
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, cytotoxic evaluation and DNA binding study of 9-fluoro-6H-indolo[2,3-b]quinoxaline derivatives†
Zhenyu Gu,
Yanci Li,
Songliang Ma,
Shenghui Li *,
Guoqiang Zhou*,
Shan Ding,
Jinchao Zhang,
Shuxiang Wang and
Chuanqi Zhou
*,
Guoqiang Zhou*,
Shan Ding,
Jinchao Zhang,
Shuxiang Wang and
Chuanqi Zhou
Key Laboratory of Chemical Biology of Hebei Province, Key Laboratory of Medicinal Chemistry and Molecular Diagnosis, Ministry of Education, College of Chemistry & Environmental Science, Hebei University, Baoding 071002, China. E-mail: lish@hbu.cn; zhougq1982@163.com; Fax: +86 0312 5079005; Tel: +86 0312 5079005
First published on 29th August 2017
Abstract
A series of novel 9-fluoro-6H-indolo[2,3-b]quinoxaline derivatives as antitumor agents were synthesized and studied. The designed compounds feature positive charges both at the 11-N position of the aromatic scaffold and the side-chain alkylamino group. The corresponding in vitro antitumor activities were evaluated against MCF-7, HeLa and A549 cancer cell lines and the DNA binding properties of these compounds were characterized by UV-vis, fluorescence, and circular dichroism (CD) spectroscopy as well as thermal denaturation. The obtained results indicate that the dicationic quaternary ammonium salt derivatives of 9-fluoro-6H-indolo[2,3-b]quinoxaline may serve as intercalators with increased DNA binding affinity. Furthermore, both the incorporation of fluorine, and alkyl amino side chains and the introduction of a positive charge at the 11-N position of the aromatic scaffold was shown to be responsible for their antitumor activity and improved DNA binding ability. Taken in concert, these findings may be valuable in the understanding of the antitumor effect of these compounds and may further provide important information for the future optimization of the DNA binding properties of this drug type.
1. Introduction
As the second leading cause of death worldwide, cancer accounts for nearly one in four of all mortality cases. It is estimated that about 10 million new cancer cases are diagnosed every year, causing a major health concern in both the developing and the developed countries.1 Despite advances in surgery and radiation therapy, chemotherapy still plays a crucial role in cancer treatment. However, the overall chemotherapeutic success is still limited by several drawbacks, including inadequate drug concentrations at the tumor site, undesirable toxicity, poor tumor cell selectivity over normal cells, and the occurrence of multiple drug resistance.2,3 Therefore, the discovery and development of selective, efficient, and safe drug types for anticancer chemotherapy remains a critical goal with high priority in medical research.Modification of existing active drug candidates, known as analogue design, is one of the most widely accepted approaches in drug discovery for the development of new drugs and improved therapeutic properties.4–9 Natural alkaloids and alkaloid derivatives, such as indoloquinoline alkaloid cryptolepine and pyridocarbazole alkaloid ellipticine, have attracted considerable attention due to their interesting biological activities demonstrated by both in vitro and in vivo studies. Their antitumor, mutagenic and cytotoxic activities were found to be due to a variety of mechanisms, including (i) DNA intercalation, (ii) inhibition of DNA topoisomerase II activity, and others.10 Unfortunately, toxicity issues have more or less limited the overall success of these drugs in pharmaceutical industry.11
In recent years, 6H-indolo[2,3-b]quinoxaline (cf. Fig. 1) have received much attention due to its important pharmacological activities, including antiviral (herpes simplex virus type 1 (HSV-1), cytomegalovirus (CMV), varicellazoster virus (VZV), etc.), cytotoxic and multidrug resistant (MDR) modulating activities, acting on ATP binding cassette transporters (ABC-transporters).12,13 6H-Indolo[2,3-b]quinoxaline combines the structural features of indoles and quinoxalines and may be regarded as an aza-analogue of the cytotoxic agents cryptolepine and ellipticine.14 The planar structure of this compound aids in the intercalation of DNA which in turn is responsible for biological activities such as cytotoxicity, antiviral activity, etc.12,13 The compound class has been well-studied and well-described in the literature, primarily by Bergman and co-workers.15–18 In recent years, several analogues of 6H-indolo[2,3-b]quinoxaline have been synthesized, e.g. B-220 (2,3-dimethyl-6-(2-dimethylaminoethyl)-6H-indolo[2,3-b]quinoxaline, Fig. 1), NCA0424, and others. Importantly, these compounds exhibited high antiviral activity and selective anticancer activity (Fig. 1).19–24 Hence, the indolo[2,3-b]quinoxaline nucleus was considered as a template for the design and development of DNA intercalating agents with cytotoxic activities in this study.
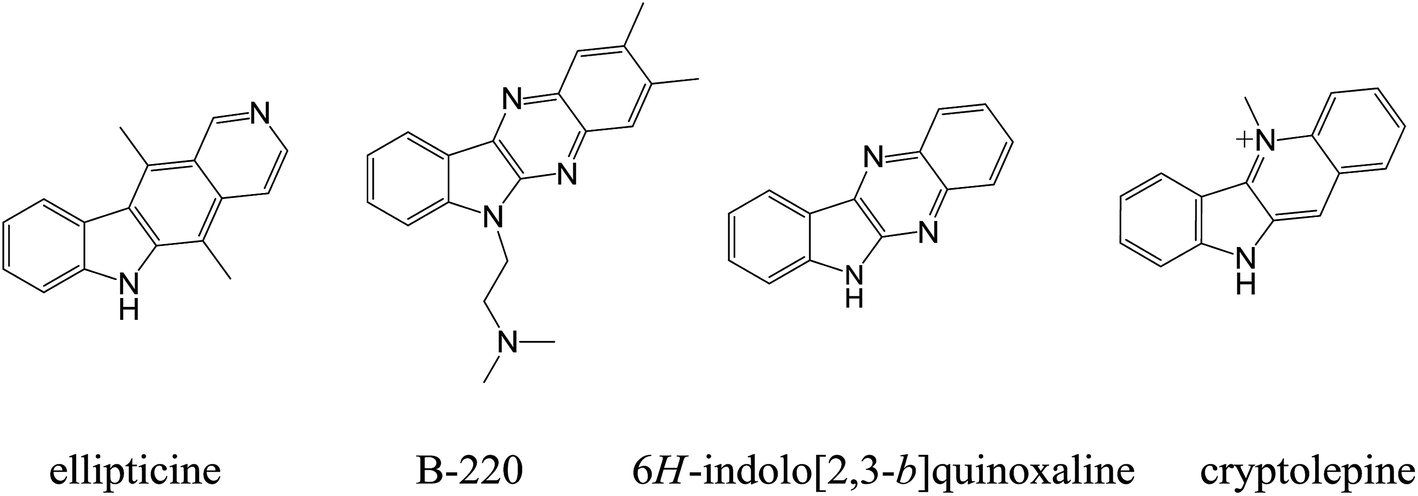 | ||
| Fig. 1 The structure of ellipticine, B-220, indolo[2,3-b]quinoxaline and cryptolepine. | ||
Owing to the special nature of fluorine as a functional group, the incorporation of fluorine usually conveys a variety of properties to certain pharmaceuticals and includes enhanced binding interactions, metabolic stability, changes in physical properties, and selective reactivities.25 Today, fluorine is present in up to 30% of pharmaceuticals. More recently, Karki et al. reported that 9-fluoro-6-(4-methylbenzyl)-6H-indolo[2,3-b]quinoxaline exhibited a potent anticancer activity against various human tumor cell lines.26 Meanwhile, it is generally appreciated that the transformation of neutral chromophores into their cationic derivatives usually enhances their affinity for DNA, thus increasing the overall antitumor activity.27,28 This is also the case for elliptinium, a cationic derivative of 9-methoxyellipiticine.29,30 Apart from the contribution of planar polycyclic aromatic systems, higher DNA binding affinity and biological activities may also be affected by the introduction of substituted groups such as amino side chains,11,31–34 sugars,35,36 or heterocycles37,38 to the chromophore. Furthermore, a cationic amino side chain may play an important role in the increase of the DNA binding affinity and biological activities due to their electrostatic interaction with the phosphate moieties of DNA. Moreover, chains of varying length, different polarity, structural rigidity, charge and steric bulk may also result in different DNA binding affinities.39
This work was designed to synthesize and study a group of novel 9-fluoro-6H-indolo[2,3-b]quinoxaline derivatives with improved DNA binding affinity and antitumor activity by attaching various alkyl amino side chains to the chromophore and introducing multiple positive charges via selective methylation at the nitrogens of the quinoxaline ring and the terminal amino-group. The DNA binding affinity of the resulting 9-fluoro-6H-indolo[2,3-b]quinoxaline derivatives was evaluated based on interaction with calf thymus (CT) DNA and the cytotoxicity of the compounds was assessed on three types of tumor cell lines, human mammary cancer (MCF-7) cells, human cervical carcinoma (HeLa) cells, and human lung cancer (A549) cells. Furthermore, the molecular structure and bioactivity relationships of the compounds were studied as discussed in subsequent sections.
