15, 4571 RSC;
(d) J.-A. Hua, Y. Zhao, Q. Liu, D. Zhao, K. Chen and W.-Y. Sun, CrystEngComm, 2014, 16, 7536 RSC;
(e) Q. Hua, Y. Zhao, G. C. Xu, M. S. Chen, Z. Su, K. Cai and W.-Y. Sun, Cryst. Growth Des., 2010, 10, 2553 CrossRef CAS.
-
(a) X.-P. Wang, L.-L. Han, Z. Wang, L.-Y. Guo and D. Sun, J. Mol. Struct., 2016, 1107, 1 CrossRef CAS;
(b) J.-A. Hua, Y. Zhao, Y.-S. Kang, Y. Lu and W.-Y. Sun, Dalton Trans., 2015, 44, 11524 RSC.
- C. A. Bauer, T. V. Timofeeva, T. B. Settersten, B. D. Patterson, V.-H. Liu, B. A. Simmons and M. D. Allendorf, J. Am. Chem. Soc., 2007, 129, 7136 CrossRef CAS PubMed.
- W. Yang, J. Feng and H. Zhang, J. Mater. Chem., 2012, 22, 6819 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: crystallographic data, PXRD and TG data. CCDC 1564034–1564038. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra08165k |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a