
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation of small-sized tungsten carbide nanorods for loading Pt with promoted electrocatalytic activity and stable anti-poisoning performance†
Chao Lv,
Gao Wu,
Kai Xi Liu,
Yi Song Zhu,
Yan Hong Yin,
Ying Yan Hu,
Ye Sheng Li*,
Xian Bin Liu and
Zi Ping Wu *
*
School of Materials Science and Engineering, Jiangxi University of Science and Technology, 86 Hong Qi Road, Ganzhou 341000, P. R. China. E-mail: ysli@jxust.edu.cn; wuziping724@jxust.edu.cn; Fax: +86-797-8312422; Tel: +86-797-8312422
First published on 15th December 2017
Abstract
The Pt-like behavior and synergistic effect of tungsten carbides (WCs) are increased with decreased size and affected by their morphology. However, achieving high specific surface areas of nanostructured WCs with a small size and controlled morphology remains a considerable challenge. In the present studies, WC nanorods with a diameter and length of approximately 50 and 100–300 nm, respectively, were prepared. The morphology of the obtained WO3·H2O was controlled by adding citric acid to the reaction solution. The secondary growth of WO3·H2O crystals was efficiently restrained because double-walled carbon nanotubes were homogenously dispersed in the solution. When the WO3·H2O nanorods were heated in air and carbonized in a nitrogen atmosphere, small-sized WC nanorods with a high specific surface area (32.2 m2 g−1) were obtained. Furthermore, the obtained catalysts displayed improved electrocatalytic activity and stable anti-poisoning performance when the WC nanorods were used as a support for loading platinum nanoparticles. All these results indicate the potential application of WC nanorods in electrochemistry and related fields.
1 Introduction
The commercialization of fuel-cell technologies is hindered by the dependence on platinum (Pt). Thus, reducing Pt loading by either increasing the utilization of Pt or replacing it with non-precious metals in fuel-cell applications is desired.1–3 Accordingly, Pt loaded on different supports4 or binary and ternary alloys with lower noble metal loading5,6 have been developed. One such alternative catalyst is tungsten carbide (WC),7,8 which displays Pt-like behavior.9 WC also exerts a synergistic effect on noble metals as electrocatalysts during methanol oxidation,10,11 the oxygen reduction reaction (ORR),12,13 and hydrogen evolution.14,15 Notably, the Pt-like behavior and synergistic effect of WC are increased with decreased size.16Several methods of preparing small-sized WC, including conventional furnace methods,17 program-controlled reduction carburization,18 microwave-assisted methods,19 and ion exchange,20 have been adopted. Studies have shown that the WC particle size can be reduced to no more than 5 nm. However, the currently available methods often result in a low specific surface area and uncontrollable morphology. The specific surface areas of commercial WC and WC prepared by other methods rarely exceed 10 m2 g−1. Controlling the nanostructure is critical to tuning the electrochemical performance when using WC as a support for loading catalysts.21 Studies on preparing WC nanorods are limited. The obtained nanostructures are usually restricted to low specific surface area and large-sized particles.22 Indeed, achieving high specific surface areas and controlled morphology remains a considerable challenge in preparing nanostructured WC.
In the present study, small-sized WC with a high specific surface area of 32.2 m2 g−1 was prepared. The rod morphology of the WC was controlled by adding organic citric acid (CA). The specific surface area of the obtained WC was higher than that of other prepared samples with a sheet or spherical morphology.18,23 The morphology of the WO3·H2O was also regulated by adding CA to the reaction solution. The secondary growth of crystals of the obtained WO3·H2O nanorods was efficiently restrained by adding a double-walled carbon nanotube (DWCNT)/ethylene glycol (EG) suspension. Consequently, small-sized WC nanorods (SWCRs) were prepared after the carbonization of WO3·H2O, which enabled adequate interaction between tungsten and the carbon sources. When the SWCRs with a high specific surface area were used as a support for loading Pt, homogenous nanoparticles uniformly scattered on or surrounding the nanorods were obtained. These features endowed the SWCR–Pt composite with improved electrocatalytic activity and stable anti-poisoning performance.
2 Experimental
2.1 Synthesis of tungstic acid (WO3·H2O) nanorods
A homogeneous DWCNT/EG suspension (0.1 mg ml−1) was synthesized according to a previous report.24 Approximately 3.96 g of sodium tungstate (Na2WO4) and 2.0 g of CA were mixed with 100 ml of DWCNT/EG suspension. Afterwards, the solution was transferred to a hydrothermal reactor. The complex solution was subsequently sealed, heated at 140 °C, and kept at this temperature for 4 h after adding 8 ml of HCl to the reactor. The obtained samples were cooled, filtered, washed with deionized water, and dried at 100 °C for 2 h in a low-vacuum system. The DWCNT/EG suspension and CA were not added to the complex to analyze their effects on the growth morphology of the obtained samples. Furthermore, EG in the reaction was replaced with deionized water.2.2 Preparation of WC nanorods
The as-received samples were carbonized with ethanol/methanol solution through a single-step method. The obtained samples were heated at 600 °C, and then oxygen flow was introduced into the reaction at a rate of 300 ml min−1 for 1 h. Subsequently, two flanges were placed on both ends of the furnace to seal the chamber. The reaction system was further heated to 950 °C as oxygen was entirely removed from the sealed chamber. Afterwards, an ethanol/methanol solution with a volume ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90 was supplied into the reactor by using a squirming pump at a supply rate of 3 ml h−1 and high-purity N2 flow at a rate of 300 ml min−1 for 1 h. Three hours later, the sealed chamber was cooled to room temperature. The carbonization process is shown in the ESI, Fig. S1.† The as-received samples were collected after the chamber was opened.
:90 was supplied into the reactor by using a squirming pump at a supply rate of 3 ml h−1 and high-purity N2 flow at a rate of 300 ml min−1 for 1 h. Three hours later, the sealed chamber was cooled to room temperature. The carbonization process is shown in the ESI, Fig. S1.† The as-received samples were collected after the chamber was opened.