2. Results and discussion
2.1. Synthesis and characterization
The synthesis of the quaternary mono- or dicationic indoloquinoxaline derivatives was carried out as depicted in Scheme 1. Isatin was obtained from commercial sources. 5-Fluoroisatin was synthesized according to a published procedure.40 9-Fluoro-6H-indolo[2,3-b]quinoxaline (2a) and 6H-indolo[2,3-b]quinoxaline (2d) were prepared by condensation of 5-fluoroisatin (or isatin) with 1,2-phenylenediamine in glacial acetic acid according to a procedure reported previously.41 The key intermediates of 6-bromoalkyl indolo[2,3-b]quinoxaline 3 were synthesized via alkylation by the use of excess dibromoalkane in tetrahydrofuran (THF) in the presence of potassium hydroxide. Amination using the corresponding cyclic secondary amines access lead to aminoalkyl-indoloquinoxalines 4. The methylation of 4 with iodomethane in different solvents (acetone or tetramethylenesulfone) selectively afforded the mono- or dicationic fluorinated indoloquinoxaline derivatives in excellent yield.42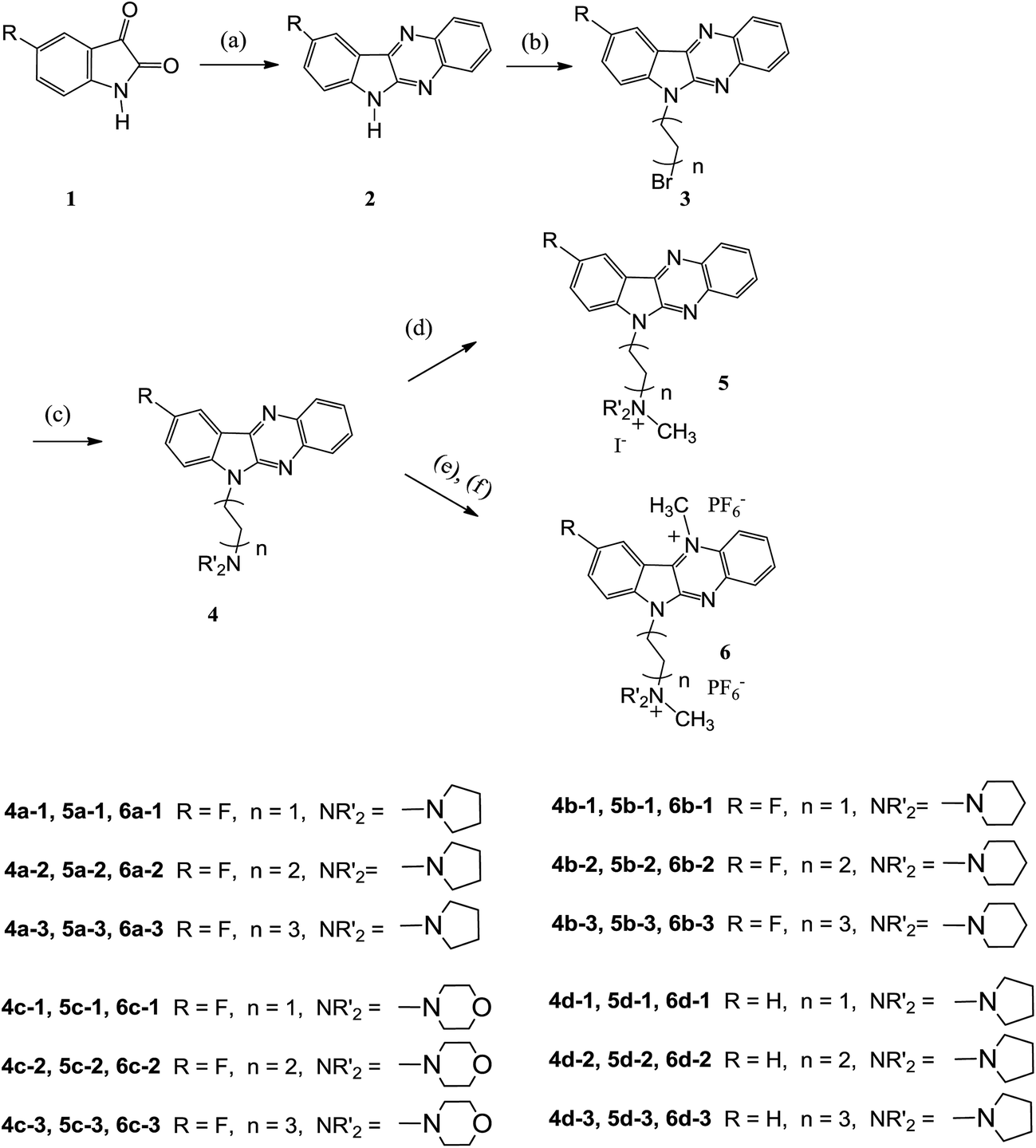 | ||
| Scheme 1 Synthesis of 9-fluoro-6H-indolo[2,3-b]quinoxaline derivatives. Reaction conditions: (a) o-phenylenediamine, acetic acid, reflux, 5 h; (b) dibromoalkane, KOH, THF, reflux, 6 h; (c) cyclic secondary amine, THF, reflux, N2; (d) CH3I, acetone; (e) CH3I, tetramethylenesulfone, 55 °C, overnight: (f) KPF6, H2O, 45 °C, 2 h. | ||
The structures of the target compounds were characterized by IR spectroscopy, mass-spectrometry, NMR-spectroscopy, and single crystal X-ray diffraction analysis of the precursor of compound 6b-1. Vibrations of aromatic C–H bonds were observed in the IR spectra of the obtained compounds at 3096–3035 cm−1, with aliphatic signals appearing at 2980–2753 cm−1. Double bond vibrations of heterocyclic fragments exhibited a band at 1621–1450 cm−1. The bands at 1236–1058 cm−1 corresponded to CH2–N vibrations. The mass spectra displayed the correct molecular ion peaks and the measured high resolution (HRMS) data was also in good agreement with the calculated values.
