
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic deoxygenation of triglycerides to green diesel over modified CaO-based catalysts†
N. Asikin-Mijan ac,
H. V. Lee*a,
J. C. Juana,
A. R. Noorsaadahb and
Y. H. Taufiq-Yap*c
ac,
H. V. Lee*a,
J. C. Juana,
A. R. Noorsaadahb and
Y. H. Taufiq-Yap*c
aNanotechnology & Catalysis Research Centre (NanoCat), Institute of Postgraduate Studies, University Malaya, 50603 Kuala Lumpur, Malaysia. E-mail: leehweivoon@um.edu.my; Fax: +603-7957 6956; Tel: +603-7967 6954
bDepartment of Chemistry, Faculty of Science, University of Malaya, 50603 Kuala Lumpur, Malaysia
cCatalysis Science and Technology Research Centre (PutraCat), Faculty of Science, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia. E-mail: taufiq@upm.edu.my; Fax: +603-89466758; Tel: +603-89466809
First published on 2nd October 2017
Abstract
Renewable fuel is a promising alternative as a petroleum replacement in view of the current worldwide demand for petroleum fuel, which is catching up with the world's petroleum supply. In the present study, gasoline and diesel-range hydrocarbons were derived from triolein via a deoxygenation process. The deoxygenation of triolein was performed using waste clamshell-derived CaO supported by active promoters (binary metal oxide systems: Ni–CaO, Zn–CaO, Fe–CaO and Co–CaO). Based on the catalytic activity study, the highest degree of deoxygenation via deCOx reaction (decarboxylation/decarbonylation) was achieved in the presence of the strong acid–base bifunctional Co–CaO catalyst. A Box-Behnken optimization study was performed to study the effect of 3 operating parameters: catalyst loading (3–7 wt%), reaction temperature (300–360 °C) and reaction time (60–120 min). The present study has indicated that the maximum hydrocarbon yield (56%) was achieved under optimized deoxygenation conditions of 5 wt% of catalyst, 340 °C within 105 min, and the interaction effect between reaction temperature and time rendered a significant effect towards the deoxygenation activity.
1 Introduction
Production of liquid biofuel is becoming more important. This is mainly due to the increase of global consumption of fossil fuels especially in the transportation sector which has contributed to the depletion of natural petroleum fossil resources. A new alternative solution is very much needed to sustain the future fuel production. Diesel is an important liquid hydrocarbon-based fuel derived primarily from fractional distillation of petroleum crude oil. This type of fuel comprises medium to long chain hydrocarbons within the range of (C13–C20) with a specific concentration of aromatic additives as octane enhancers.1 Diesel is industrially preferable as it converts 20% more fuel energy to actual energy compared to gasoline (range (C8–C12)) and subsequently produces less waste heat. The higher cetane number of diesel also results in shorter ignition delaying time which will help to achieve complete combustion. Moreover, it also lasts longer and safer as diesel not release as much fume as gasoline. The global demand on diesel has increased tremendously while world crude oil reserve is continuously depleting. An alternative route for diesel production from non-fossil sources is a necessity. Deoxygenation of vegetable oil is one of the suggested alternative routes to produce green-diesel. Deoxygenation of vegetable oil is one of the suggested alternative routes to produce renewable green diesel. The deoxygenation of vegetable-based feedstock is typically related to cracking of hydrocarbon chain with the hydrocarbon chain being broken and the oxygen being removed in the form of CO2/CO via decarboxylation/decarbonylation (deCOx) reactions.2 The produced green diesel presented similar properties to conventional petroleum derived fuels and deoxygenation also is an economical process as there is no H2 is used as compared to well-established refinery process (hydrodeoxygenation). Hydrodeoxygenation, is a process involves an exothermic reaction which removes oxygen in the form of water in the presence of H2.3The studies on the effect