2.3 Pt deposition on WC nanorods
Chloroplatinic acid (H2PtCl6) with EG solution was heated to 140 °C in a three-necked flask (500 ml) in a reflux system for about 5 h. After cooling the flask to room temperature, the obtained samples were filtered, washed with deionized water, and dried in a low vacuum device at room temperature. The Pt particle loading on the supports was controlled to about 15 wt%.2.4 Material characterization
High-resolution transmission electron microscopy (HRTEM, JEOL 2100F, the accelerating voltage is 200 kV) and field-emission scanning electron microscopy (SEM, Zeiss Supra55 Sapphire) were used to examine the microscopic structures of the prepared samples. Selected area electron diffraction analysis (SAED) and energy-dispersive X-ray spectroscopy (EDS) were used to measure the chemical composition of the sample from a selected area. X-ray diffraction (XRD, Bruker, D8 Advance) was used to analyze the crystal structure of the as-received samples. A BELSORP instrument (BEL Inc., Japan) was used to obtain N2 adsorption/desorption isotherms of the prepared samples at 77 K (details of the analytical processes are presented in ref. 25). Pore-size distributions were determined from the desorption branch of the N2 isotherms through the Barrett–Joyner–Halenda method.262.5 Measurements of electrochemical performance
All electrochemical measurements were conducted in a three-electrode cell using an EG and G potentiostat (CHI 760E). The working electrode (diameter 3 mm) was the catalyst and 5% Nafion mixed in ethanol and spread on a glass carbon cylinder. The catalyst loading on the electrode was controlled to be about 0.2 mg cm−2. The counter electrode was a graphite rod, and the reference electrode was a saturated calomel electrode (SCE). The electrolyte was prepared from H2SO4 (0.5 M) or H2SO4 (0.5 M)/CH3OH (1.0 M) solution. Methanol electrooxidation activity was measured in a mixed N2-saturated solution containing H2SO4 (0.5 M) and CH3OH (1 M). Cyclic voltammetry (CV) profiles were obtained at a scan rate of 100 mV s−1 for potentials against the SCE ranging from −0.3 V to 0.999 V. Linear sweep voltammetry at a scan rate of 5 mV s−1 was conducted in H2SO4. The working electrode was mounted onto a rotating ring disc at different rotating speeds during the test. To study the tolerance to CO toxicosis, CO stripping experiments were performed in a 0.5 M H2SO4 solution. At the beginning, the electrolyte was purged with N2 and then the electrode potential was fixed at −0.2 V vs. SCE for CO adsorption, along with continuous CO bubbling for 30 min. Finally, the stripping test was performed after purging the solution with N2 for 30 min to remove the dissolved CO. Before all the measurements, the working electrodes were cycled in N2-saturated 0.5 M H2SO4 solution from −0.3 V to 1.0 V at a scan rate of 100 mV s−1 in order to remove surfactant residues from the surface. A commercial Pt–C catalyst from Johnson Matthey with the same Pt loading was also used for comparison with the obtained samples.3 Results and discussion
Our group has recently prepared WO3 and WC nanosheets in a round flask with a reflux device by using an in situ DWCNT template.18 With an increased molar ratio of tungsten precursor and DWCNTs, the sheet morphology of the obtained samples transformed into spherical particles.23 The results indicated that DWCNTs in the solution significantly influence the size and morphology of the deposited samples based on the reaction of Na2WO4 and HCl, but the specific surface area of the obtained samples was insufficiently high. In the present study, the prepared solution with added CA was transferred into a hydrothermal reactor.The microstructures of the received WO3·H2O after heating were examined using SEM and HRTEM. Fig. 1 depicts the morphology of the as-prepared WO3·H2O. The results indicated the presence of a chemical reaction between Na2WO4 and HCl, and the formation of WO3·H2O nanorods (Fig. 1a and b). In the absence of the CA and DWCNT/EG suspension in the reaction, random particles were obtained, as shown in Fig. 2a. Complex molecules were formed upon adding CA to EG.27,28 When the complex molecules were deposited onto the crystal seeds, the growth of WO3·H2O nanorods could be controlled. Subsequently, the obtained crystal seeds of the WO3·H2O nanorods grew along the complex molecules (Fig. 2b). Consequently, WO3·H2O with a nanorod morphology was prepared, as shown in Fig. 1a. When the CA and DWCNT/EG suspension was added to the reaction, the crystal seeds grew along the complex molecules. Furthermore, the dispersed nanotubes prevented the union of the crystal grains of the WO3·H2O nanorods (Fig. 2c). As a result, the secondary growth of crystal grains of the WO3·H2O nanorods was efficiently restrained. Fig. 1b shows that the small-sized nanorods and DWCNT bundles were randomly distributed.