Two “parts” corresponding to the aromatic and aliphatic moieties of the molecules were observed in 1H NMR-spectra of the synthesized indoloquinoxalines derivatives. The spectral ranges for the aliphatic regions were different due to the presence of terminal amino groups. In the 13C NMR spectrum of the 9-fluoro-6H-indolo[2,3-b]quinoxaline derivatives, the carbons in the fluorinated aromatic ring resonated as doublets due to spin–spin coupling with the fluorine atom. The carbon–fluorine spin–spin coupling constants of 1JCF, 2JCF, 3JCF and 4JCF have been determined as 234.0–240.0 Hz, 22.0–25.5 Hz, 9.0–10.5 Hz and 3.0–4.5 Hz, respectively.
2.2. Structural studies
An X-ray crystal structure determination was performed to confirm the structure of the precursor of 6b-1 in the solid phase. A summary of data collection and refinement parameters is provided in Table 1. Based on crystallographic data, the molecular structure of the precursor of compound 6b-1 is shown in Fig. 2. The molecule adopted an almost planar configuration with the piperidinyl ring in the expected chair conformation. The dihedral angle between the two planes intersecting along the N,N-axis in N,N-dihydropyrazine was 0.0038° (109). The position of methylation on the nucleus was unambiguously observed at N3 instead of N2. Another methylated position was found at N4. The bond length of N3–C23 was 1.4738 Å (60), while the bond length of N4–C22 was 1.510 Å (6) and the bond length of C4–F1 was 1.3761 Å (43) (Table 2).| Formula | C23H29FI2N4O |
| Fw | 650.30 |
| T (K) | 296(2) |
| Cryst syst | Triclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a (Å) | 7.840(3) |
| b (Å) | 12.067(5) |
| c (Å) | 14.220(6) |
| V (nm3) | 1312.3(9) |
| Z | 2 |
| Dc (Mg m−3) | 1.646 |
| F (000) | 636 |
| Cryst dimens (mm) | 0.35 × 0.15 × 0.13 |
| θ Range (deg) | 2.18–26.00 |
| hkl ranges | −9 < h < 7 |
| −14 < k < 14 | |
| −17 < l < 15 | |
| Data/parameters | 5085/290 |
| Goodness-of-fit on F2 | 1.072 |
| Final R indices [I > 2σ(I)] | R1 = 0.0342 |
| wR2 = 0.0831 |
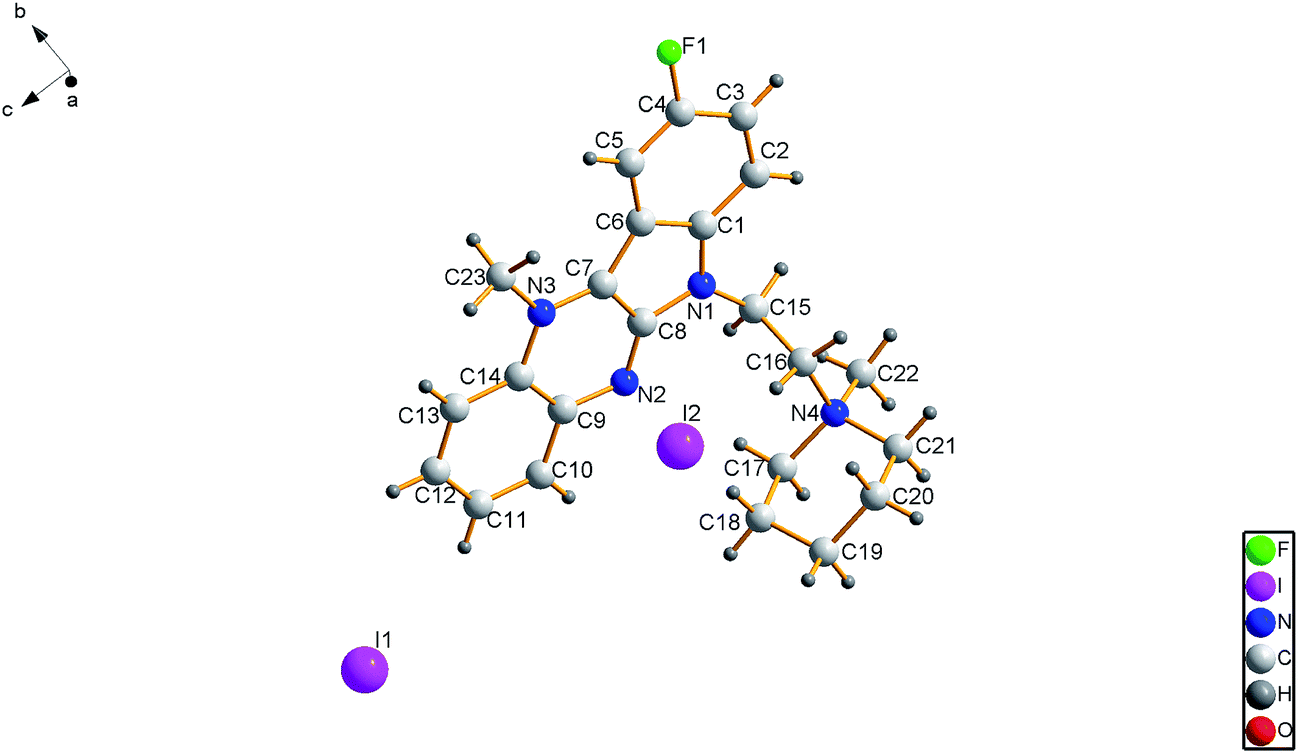 | ||
| Fig. 2 ORTEP view of the precursor of 6b-1. | ||
| Length/Å or angle/° | |||
|---|---|---|---|
| F(1)–C(4) | 1.376(5) | C(5)–C(4)–F(1) | 118.2(4) |
| N(1)–C(8) | 2.281(4) | F(1)–C(4)–C(3) | 117.1(4) |
| N(2)–C(8) | 1.310(5) | C(8)–N(1)–C(1) | 108.9(3) |
| N(2)–C(9) | 1.360(5) | C(8)–N(2)–C(9) | 114.5(3) |
| N(3)–C(7) | 1.343(4) | C(7)–N(3)–C(23) | 119.3(3) |
| N(3)–C(14) | 1.399(5) | C(7)–N(3)–C(14) | 119.0(3) |
| N(3)–C(23) | 1.473(5) | C(14)–N(3)–C(23) | 121.7(3) |
| N(4)–C(22) | 1.510(5) | C(17)–N(4)–C(16) | 112.6(3) |
| N(4)–C(17) | 1.520(5) | C(22)–N(4)–C(21) | 108.3(3) |
| N(4)–C(16) | 1.521(5) | C(17)–N(4)–C(21) | 110.1(3) |
| N(4)–C(21) | 1.526(5) | C(22)–N(4)–C(16) | 109.6(3) |
2.3. Cytotoxicity studies
The antitumor activities of aminoalkyl-indoloquinoxalines 4 as well as the quaternary mono- and dicationic indoloquinoxaline derivatives 5 and 6 were evaluated in vitro against tumor cell lines MCF-7, HeLa and A549, respectively, as listed in Tables 3–5. 9-Fluoro-6H-indolo[2,3-b]quinoxaline (FIQX) and cisplatin were used as positive controls. According to the data shown in Tables 3–5, most of the synthesized quaternary dicationic FIQX derivatives were found to be more active than the corresponding quaternary monocationic FIQX derivatives and the neutral 6H-indolo-[2,3-b]quinoxaline derivatives.| Compounds | IC50 (μM)a | ||
|---|---|---|---|
| MCF-7 | Hela | A549 | |
| a IC50 values were calculated from the linear regression of the dose–log response curves. Values are mean ± S.D. of at least three independent experiments.b Ref. 43.c Ref. 44.d Ref. 45. | |||
| 4a-1 | 3.68 ± 0.36 | 5.69 ± 0.48 | 4.32 ± 0.57 |