of alkali metal and alkaline earth metal, especially on Mg4 and Ca metals were widely investigated to improve the quality of diesel via deoxygenation reaction due to their unique basic properties. It was found CaO helped in oxygen removal by being able to absorb more CO2 in gas phase.5 Moreover, application of basic over calcined dolomite (MgO–CaO) catalyst also showed reduction of tar formation with an increase of H/C ratio.5 In brief, these observations indicated that MgO and CaO presence will produce a better route for oxygen removal in the form of CO2 through catalytic deoxygenation. Transition metal oxides (TMOs) are important materials and widely applied for various processes such as oxidation, hydrodeoxygenation, selective/reduction, ammoxidation, metathesis and etc.6 TMOs is a good promoter in catalyst synthesis which acted as an alternative catalyst family to noble metals such as Pt and Pd in the field of catalytic cracking activity. Promoter played an important role in tuning product selectivity towards monofunctional hydrocarbon intermediates, which then further proceeded to desired hydrocarbon-like chain.7 Application of noble metals in catalytic cracking activity is still being widely explored and studied but it is still unattractive due to the high cost constrain.8 Thus, it is more reasonable to develop a new inexpensive catalyst. Several literatures had reported that the rule over transition metal catalysts in enhancing the formation of unsaturated hydrocarbon from saturated fatty acid and fatty acid ester. Several studies had reported on the application of bimetallic fraction cooperation of Co/Pt, Co/Mo, Ni/pristine and Fe/MSN(mesoporous silica nanomaterials)to enhance the effectiveness of converting model triglycerides compound into desired hydrocarbons.9,10 These reports had successfully proved that TMOs are active and selective in the mentioned reactions because of its unique properties such as existence of both basic and acid properties, cationic and anionic vacancies and high mobility of lattice oxygen.11
The optimization study via OVAT (one-variable-at-a-time) approach for deoxygenation reaction under H2-free condition through investigation of different parameters (i.e. reaction temperature, reaction time and catalyst loading) was well established and supported by several researchers.12–14 Conventionally, OVAT technique is an optimization process that is performed with the variation of one component at a time, where the response is a function to a single parameter.15 OVAT technique excludes the interactive effects among the parameter and eventually, it does not depict the integration and relationship between parameters in the process. Thus, in order to overcome this weakness, the optimization study can be carried out by using response surface methodology (RSM), which is among the most relevant multivariate techniques. Utilization of RSM optimization to study the deoxygenation activity and the interaction effects between different deoxygenation parameter is necessary as this study has not been thoroughly investigated. To date, only a literature was found to report on the deoxygenation of activity over SiO2–Al2O3 supported catalyst using RSM optimization study. But, the study focused on the usage of acidic catalyst.15
Thus, the present study highlights the usage of solid base catalysts (Ni–CaO, Zn–CaO, Fe–CaO, Co–CaO), prepared from low cost and toxic-free waste clamshell (meretrix–meretrix) for deoxygenation of triolein. The physicochemical properties of CaO-based catalysts were further investigated by XRF, XRD, SEM, FESEM-EDX mapping, BET and TPD analysis. Detail study on the chemical composition and product distribution of deoxygenized liquid products were discussed based on FTIR, GC-FID, GC-MS and CHNOS analysis. Furthermore, optimization of triolein deoxygenation process was evaluated via RSM approaches by manipulating the three reaction variables: (i) temperature, (ii) reaction time and (iii) catalyst concentration. The recoverability and stability profile of CaO-based catalyst was also being evaluated.