 | ||
| Fig. 1 The morphology of the WO3·H2O nanorods prepared by the addition of CA (a) and CA in a DWCNT/EG suspension (b), and TEM (c) and HRTEM (d) images of the obtained sample in (b). | ||
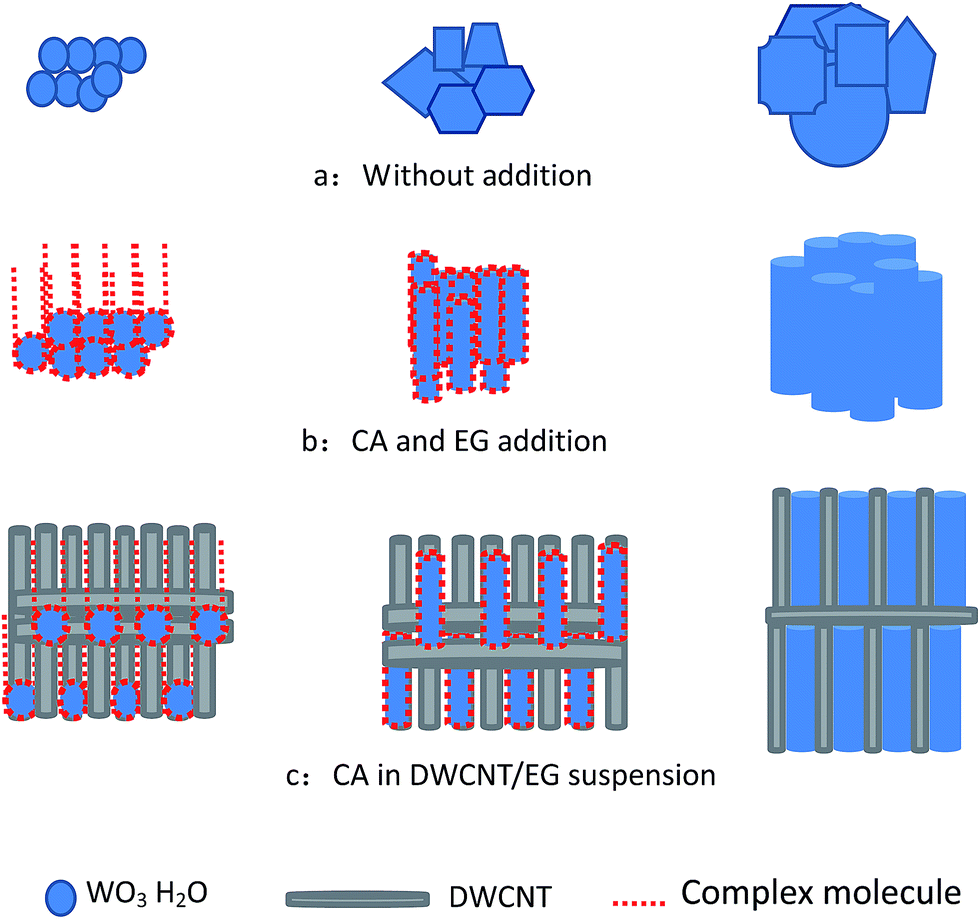 | ||
| Fig. 2 Schematic of the formation of small-sized WO3·H2O by the addition of CA in a DWCNT/EG suspension. | ||
The sample morphology was also studied by TEM and HRTEM. Large quantities of nanorods are shown in Fig. 1c. The morphologies of the nanorods with a diameter and length of 20 and 100 nm, respectively, were clearly observed in the magnified image. The lattice fringes displayed in Fig. 1d indicated that the obtained sample possessed a highly crystalline structure. The lattice spacing was 0.35 nm, which corresponds to the (111) interplanar distance of WO3·H2O, thereby indicating the occurrence of the reaction Na2WO4 + 2HCl → WO3·H2O↓ + 2NaCl.
In addition, multi-walled CNTs (ESI Fig. S2†) dispersed in EG with the same concentration were also added into the reaction. The results displayed that a rod morphology can be prepared, but the distribution of the rod diameter is not homogenous. The reason for this may be due to their large diameter and low aspect ratio, and less nanotubes are distributed in a similar space compare to CNTs with a small diameter. This result means that the secondary growth of the crystal grains of the WO3·H2O nanorods could be restrained, but other areas could not be restrained. If the multi-walled CNTs were replaced with DWCNTs in the study, the rod morphology of WO3·H2O was more homogenous, as shown in Fig. 1a and b. The results demonstrate the DWCNTs are key for the preparation of the WO3·H2O nanorods.
Fig. 3 shows the morphology of the WO3·H2O sample after heating in air at 600 °C and carbonizing in N2 with ethanol and methanol solution in a sealed reaction chamber at 950 °C. The typical SEM images of the carbonized WC with added CA (Fig. 3a) and CA in DWCNT/EG suspension (Fig. 3b) display different morphologies. The sample obtained without DWCNT/EG suspension addition showed a morphology in which the nanorods conglomerated with one another. When the DWCNT/EG suspension was added to the reaction solution, small-sized samples were clearly observed, as shown in Fig. 3b.
 | ||
| Fig. 3 Typical SEM images of the carbonized WC with a small size by the addition of CA (a) and CA in a DWCNT/EG suspension (b), and SEM (c) and HRTEM (d) images of the sample from (b). | ||
These samples were further characterized by SEM and TEM. As shown in Fig. 3c, the image of the received samples indicated that the sample possessed a rod morphology with a diameter and length of approximately 50 and 100–300 nm. Additionally, the rods became thicker and longer than those of the sample before carbonization. The large size of the carbonized samples may be attributed to the growth of small rods at the carbonization temperature. The lattice fringes displayed in Fig. 3d indicate the high crystallinity of the rod samples. The lattice spacing is 0.28 nm, corresponding to the (001) interplanar distance of WC. This result indicates that WO3 can be obtained after heating WO3·H2O and when the 2WO3 + 5C → 2WC + 3CO2 reaction occurred. Thus, the WC nanorods (WCRs) conglomerated with one another, and SWCRs were obtained.
Samples before and after carbonization were analyzed by XRD based on our previous thermoanalysis results to clarify the carbonization process of WO3·H2O.18 Fig. S3a in the ESI† shows the diffraction peaks indexed as different planes of WO3·H2O. The lattice spacing shown in Fig. 1d is consistent with the XRD data. The XRD results shown in Fig. S3b in the ESI† further indicate that WO3·H2O was carbonized, consistent with the HRTEM image in Fig. 3d. Therefore, the DWCNTs in the received WO3·H2O were completely burned. Subsequently, a full carbonization reaction occurred when a sufficient number of carbon atoms diffused from the surface to within the WO3·H2O sample, and thereby pure SWCRs (Fig. 3c) were obtained.