| 4a-2 | 8.11 ± 0.52 | 7.65 ± 1.24 | 6.36 ± 0.88 |
| 4a-3 | 4.76 ± 0.31 | 4.25 ± 0.59 | 4.13 ± 0.33 |
| 4b-1 | 45.58 ± 5.99 | 19.38 ± 2.10 | 25.70 ± 3.53 |
| 4b-2 | 34.33 ± 4.65 | 13.14 ± 1.46 | 9.01 ± 0.89 |
| 4b-3 | 28.89 ± 3.37 | 8.61 ± 1.38 | 10.23 ± 1.05 |
| 4c-1 | 42.68 ± 4.76 | 27.72 ± 3.48 | 53.41 ± 6.64 |
| 4c-2 | 11.85 ± 0.98 | 15.76 ± 1.04 | 15.00 ± 2.21 |
| 4c-3 | 5.48 ± 0.43 | 15.49 ± 2.11 | 19.70 ± 1.98 |
| 4d-1 | 3.69 ± 0.32 | 6.63 ± 0.55 | 4.36 ± 0.29 |
| 4d-2 | 10.50 ± 1.76 | 16.97 ± 2.10 | 7.45 ± 0.68 |
| 4d-3 | 6.44 ± 0.65 | 9.58 ± 1.03 | 3.38 ± 0.44 |
| FIQX | 15.44 ± 1.46 | 20.83 ± 2.22 | 24.99 ± 3.19 |
| Cisplatin | 8.25 ± 0.53b | 11.06 ± 1.08c | 8.61 ± 0.39d |
| Compounds | IC50 (μM)a | ||
|---|---|---|---|
| MCF-7 | Hela | A549 | |
| a IC50 values were calculated from the linear regression of the dose–log response curves. Values are mean ± S.D. of at least three independent experiments.b Ref. 43.c Ref. 44.d Ref. 45. | |||
| 5a-1 | 22.31 ± 3.12 | 22.64 ± 2.43 | 20.28 ± 3.04 |
| 5a-2 | 31.57 ± 3.67 | 23.86 ± 2.75 | 75.99 ± 6.98 |
| 5a-3 | 16.12 ± 1.31 | 30.43 ± 3.66 | 17.82 ± 2.15 |
| 5b-1 | 29.65 ± 3.34 | 22.88 ± 4.02 | 17.83 ± 1.60 |
| 5b-2 | 16.58 ± 0.89 | 24.96 ± 3.57 | 16.78 ± 2.31 |
| 5b-3 | 12.45 ± 0.81 | 27.28 ± 2.64 | 16.25 ± 1.42 |
| 5c-1 | 12.92 ± 1.34 | 18.09 ± 1.73 | 12.33 ± 0.51 |
| 5c-2 | 14.38 ± 2.21 | 30.77 ± 4.03 | 22.35 ± 2.59 |
| 5c-3 | 19.19 ± 1.47 | 21.62 ± 2.85 | 26.84 ± 3.07 |
| 5d-1 | 8.70 ± 0.82 | 8.56 ± 0.67 | 7.80 ± 1.20 |
| 5d-2 | 14.75 ± 2.18 | 9.21 ± 0.62 | 12.81 ± 0.97 |
| 5d-3 | 8.66 ± 0.42 | 10.31 ± 1.23 | 11.41 ± 0.86 |
| FIQX | 15.44 ± 1.46 | 20.83 ± 2.22 | 24.99 ± 3.19 |
| Cisplatin | 8.25 ± 0.53b | 11.06 ± 1.08c | 8.61 ± 0.39d |
| Compounds | IC50 (μM)a | ||
|---|---|---|---|
| MCF-7 | Hela | A549 | |
| a IC50 values were calculated from the linear regression of the dose–log response curves. Values are mean ± S.D. of at least three independent experiments.b Ref. 43.c Ref. 44.d Ref. 45. | |||
| 6a-1 | 3.14 ± 0.33 | 5.05 ± 0.41 | 6.31 ± 0.70 |
| 6a-2 | 3.54 ± 0.26 | 3.87 ± 0.35 | 5.68 ± 0.42 |
| 6a-3 | 1.80 ± 0.24 | 4.22 ± 0.63 | 5.25 ± 0.39 |
| 6b-1 | 2.89 ± 0.38 | 4.08 ± 0.37 | 6.09 ± 0.91 |
| 6b-2 | 1.94 ± 0.29 | 6.04 ± 0.67 | 8.45 ± 1.28 |
| 6b-3 | 1.60 ± 0.31 | 3.34 ± 0.55 | 6.86 ± 1.30 |
| 6c-1 | 7.23 ± 1.06 | 4.73 ± 0.49 | 6.82 ± 0.88 |
| 6c-2 | 4.47 ± 0.57 | 10.43 ± 1.38 | 6.96 ± 0.59 |
| 6c-3 | 3.01 ± 0.25 | 8.31 ± 0.66 | 9.70 ± 0.74 |
| 6d-1 | 3.73 ± 0.44 | 22.15 ± 2.61 | 13.33 ± 1.81 |
| 6d-2 | 3.99 ± 0.35 | 16.12 ± 1.55 | 9.34 ± 1.02 |
| 6d-3 | 2.02 ± 0.30 | 9.69 ± 0.77 | 6.20 ± 0.67 |
| FIQX | 15.44 ± 1.46 | 20.83 ± 2.22 | 24.99 ± 3.19 |
| Cisplatin | 8.25 ± 0.53b | 11.06 ± 1.08c | 8.61 ± 0.39d |
As shown in Table 3, most of the neutral 6H-indolo[2,3-b]quinoxaline derivatives with various alkyl amino side chains showed promising cytotoxicity properties against the tested carcinoma cell lines with low IC50 values. Compounds, which contain pyrrolidine as terminal amino-group, i.e. compound 4a-1, 4a-2 and 4a-3, exhibited potent antiproliferative activities, comparable to the compounds containing piperidinyl and morpholinyl as terminal amino group. Besides, the introduction of fluorine into the aromatic core of 6H-indolo[2,3-b]quinoxaline seems to enhance the antiproliferative activities of the corresponding compounds as judged by a comparison of the fluorinated indoloquinoxaline derivatives 4a-1, 4a-2 and 4a-3 as well as the non-fluorinated compounds 4d-1, 4d-2 and 4d-3.
As for the monocationic quaternary ammonium salts of 6H-indolo[2,3-b]quinoxaline (5), the N-methylation of the terminal amino-group resulted in an apparent decrease of cytotoxicity against MCF-7, HeLa and A549 cells, except for compound 5c-1.