The specific surface areas and corresponding pore-size distribution of the WCRs and SWCRs are illustrated in the N2 adsorption–desorption isotherms shown in Fig. 4. The specific surface areas of the WCRs and SWCRs from the Brunauer–Emmet–Teller method were 15.5 and 32.2 m2 g−1. The total pore volumes of the samples were 3.6 and 7.4 cm3 g−1 (P/P0 = 0.966), suggesting that the high specific surface areas and pore volumes of the WCRs and SWCRs were due to CA addition.29 The results demonstrate that the WC nanorod morphology was influenced by CA, which significantly increased the specific surface areas and pore volumes, which both further increased with the homogenously dispersed DWCNTs that restrained the crystal grain growth. This effect benefited the loading of nanoparticles when the nanorods were used as electrocatalyst supports.
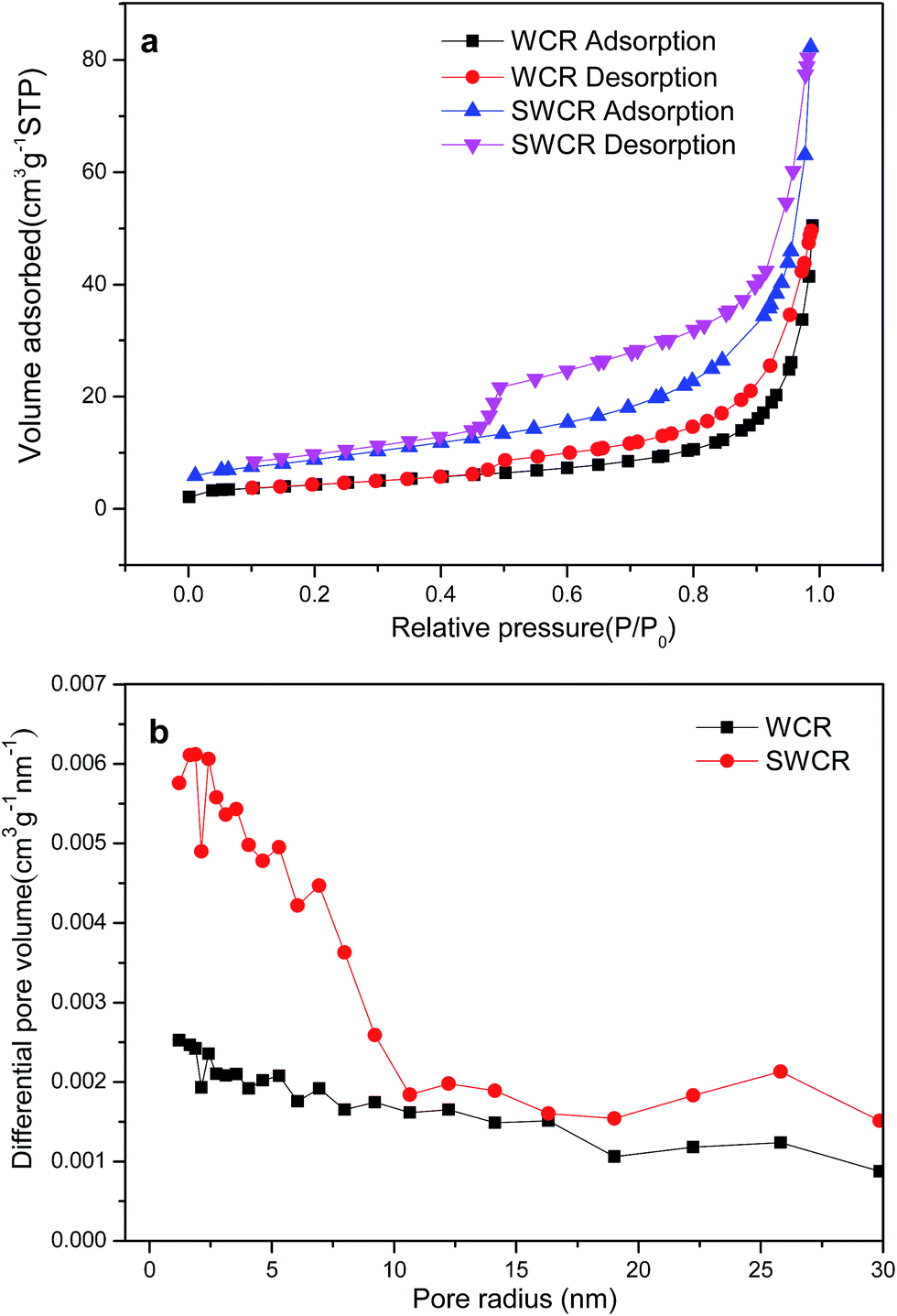 | ||
| Fig. 4 N2 adsorption–desorption isotherms (a) and pore radius distributions (b) of the obtained SWCRs and WCRs. | ||
The TEM and HRTEM images of the SWCR–Pt composites obtained from the homogeneous EG systems are shown in Fig. 5. The nanoparticles (diameter, 3–5 nm), whose growth was accelerated uniformly on the SWCR supports, were homogeneously distributed as shown in Fig. 5a. Isolated nanorods approximately 50 nm in diameter, which are larger than the other particles, were also observed. Almost all nanoparticles were scattered on or around the nanorods.