As for the dicationic quaternary ammonium salts of 6H-indolo[2,3-b]quinoxaline, the preliminary results demonstrated that most of the compounds were more toxic against MCF-7 than HeLa and A549 cells. Firstly, this compound series exhibited potent antiproliferative activities compared to the corresponding monocationic quaternary ammonium salts derivatives of 6H-indolo[2,3-b]quinoxaline. For example, compounds 6a-3, 6b-3 and 6c-3 exhibited an increased antiproliferative activity against MCF-7, with IC50 values of 1.80, 1.60 and 3.01 μM, while IC50 values for 5a-3, 5b-3 and 5c-3 of 16.12, 12.45 and 19.19 μM were determined, respectively. It may be concluded that N-methylation at 11-position of the indoloquinoxaline ring is essential for the cytotoxicity of 6H-indolo[2,3-b]quinoxaline derivatives. Secondly, the fluorinated dicationic quaternary ammonium salts of 6H-indolo[2,3-b]quinoxaline also showed an increased activity compared to the non-fluorinated derivatives. For example, the IC50 values of compounds 6a-1, 6a-2 and 6a-3 were 3.14, 3.54 and 1.80 μM, while the IC50 values of compounds 6d-1, 6d-2 and 6d-3 were 3.73, 3.99 and 2.02 μM, respectively. Additionally, the fluorinated dicationic quaternary ammonium salt derivatives 6a–6c exhibited higher activities than cisplain against all tested cell lines, however, compound 6c-3 proved to be an exception. Thirdly, the anti-tumor activity results against MCF-7 cells suggested that the dicationic quaternary ammonium salts of 6H-indolo[2,3-b]quinoxaline with the largest alkyl chains, such as compounds 6a-3, 6b-3, 6c-3 and 6d-3, were more active than compounds with shorter alkyl chains as linkers attached between the terminal amino group and the indolo[2,3-b]quinoxaline nucleus.
2.4. DNA-binding property
To evaluate the DNA-binding properties of the cryptolepine analogues, UV-vis absorption spectra, fluorescent spectra, and circular dichroism (CD) spectra were recorded and a thermal denaturation study of the selected compounds 5a-1, 5a-2, 6a-1, 6d-1 and 6b-3 was carried out with CT DNA.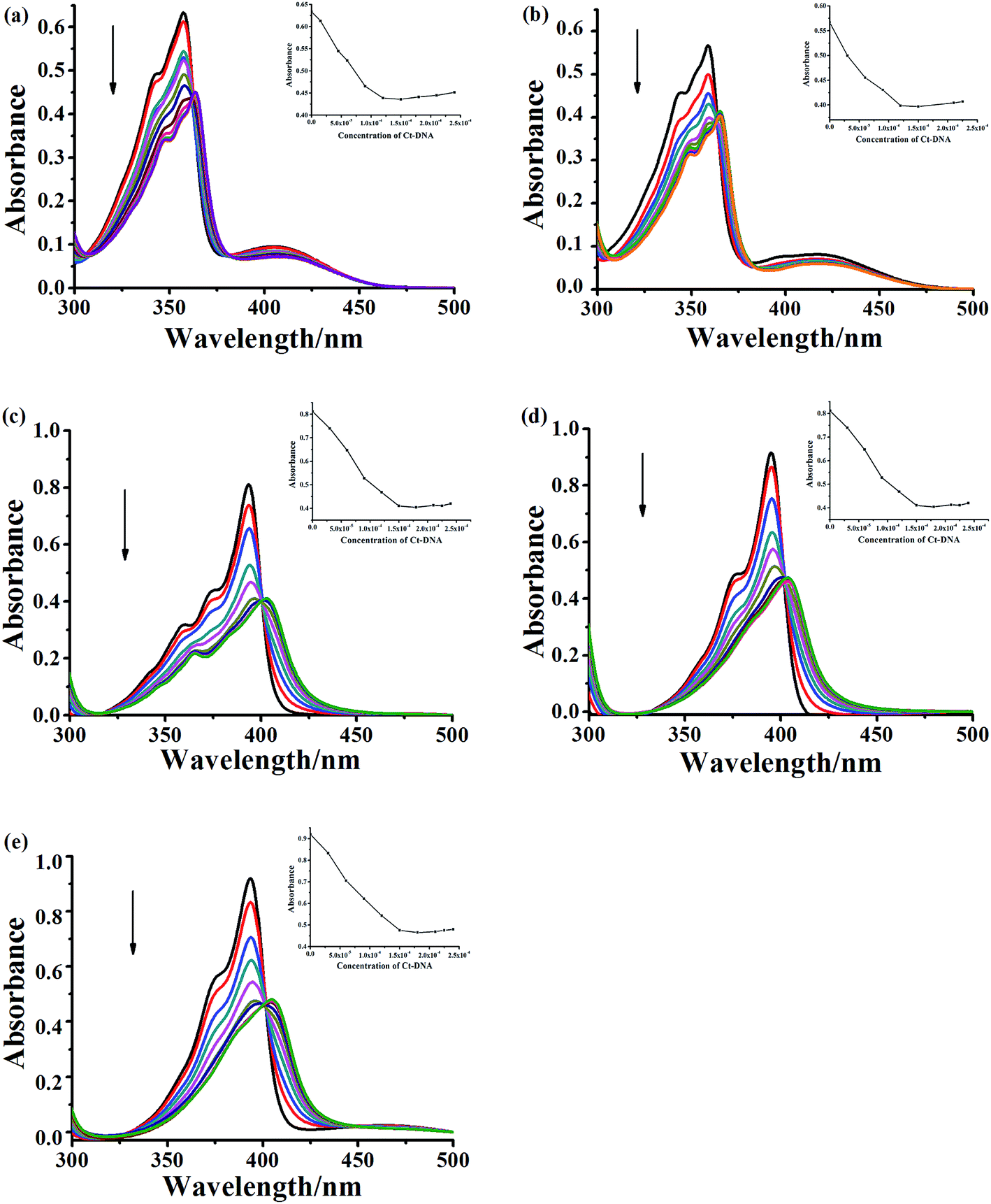 | ||
| Fig. 3 UV-vis spectral changes of compound 5a-1 (a), 5a-2 (b), 6a-1 (c), 6d-1 (d) and 6b-3 (e) at the concentration of 3.0 × 10−5 M upon addition of CT DNA (arrow: 0–210 μM) in phosphate buffer (1 mM, pH 7.4) containing 20 mM NaCl at 25 °C. | ||
To further evaluate and compare the DNA binding performance of these compounds, the binding constants were determined from the following equation:
| [DNA]/(εa − εf) = [DNA]/(εb − εf) + 1/Kb(εb − εf) |
In addition, other variation in the structure, such as the linker length, terminal amino-group and introduction of fluorine atom in the indoloquinoxaline moiety, are shown to have an immediate significant influence on the DNA binding ability. For monocationic quaternary ammonium salts, the binding constants decrease as the linker length increases (5a-1 > 5a-2). The compounds with pyrrolidine at the end of side chain show more stronger CT DNA binding affinity than that of compound decorated with piperidine (6a-1 > 6b-3). The introduction of fluorine atom into the aromatic core could also enhance DNA binding affinity (6a-1 > 6d-3).
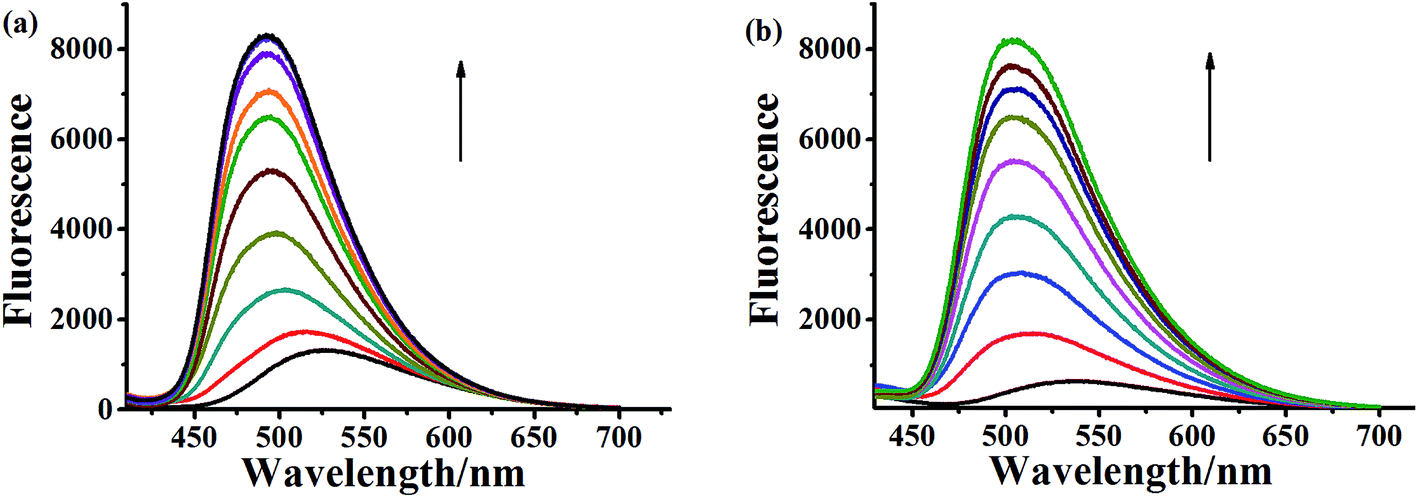 | ||
| Fig. 4 Fluorescence spectral changes of compound 5a-1 (a) and 5a-2 (b) at the concentration of 3.0 × 10−5 M upon addition of CT DNA (arrow: 0–210 μM) in phosphate buffer (1 mM, pH 7.4) containing 20 mM NaCl at 25 °C. | ||
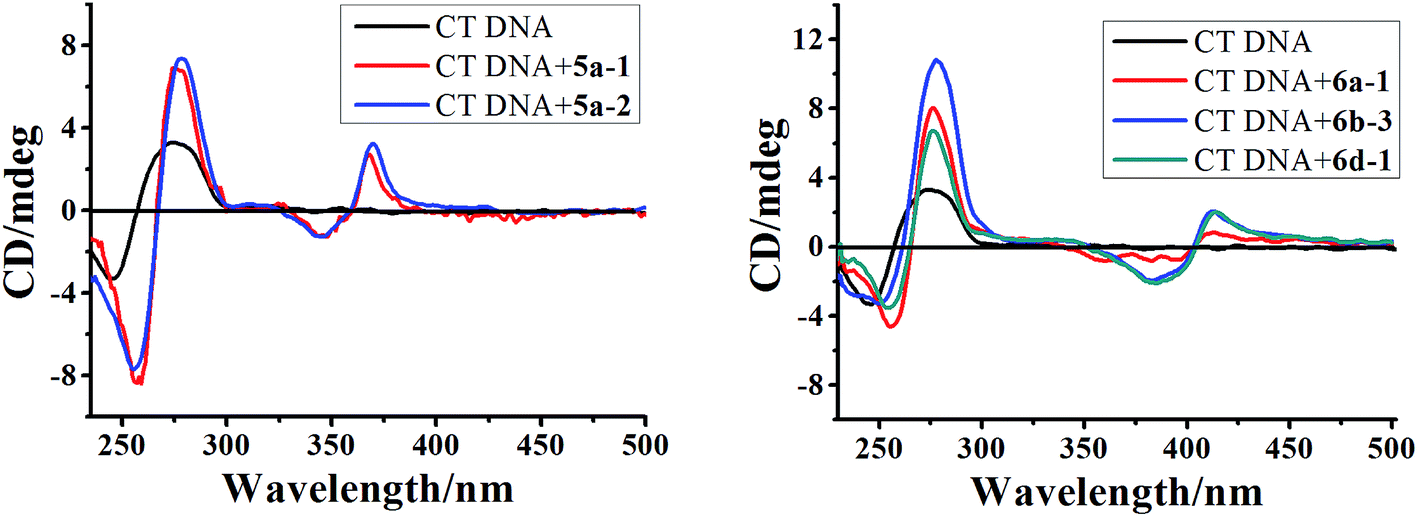 | ||
| Fig. 5 CD spectra of CT DNA (6.0 × 10−5 M) in the absence and presence of 5a-1, 5a-2, 6a-1, 6b-3 and 6d-1 (5.0 × 10−5 M) in phosphate buffer (1 mM, pH 7.4) containing 20 mM NaCl at 25 °C. | ||
Further evidence for the binding modes of these compounds was obtained from the ICD spectra in the range of 325–450 nm corresponding to an indoloquinoxaline chromophore. The ICD signal, displayed by these achiral molecules as a result of interaction with the chiral DNA, usually depends on the position and orientation with respect to the polynucleotide. Generally, adducts displaying intercalation binding characteristics were found to exhibit a positive or negative ICD.50 Intriguingly, ICD spectra of the tested compounds and CT DNA exhibited a weaker and biphasic ICD signal. The biphasic shape of the CD spectrum, i.e. a negative band at lower wavelengths and a positive band at higher wavelengths, is characteristic of chromophores with a right-hand orientation, as expected for dyes bound to B-form DNA.51 The exciton CD observed in the drug absorption region is generally considered as evidence of groove binding or external stacking.44 However, the groove binding mode should be ruled out in this case due to the following consideration. Firstly, the ICD of groove binders is generally an order of magnitude stronger than those of intercalators and usually carries a positive sign if DNA is in B-form.48,52 Secondly, in the absorption titration we have demonstrated that these drugs mixed with CT DNA still contain a partially free chromophore which did not interfere with the DNA helix. This in turn may be due to the notion that an external binding mode may exist simultaneously. Thirdly, considering the positive nature of these compounds and negatively charged polyphosphate skeleton of DNA, the electrostatic interactions between the intercalator and DNA has to be considered. Taken in concert, these results, together with light absorption characteristics, may suggest multiple binding modes including intercalation as major and external binding as minor mode.