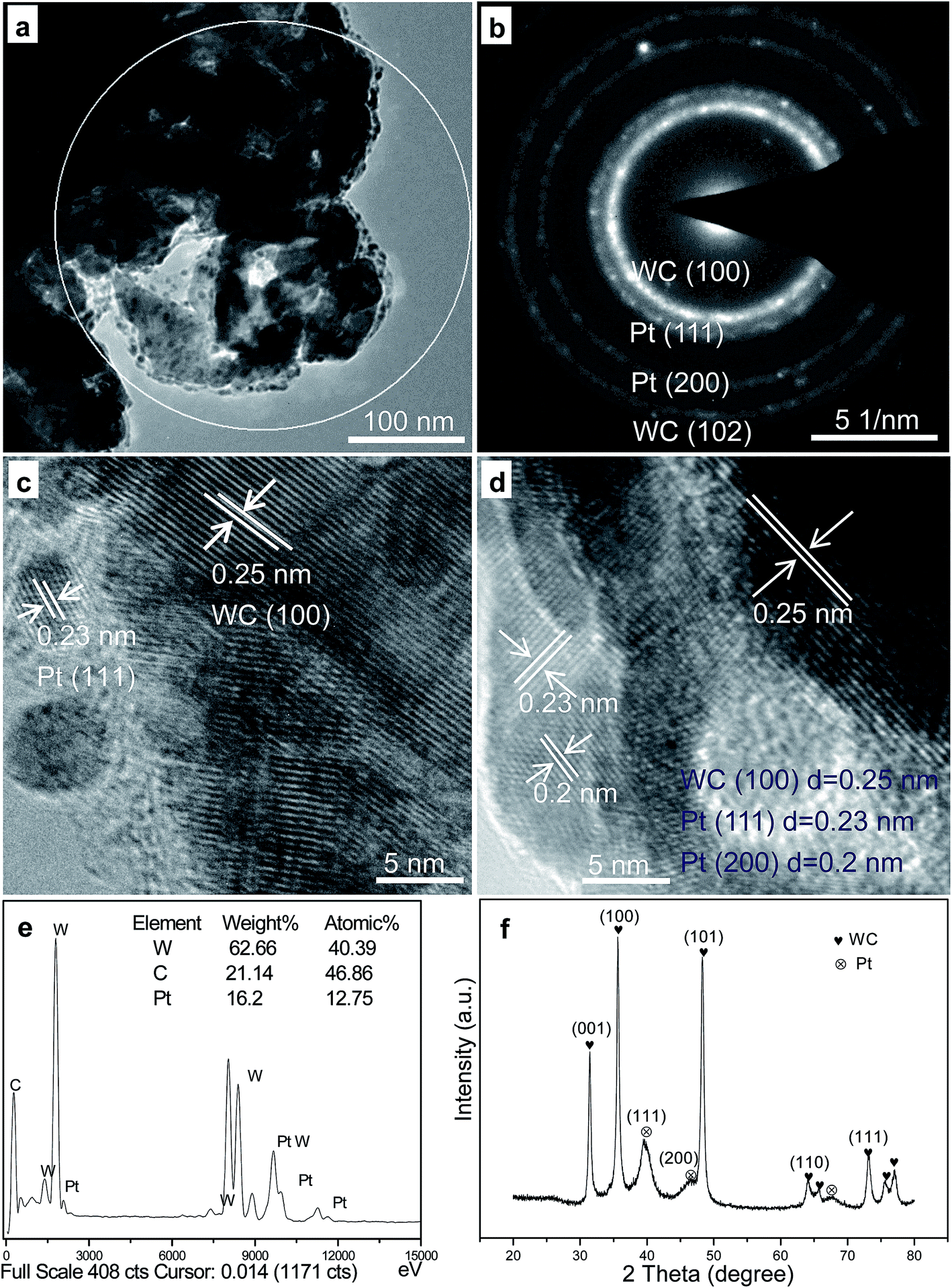 | ||
| Fig. 5 Typical TEM (a) and HRTEM (c and d) images and the SAED (b), EDS (e) and XRD (f) of the obtained SWCR–Pt catalyst. | ||
Moreover, the SAED result in Fig. 5b (the selected area in Fig. 5a) indicated that the small-sized particles were Pt nanoparticles, which decomposed from the H2PtCl6 precursors. The large-sized rods were SWCRs. The selected area in Fig. 5a was magnified to describe in detail the isolated SWCRs and Pt nanoparticles, as shown in Fig. 5c and d. Various lattice fringes were clearly observed, and SWCRs were confirmed by the lattice spacing of approximately 0.25 nm. In addition, the small-sized nanoparticles were Pt nanoparticles, as demonstrated by the lattice spacings of approximately 0.23 and 0.2 nm. WC and Pt phases (Fig. 5c and d) with highly crystalline structures were also observed.
EDS (Fig. 5e) and XRD (Fig. 5f) data indicated that the small dots were Pt nanoparticles, and that the Pt loading was approximately 15 wt% (the selected area in Fig. 5a). The diameter of the Pt nanoparticles was similar to that calculated from the Scherrer formula30 according to the XRD data (Fig. 5f). WC and Pt were evidently confirmed by the XRD patterns, and the results corresponded to the HRTEM images shown in Fig. 5.
The electrocatalytic performance in fuel-cells can be improved by excellent electrochemical activity and high power density. The obtained catalysts' electrochemical activities in H2SO4 are illustrated in Fig. 6a. The SWCR–Pt catalysts demonstrated obviously stronger peaks (0.036 A cm−2) of hydrogen desorption/adsorption and higher electrochemical activity than WCR–Pt (0.017 A cm−2). In addition, the electrochemical activity of the SWCR–Pt catalyst is much higher than that of a commercial Pt–C catalyst from Johnson Matthey.
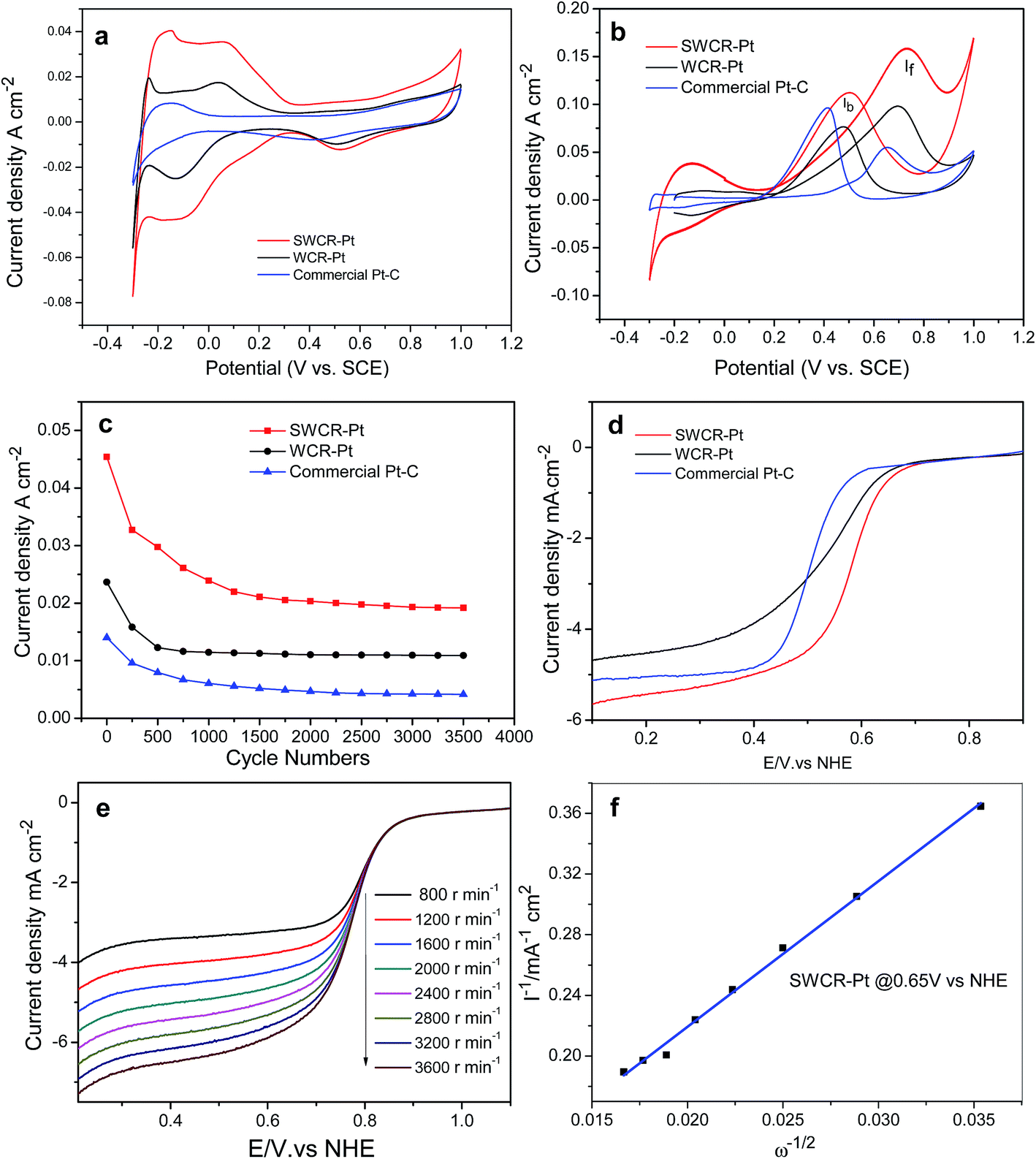 | ||
| Fig. 6 CV curves of the SWCR–Pt, WCR–Pt and commercial Pt–C catalysts at a scanning rate of 100 mV s−1 in 0.5 M H2SO4 (a) and 0.5H2SO4/1.0 M CH3OH (b) solutions. The stability with a scan rate of 100 mV s−1 in 0.5 M H2SO4 (c). The ORR curves (d) of the different catalysts, and the ORR curves (e) of SWCR–Pt at different rotation speeds in O2-saturated 0.5 M H2SO4 solution at room temperature with a scan rate of 5 mV s−1 and the corresponding Koutecky–Levich plots (f) at different potentials. Note: the WC–Pt material loading on the working electrode was controlled to be 0.2 mg cm−2. | ||
In direct methanol fuel-cells, methanol oxidation is the limiting reaction step. In the present study, the methanol oxidation of SWCR–Pt, WCR–Pt and the commercial Pt–C catalyst from Johnson Matthey was also investigated by CV in H2SO4 (0.5 M)/CH3OH (1.0 M) solutions (Fig. 6b). The main methanol oxidation peak of WCR–Pt was located at 0.72 V at a current density of 0.097 A cm−2 in the anodic sweep. The other methanol oxidation peak of the sample was at 0.48 V with a current density of 0.076 A cm−2 in the cathodic sweep. However, the current density of the SWCR–Pt catalyst increased significantly (the corresponding values were 0.160 and 0.110 A cm−2). The results demonstrate that the highly dispersed nanorods used as supports promoted the electrocatalytic activity of the catalyst when Pt nanoparticles were loaded onto the SWCR surfaces. The value of the current density of SWCR–Pt was also much higher than those of previously prepared WC materials with different morphologies10,11 under similar measurement conditions. The current density of the commercial Pt–C catalyst with a similar Pt loading from Johnson Matthey has also been measured. The value of current density is also less than that of the SWCR–Pt catalyst.