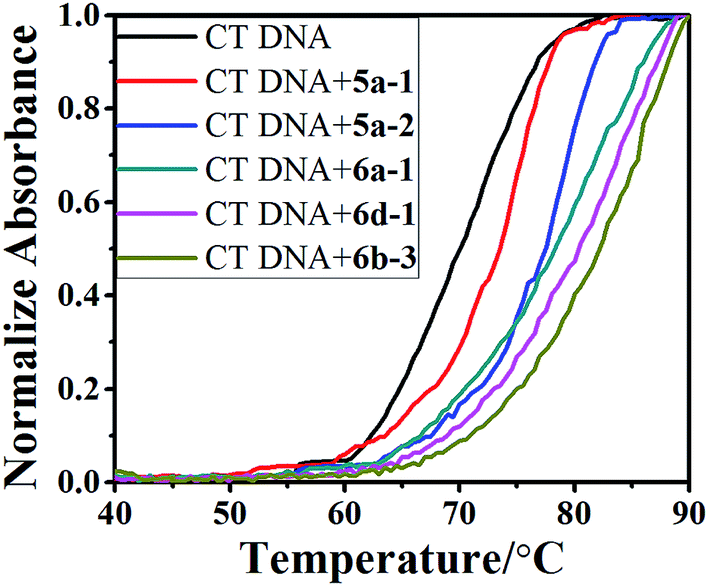 | ||
| Fig. 6 DNA melting curves for CT DNA (5.0 × 10−5 M) in the absence and presence of 5a-1, 5a-2, 6a-1, 6d-1 and 6b-3 with concentration of 5.0 × 10−6 M in phosphate buffer (1 mM, pH 7.4) containing 20 mM NaCl. | ||
| Compound | Tm (°C) | ΔTm (°C) |
|---|---|---|
| CT DNA | 69.4 | 0 |
| 5a-1 | 73.5 | 4.1 |
| 5a-2 | 77.8 | 8.4 |
| 6a-1 | 79.3 | 9.9 |
| 6b-3 | 81.1 | 11.7 |
| 6d-1 | 80.2 | 10.8 |
3. Conclusions
A series of novel 9-fluoro-6H-indolo[2,3-b]quinoxaline derivatives functionalized with amino side chains and their monocationic/dicationic quaternary ammonium salts were designed and synthesized and their interactions with CT DNA were studied. The incorporation of fluorine, alkyl amino side chains and the introduction of positive charge at 11-N position of the aromatic scaffold were found to significantly influence the antitumor activity and further demonstrated to enhance the binding affinity of the compounds with DNA. The antitumor activity and the DNA binding affinity of most of the synthesized quaternary dicationic FIQX derivatives were found to be significantly higher compared to the corresponding monocationic FIQX derivatives and the neutral 6H-indolo[2,3-b]quinoxaline derivatives. The synthesized dicationic FIQX derivatives with pyrrolidine or piperidinyl as terminal amino-groups exhibited a higher inhibitory activity. The molecular structure–DNA binding relationships assessed in this study may be useful for the design of 6H-indolo[2,3-b]quinoxaline derivatives with desired DNA binding properties and promising antitumor abilities.4. Experimental section
4.1. Materials
All chemicals and reagents were of analytical grade. RPMI-1640 medium, trypsin and fetal bovine serum were purchased from Gibco (Grand Island, NY, USA). CT DNA, MTT, benzylpenicillin and streptomycin were purchased from Sigma-Aldrich (St. Louis, MO, USA). Three different human carcinoma cell lines, MCF-7, HeLa and A549, were obtained from American Type Culture Collection.4.2. Instrumentation and measurements
The melting points were determined on an XT-4 microscopic melting-point spectrometer and are shown uncorrected. Elemental analysis was performed on an Elementar Vario EL III elemental analyzer. Infrared (IR) spectra were measured on a Perkin-Elmer Model-683 spectrophotometer, with samples analyzed as KBr pellets. The 1H and 13C NMR spectra were recorded on a Bruker AVIII 600 NMR spectrometer. Mass spectra were measured using an Agilent 1200-6310 LC-MS apparatus. High-resolution mass spectra (HRMS) were recorded on a Bruker Apex Ultra 7.0T FTMS mass spectrometer. UV-vis absorption spectra were recorded on a Shimadzu UV-3600 UV-vis spectrophotometer and fluorescent spectra were measured on a Hitachi F-7000 luminescence spectrophotometer. The optical density (OD) was measured on a microplate spectrophotometer (Bio-Rad Model 680, USA). The yield, IR, 1H NMR, 13C NMR and high-resolution electrospray ionization (HRESI) MS spectral data of the obtained compounds are provided in the ESI section.†4.3. Synthesis of compounds
4.4. Procedure for in vitro cytotoxictity assessment
In vitro cytotoxictity of the obtained compounds was evaluated by a MTT colorimetric assay.54 Three different human carcinoma cell lines, MCF-7, HeLa and A549, were cultured in Dulbecco's modified Eagel's medium (DMEM, Gibco, USA) solution supplemented with 10% fetal bovine serum, 100 U mL−1 of penicillin, and 100 μg mL−1 of streptomycin. The cancer cells were seeded at a density of 1.5 × 104 cells per well into a 96-well microplate pre-incubated in a 5% CO2/95% air-humidified atmosphere at 37 °C for 12 h to acquire adherent cells.Prior to dilution, a stock solution of the compounds (5 mM) was prepared in DMSO. The 50% inhibitory concentration (IC50) was determinated over a range of concentrations (0.1–100 μM). In all experiments, the concentration of DMSO was less than 0.1% (v/v) to avoid DMSO toxicity.
Wells containing culture medium without cells were used as blanks. Wells containing culture medium, FIQX and cisplatin were used as positive control. After incubation for 48 h with the test compounds, 20 μL of MTT solution (5 mg mL−1) was added to each well and the mixtures were incubated for an additional 4 h. Then, 100 μL of DMSO was added to solubilize the MTT formazan and the OD of each well was measured on a microplate spectrophotometer at a wavelength of 570 nm. The IC50 value was determined from plot of the %-viability against a defined dose of added compounds. Three independent experiments were performed in quintuplicate.
4.5. Procedure for DNA binding studies
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Nature Science Fund of Hebei Province (B2015201213, B2015201069), the Key Basic Research Special Foundation of Science Technology Ministry of Hebei Province (15962602D), the Key Research Project Foundation of Department of Education of Hebei Province (Grant No. ZH2012041).References
- R. L. Siegel, K. D. Miller and A. Jemal, Cancer Statistics, Ca-Cancer J. Clin., 2015, 65, 5–29 CrossRef PubMed.
- B. G. Jackson, Science, 2000, 287, 1969–1973 CrossRef.
- American Cancer Society, Inc., Surveillance Research. Cancer Faiths and Figures, 2005, http://www.geocites.com.
- J. Fischer and C. R. Ganellin, Analogue-based drug discovery, Wiley-VCH, Weinheim, 2006, pp. 1–24 Search PubMed.
- X. B. Zhou, M. Chen, Z. Y. Zheng, G. Y. Zhu, Z. H. Jiang and L. P. Bai, RSC Adv., 2017, 7, 26921–26929 RSC.
- B. Zhang, R. Y. Guo, Y. Z. Hu, X. W. Dong, N. M. Lin, X. Y. Dai, H. H. Wu, S. L. Ma and B. Yang, RSC Adv., 2017, 7, 31899–31906 RSC.
- Y. Chen, H. Z. Lin, J. Zhu, K. Gu, Q. Li, S. Y. He, X. Lu, R. X. Tan, Y. Q. Pei, L. Wu, Y. Y. Bian and H. P. Sun, RSC Adv., 2017, 7, 33851–31867 RSC.
- T. Zawadowski and M. Klimaszewska, Acta Pol. Pharm., 1995, 52, 249–252 CAS.
- S. T. Hazeldine, L. Polin, J. Kushner, K. White, T. H. Corbett and J. P. Horwitz, Bioorg. Med. Chem., 2006, 14, 2462–2467 CrossRef CAS PubMed.
- P. J. Aragon, A. D. Yapi, F. Pinguet, J. M. Chezal, J. C. Teulade and Y. Blache, Chem. Pharm. Bull., 2007, 55, 1349–1355 CrossRef CAS PubMed.
- L. M. Wilhelmsson, N. Kingi and J. Bergman, J. Med. Chem., 2008, 51, 7744–7750 CrossRef CAS PubMed.
- N. S. H. N. Moorthy, E. Manivannan, C. Karthikeyan and P. Trivedi, Mini-Rev. Med. Chem., 2013, 13, 1415–1420 CrossRef PubMed.
- N. S. H. N. Moorthy, C. Karthikeyan and P. Trivedi, J. Enzyme Inhib. Med. Chem., 2010, 25, 394–405 CrossRef CAS PubMed.
- M. Stiborová, J. Poljaková, E. Martínková, L. Borek-Dohalska, T. Eckschlager, R. Kizek and E. Frei, Interdiscip. Toxicol., 2011, 4, 98–105 Search PubMed.
- A. M. A. Osman, E. B. Pedersen and J. Bergman, Nucleosides, Nucleotides Nucleic Acids, 2013, 32, 98–108 CAS.