The high electrochemical activity of SWCR–Pt may be attributed to the dispersed nanorod characteristics. The presence of small-sized SWCRs distributed in solution prevented the union of the crystal seeds of loaded Pt. Thus, the formation of secondary seed growth was restricted, which resulted in the direct deposition and growth of Pt seeds that used SWCRs as a support. The SWCRs also significantly influenced the morphology and size of the deposited Pt nanoparticles obtained from H2PtCl6 and EG. Therefore, the catalytic and synergistic performances of the SWCR–Pt catalyst during hydrogen desorption, adsorption, and methanol oxidation may have resulted from the self-regulation mechanism in the SWCRs. The anti-poisoning performance of the SWCRs was also investigated. The catalyst showed a higher ratio of If/Ib (in which If and Ib are the forward and back current densities, respectively) (1.45) than the WCR–Pt as shown in Fig. 6b in the first cycle. This finding suggested that the SWCRs affected the anti-poisoning performance of the catalyst. The stability of the anti-poisoning property of the SWCR–Pt was further studied. If/Ib gradually decreased with an increasing number of cycles (ESI, Fig. S4†) and decreased to 0.98 after 400 cycles (ESI, Fig. S4a and Table S1†). The reason for this may be the growth of Pt particles and oxidation of the support. The results demonstrated that the CA and DWCNT/EG suspension in the reaction solution increased the stability of the anti-poisoning performance of WC.23 The anti-poisoning performance of SWCR–Pt may be attributed to the dispersed nanorod morphology. The presence of small-sized SWCRs distributed in the solution significantly influenced the morphology, size and distribution of the deposited Pt nanoparticles. Furthermore, methanol molecules were effectively oxidized on the SWCR–Pt catalyst during the forward potential scan. The If/Ib of WCR–Pt (ESI, Fig. S4b†) and the commercial Pt–C from Johnson Matthey (ESI, Fig. S4c†) was also explored. The If/Ib of WCR–Pt was larger than 1, demonstrating the good anti-poisoning performance of the catalyst, but it is less than that of SWCR–Pt. The If/Ib of the commercial Pt–C catalyst from Johnson Matthey was less than 1, demonstrating the poor anti-poisoning performance of the catalyst. This phenomenon resulted in the generation of relatively less poisoning species than when WCR–Pt and the commercial Pt electrocatalyst were used.31 The CO oxidation ability of SWCR–Pt, WCR–Pt and the commercial Pt–C catalyst was also evaluated by a CO-stripping experiment. Fig. S5 in the ESI† presents the CV curves of the CO-adsorbed SWCR–Pt, WCR–Pt and commercial Pt–C. It can be seen that the onset potential of CO electrooxidation on SWCR–Pt is 0.6 V, which is lower than that of WCR–Pt and the commercial Pt–C, indicating the SWCR–Pt has better electrocatalytic activity toward CO oxidation. The peak area of CO oxidation on SWCR–Pt is smaller than that on WCR–Pt and the commercial Pt–C, because the Pt surface area of SWCR–Pt available is smaller than that on the others for CO adsorption. The stability of the electrocatalytic performance of the SWCR–Pt in a 0.5 M H2SO4 solution with a scan rate of 100 mV s−1 was further studied. The results in Fig. 6c indicate that the current density of SWCR–Pt was promoted more significantly than when the WCR–Pt catalyst and commercial Pt–C catalyst from Johnson Matthey with the same Pt loading were used for the entire time course. The high electrocatalytic performance of SWCR–Pt may be attributed to the distinct rod morphology and small size, which possibly resulted in a synergetic effect between WC and Pt.32 The similar trend of the curves demonstrated that the catalyst has a similar stability performance to the WCR–Pt catalyst and commercial Pt–C catalyst. In addition, the current density of the catalysts decreased significantly after 500 cycles, and the performance was stable after 1000 cycles. The reason for this may be attributed to the growth and loss of the Pt particles after uninterrupted cycling. The supports can also be oxidized by the environment with long-term cycling, which further leads to a decrease in current density. The ORR performances of SWCR–Pt and WCR–Pt were also determined in an O2-saturated H2SO4 solution (0.5 M) at a scan rate of 5 mV s−1 and 2000 rpm−1 (Fig. 6d). The onset potential of SWCR–Pt was lower than that of WCR–Pt and the commercial Pt–C catalyst. In addition, the half-potential values of SWCR–Pt were higher than those of WCR–Pt and the commercial Pt–C catalyst. These results indicate that the introduction of the CA and DWCNT/EG suspension in the solution significantly reduced the ORR overpotential. Fig. 6e shows the ORR curves of SWCR–Pt at rotation speeds of 800, 1200, 1600, 2000, 2400, 2800, 3200, and 3600 rpm−1. Evidently, the catalytic current was promoted with an increased rotation rate of the electrode. High values of current at high rotation rates also demonstrate a high catalyst turnover rate for the ORR at the electrode. The number (n) of electrons transferred per oxygen molecule was obtained using the Koutecky–Levich equation:33
| 1/I = 1/Ik + 1/Bω1/2 | (1) |
485C mol−1), A is the area of the electrode (0.1256 cm2), C is the bulk concentration of O2 (1.1 × 10−5 mol cm−3), D is the oxygen–diffusion coefficient (1.4 × 10−5 cm2 s−1), and ν is the kinetic viscosity of the electrolyte (0.01 cm2 s−1). The plots of I−1 against ω−1/2, i.e., the Koutecky–Levich plots at typical potentials of 0.65 V, are shown in Fig. 6f. The number of electrons (n) transferred by SWCR–Pt obtained based on eqn (1) was 3.98. This value indicated direct reduction of O2 to H2O on SWCR–Pt in a four-electron reduction reaction, which was more effective than that of previously studied Pt/C catalysts.34
4 Conclusions
In conclusion, WO3·H2O with a rod morphology and a diameter and length of 20 and 100 nm, respectively, was prepared via a hydrothermal approach. CA addition to the reaction solution controlled the morphology of the obtained WO3·H2O. The secondary crystal grain growth of WO3·H2O was also efficiently restrained due to the DWCNTs homogenously dispersed in the solution. When the WO3·H2O nanorods were heated in air and carbonized in N2 inside a sealed reaction chamber, the CA and DWCNTs in the tungsten precursor were burned. Consequently, small-sized SWCRs with a high specific surface area of 32.2 m2 g−1 were prepared. When the SWCRs were used for loading noble metals, the prepared SWCR–Pt catalyst showed promoted electrochemical activity and stable anti-poisoning performance. Therefore, the distinct SWCRs have potential applications in electrochemistry and related fields, considering their distinct rod morphologies and nanosize.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51264010), Department of Science & Technology of Jiangxi Province (20153BCB23011, GJJ150617, GJJ160596, 20171ACB21043), and Program for Excellent Young Talents, JXUST.Notes and references
- S. Rösler, J. Obenauf and R. Kempe, J. Am. Chem. Soc., 2015, 137, 7998–8001 CrossRef PubMed.