- N. Patel, J. Bergman and A. H. Gräslund, Eur. J. Biochem., 1991, 197, 597–604 CrossRef CAS PubMed.
- I. Zegar, A. Gräslund, J. Bergman, M. Eriksson and B. Nordén, Chem.-Biol. Interact., 1989, 72, 277–293 CrossRef CAS PubMed.
- J. Bergman, C. Damberg and H. Valberg, Recl. Trav. Chim. Pays-Bas, 1996, 115, 31–36 CrossRef CAS.
- U. Sehlstedt, P. Aich, J. Bergman, H. Vallberg, B. Nordén and A. Gräslund, J. Mol. Biol., 1998, 278, 31–56 CrossRef CAS PubMed.
- J. Harmenberg, A. Åkesson-Johansson, A. Gräslund, T. Malmfors, J. Bergman, B. Wahren, S. Åkerfeldt, L. Lundblad and S. Cox, Antiviral Res., 1991, 15, 193–204 CrossRef CAS PubMed.
- K. Hirata, J. Araya, S. Nakaike, K. Kitamura and T. Ishida, Chem. Pharm. Bull., 2001, 49, 44–48 CrossRef CAS PubMed.
- T. Ishida, Y. Mihara, Y. Hama, A. Hanatani, M. Tarui, M. Doi, S. Nakaike and K. Kitamura, Chem. Pharm. Bull., 1998, 46, 739–743 CrossRef CAS PubMed.
- S. P. Nikumbh, A. Raghunadh, V. N. Murthy, R. Jinkala, S. C. Joseph, Y. L. N. Murthy, B. Prasad and M. Pal, RSC Adv., 2015, 5, 74570–74574 RSC.
- S. P. Nikumbh, A. Raghunadh, T. S. Rao, V. N. Murthy, S. C. Joseph, Y. L. N. Murthy and M. Pal, RSC Adv., 2016, 6, 23489–23497 RSC.
- K. W. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef PubMed.
- S. S. Karki, R. Hazare, S. Kumar, V. S. Bhadauria, J. Balzarini and E. De Clercq, Acta Pharm., 2009, 59, 431–440 CrossRef CAS PubMed.
- M. F. Braña, M. Cacho, A. Gradillas, B. de Pascual-Teresa and A. Ramos, Curr. Pharm. Des., 2001, 7, 1745–1780 CrossRef.
- D. Monchaud and M. P. Teulade-Fichou, Org. Biomol. Chem., 2008, 6, 627–636 CAS.
- G. W. Gribble, Synthesis and antitumor activity of ellipticine alkaloids and related compounds, in The Alkaloids, ed. A. Brossi, Academic Press, New York, 1990, vol. 39, pp. 239–343 Search PubMed.
- V. K. Kansal and P. Potier, Tetrahedron, 1986, 32, 2389–2408 CrossRef.
- M. O. Shibinskaya, S. A. Lyakhov, A. V. Mazepa, S. A. Andronati, A. V. Turov, N. M. Zholobak and N. Y. Spivak, Eur. J. Med. Chem., 2010, 45, 1237–1243 CrossRef CAS PubMed.
- M. O. Shibinskaya, A. S. Karpenko, S. A. Lyakhov, S. A. Andronati, N. M. Zholobak, N. Y. Spivak, N. A. Samochina, L. M. Shafran, M. J. Zubritsky and V. F. Galat, Eur. J. Med. Chem., 2011, 46, 794–798 CrossRef CAS PubMed.
- P. B. Arimondo, B. Baldeyrou, W. Laine, C. Bal, F. A. Alphonse, S. Routier, G. Coudert, J. Y. Mérour, P. Colson, C. Houssier and C. Bailly, Chem.-Biol. Interact., 2001, 138, 59–75 CrossRef CAS PubMed.
- M. O. Shibinskaya, N. A. Kutuzova, A. V. Mazepa, S. A. Lyakhov, S. A. Andronati, M. J. Zubritsky, V. F. Galat, J. Lipkowski and V. C. Kravtsov, J. Heterocycl. Chem., 2012, 49, 678–682 CrossRef CAS.
- K. M. Driller, S. Libnow, M. Hein, M. Harms, K. Wende, M. Lalk, D. Michalik, H. Reinke and P. Langer, Org. Biomol. Chem., 2008, 6, 4218–4223 CAS.
- M. C. Wamberg, A. A. Hassan, A. D. Bond and E. B. Pedersen, Tetrahedron, 2006, 62, 11187–11199 CrossRef CAS.
- S. Avula, J. R. Komsani, S. Koppireddi, R. Yadla, A. K. R. Kanugula and S. Kotamraju, Med. Chem. Res., 2013, 22, 3712–3718 CrossRef CAS.
- A. H. Abadi, Arch. Pharm., 1998, 331, 352–358 CrossRef CAS PubMed.
- J. H. Tan, Q. X. Zhang, Z. S. Huang, Y. Chen, X. D. Wang, L. Q. Gu and J. Y. Wu, Eur. J. Med. Chem., 2006, 41, 1041–1047 CrossRef CAS PubMed.
- X. Q. Chen and J. M. Gu, App. Chem. Ind., 2007, 36, 901–902 CAS.
- V. Q. Yen, N. P. Buu-Hoi and N. D. Xuong, J. Org. Chem., 1958, 23, 1858–1861 CrossRef CAS.
- P. Helissey, S. Desbene-Finck and S. Giorgi-Renault, Eur. J. Org. Chem., 2005, 410–415 CrossRef CAS.
- Z. F. Chen, S. P. Zhang, J. Zhang and Z. Z. Zhu, New J. Chem., 2017, 41, 6760–6768 RSC.
- S. J. T. Rezaei, V. Amani, M. R. Nabid, N. Safari and H. Niknejad, Polym. Chem., 2015, 6, 2986 RSC.
- R. Raveendran, J. P. Braude, E. Wexselblatt, V. Novohradsky, O. Stuchlikova, V. Brabec, V. Gandin and D. Gibson, Chem. Sci., 2016, 7, 2381–2391 RSC.
- Y. J. Lu, T. M. Ou, J. H. Tan, J. Q. Hou, W. Y. Shao, D. Peng, N. Sun, X. D. Wang, W. B. Wu, X. Z. Bu, Z. S. Huang, D. L. Ma, K. Y. Wong and L. Q. Gu, J. Med. Chem., 2008, 51, 6381–6392 CrossRef CAS PubMed.
- R. Palchaudhuri and P. J. Hergenrother, Curr. Opin. Biotechnol., 2007, 18, 497–503 CrossRef CAS PubMed.
- J. H. Tan, Y. J. Lu, Z. S. Huang, L. Q. Gu and J. Y. Wu, Eur. J. Med. Chem., 2007, 42, 1169–1175 CrossRef CAS PubMed.
- M. V. Keck and S. J. Lippard, J. Am. Chem. Soc., 1992, 114, 3386–3390 CrossRef CAS.
- P. Pradhan, B. Jernström, A. Seidel, B. Nordén and A. Gräslund, Biochemistry, 1998, 37, 4664–4673 CrossRef CAS PubMed.
- K. M. Sovenyhazy, J. A. Bordelon and J. T. Petty, Nucleic Acids Res., 2003, 31, 2561–2569 CrossRef CAS PubMed.
- B. Norden and T. Kurucsev, J. Mol. Recognit., 1994, 7, 141–156 CrossRef CAS PubMed.
- Y. Cao and X. W. He, Spectrochim. Acta, Part A, 1998, 54, 883–892 CrossRef.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1541589. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra08138c |
| This journal is © The Royal Society of Chemistry 2017 |