- T. Maiyalagan, P. Kannan and M. J. Niedziolka, Anal. Chem., 2014, 86, 7849–7857 CrossRef CAS PubMed.
- B. Y. Xia, W. T. Ng, H. B. Wu, X. Wang and X. W. Lou, Angew. Chem., Int. Ed., 2012, 124, 7325–7328 CrossRef.
- C. K. Poh, S. H. Lim, Z. Q. Tian, L. F. Lai, Y. P. Feng, Z. X. Shen and J. Y. Lin, Nano Energy, 2013, 2, 28–39 CrossRef CAS.
- M. Shao, Q. W. Chang, J. P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- M. Millán, H. Zamora, M. A. Rodrigo and J. Lobato, ACS Appl. Mater. Interfaces, 2017, 9, 5927–5936 Search PubMed.
- H. Meng and P. K. Shen, J. Phys. Chem. B, 2005, 109, 22705–22709 CrossRef CAS PubMed.
- Z. Y. Chen, C. A. Ma, Y. Q. Chu, J. M. Jin, X. Lin, C. Hardacre and W. F. Lin, Chem. Commun., 2013, 49, 11677–11679 RSC.
- R. B. Levy and M. Boudart, Science, 1973, 181, 547–549 CAS.
- M. D. Obradović, B. M. Babić, V. R. Radmilović, N. V. Krstajić and S. L. J. Gojković, Int. J. Hydrogen Energy, 2012, 37, 10671–10679 CrossRef.
- J. S. Moon, Y. W. Lee, S. B. Han and K. W. Park, Int. J. Hydrogen Energy, 2014, 39, 7798–7804 CrossRef CAS.
- Y. Zhang, J. B. Zang, S. P. F. Tian, C. Han and Y. H. Wang, Appl. Surf. Sci., 2017, 412, 327–334 CrossRef CAS.
- J. Z. Sun, J. Shi, J. L. Xu, X. T. Chen, Z. H. Zhang and Z. Q. Peng, J. Power Sources, 2015, 279, 334–344 CrossRef CAS.
- T. G. Kelly, S. T. Hunt, D. V. Esposito and J. G. Chen, Int. J. Hydrogen Energy, 2013, 38, 5638–5644 CrossRef CAS.
- A. V. Nikiforov, I. M. Petrushina, E. Christensen, N. V. Alexeev, A. V. Samokhin and N. J. Bjerrum, Int. J. Hydrogen Energy, 2012, 37, 18591–18597 CrossRef CAS.
- Z. X. Yan, F. Li, J. M. Xie and X. L. Miu, RSC Adv., 2015, 5, 6790–6796 RSC.
- G. D. Rieck, Tungsten and its compounds, Pergamon Press, England, 1967 Search PubMed.
- Z. P. Wu, M. Zhao, J. W. Hu, W. B. Zhang, Y. H. Yin, Y. Y. Hu, Y. S. Li, J. G. Yang, Q. F. Xu and A. Krishamurthy, RSC Adv., 2014, 4, 47414–47420 RSC.
- P. K. Shen, S. Yin, Z. Li and C. Chen, Electrochim. Acta, 2010, 55, 7969–7974 CrossRef CAS.
- Z. X. Yan, M. Cai and P. K. Shen, Sci. Rep., 2013, 3, 1646 CrossRef PubMed.
- Y. Yan, L. Zhang, X. Y. Qi, H. Song, J. Y. Wang, H. Zhang and X. Wang, Small, 2012, 8, 3350–3356 CrossRef CAS PubMed.
- S. V. Pol, V. G. Pol and A. Gedanken, Adv. Mater., 2006, 18, 2023–2027 CrossRef CAS.
- B. J. Han, Z. Z. Huang, G. Wu, C. Y. Zhou, Y. S. Li, Q. H. Wang, Y. L. Zhang, Y. H. Yin and Z. P. Wu, RSC Adv., 2016, 6, 75178–75185 RSC.
- Z. P. Wu, B. Y. Xia, X. X. Wang and J. N. Wang, J. Power Sources, 2010, 195, 2143–2148 CrossRef CAS.
- B. C. Lippens and J. H. D. Boer, J. Catal., 1965, 4, 319–323 CrossRef CAS.
- Z. Slanina and J. F. Crifo, Int. J. Thermophys., 1992, 13, 465–476 CrossRef CAS.
- M. Hlaïbi, S. Chapelle, M. Benaïssa and J. F. Verchère, Inorg. Chem., 1995, 34, 4434–4440 CrossRef.
- V. M. L. Whiffen, K. J. Smith and S. K. Straus, Appl. Catal., A, 2012, 419–420, 111–125 CrossRef CAS.
- E. Antolini and E. R. Gonzalez, Appl. Catal., B, 2010, 196, 245–266 CrossRef.
- A. L. Patterson, Phys. Rev., 1939, 978–982 CrossRef CAS.
- D. Xu, Z. P. Liu, H. Z. Yang, Q. S. Liu, J. Zhang, J. Y. Fang, S. Z. Zou and K. Sun, Angew. Chem., Int. Ed., 2009, 48, 4217–4221 CrossRef CAS PubMed.
- D. S. Wang and Y. D. Li, Adv. Mater., 2011, 23, 1044–1060 CrossRef CAS PubMed.
- M. S. Ahmed, D. Kim and S. Jeon, Electrochim. Acta, 2013, 92, 168–175 CrossRef CAS.
- M. Ammam and E. B. Easton, J. Power Sources, 2013, 236, 311–320 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08065d |
| This journal is © The Royal Society of Chemistry 2017 |