2 Experimental
2.1 Material
The CaO was prepared from waste clamshell based on previous study.16 The nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O) with purity >99% was obtained from R&M Company. Iron(III) nitrate nanohydrate (Fe(NO3)3·9H2O) with 99% purity was purchased from Analar, zinc(II) nitrate hexahydrate (Zn(NO3)2·6H2O) with 98% purity, cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O) with 99.9% purity, triolein (glyceryl trioleate) with 65% purity, standard solution of alkane and alkene (C8–C20) and internal standard 1-bromohexane with purity >98% (GC grade) were purchased from sigma Aldrich. The n-Hexane (GC grade) with purity >98% from Merck was used for dilution.2.2 Catalyst development
Calcium-based catalysts were synthesized using wet impregnation method. 5 g of clamshell-derived calcium oxide (CaO) was impregnated with an aqueous solution of metal salts containing 20 wt% concentration of nickel(II) nitrate hexahydrate and the mixture was stirred for 6 h. The mixture was then dried in the oven at the temperature of 100 °C for 24 h. The dried samples were grounded into fine powder before thermally activated at the temperature of 900 °C for 2 h under atmosphere condition. The 900 °C calcination temperature is in agreement with Taufiq's group study.17 The procedures above were repeated using cobalt(II) nitrate hexahydrate, iron(III) nitrate 9-hydrate and zinc(II) nitrate hexahydrate. The samples were denoted as Ni–CaO, Zn–CaO, Fe–CaO and Co–CaO.2.3 Catalyst characterization
X-ray fluorescence (XRF) spectrometry was used to trace elemental composition in the catalyst. In this study, the XRF (Philips PWI404) was equipped with a scandium anode tube. The powder X-ray diffraction (XRD) analysis was carried out to identify the crystallography of the mixed metal oxide catalysts. The XRD analyses were performed using Shimadzu diffractometer model XRD-6000. The specific surface area and pore distribution of the catalysts were determined by Brunauer-Emmet-Teller (BET) method with N2 adsorption/desorption analyzer using Thermo-Finnegan Sorpmatic 1990 series. The catalysts were degassed overnight at 150 °C to remove moisture and foreign gases on the surfaces of the catalysts. Adsorption and desorption processes of N2 on the catalysts surfaces were analyzed in a vacuum chamber at −196 °C. The basicity and basic strength distribution of the catalysts were studied by using temperature-programmed desorption with CO2 as probe molecule. TPD-CO2 experiment was performed using a Thermo Finnegan TPDRO 1100 apparatus equipped with a thermal conductivity detector. Catalysts (0.05 g) were pre-treated with N2 gas flow for 30 min at 250 °C to remove the moisture in the sample. This was followed by CO2 gas flow for 1 h under ambient temperature to allow adsorption of CO2 onto the surfaces. The excess CO2 was subsequently flushed with N2 gas flow at 20 mL min−1 for 30 min. The desorption of CO2 from the basic sites of the catalysts was detected by thermal conductivity detector (TCD) in helium gas flow (30 mL min−1) from 50 °C to 900 °C and hold for 30 min. The acidity of the catalyst was determined by using ammonia (NH3) as probe gas. The adsorption and desorption of NH3 utilized the same procedures as in TPD-CO2 method. The amount of basicity/acidity of the catalyst were determined using peak area of CO2/NH3 desorption peaks. The temperatures of the peak maximum (Tmax) at which the desorption of CO2/NH3 had occurred were used to study the characteristics and the basic/acid site distribution of the active sites for the catalysts. The morphology of the catalysts were then investigated by using scanning electron microscope (SEM) (SEM JOEL 6400). The determination of elemental compositions such as Ca, Ni, Zn, Fe and Co on the surface of catalysts were determined by FESEM-EDX mapping analysis using LEO 1455 VP electron microscope via Rayny EDX-720 spectrometer.2.4 Activity test
Catalytic deoxygenation of triolein was carried out in a 250 mL two-necked round bottom flask equipped with magnetic stirrer bar, heating mantle and connected to a distillation system. Approximately 20 g of triolein and 5 wt% of synthesized catalyst was added into the flask. The deoxygenation reaction of triolein was performed by heating the reaction medium to 350 °C under inert condition (N2 flow = 20 cc per min) for 60 min. The cracked/deoxygenated product was then collected in a receiver flask and at the end of the test, the reactor was cooled to room temperature using an external water circulation cooling system and the liquid products was further weighted and analyzed using acid number (AN) test, Fourier transform infrared spectroscope (FTIR), gas chromatography flame ionization detector (GC-FID) and gas chromatography mass spectrometry (GCMS). All the reactions were repeated 3 times and the reported data described as the mean values of the 3 repetitions. The analyses of mass balance were conducted for all deoxygenation reactions. The solid catalysts were separated by mixing the liquid residue inside the flask with hexane to determine the mass of the retained products (char + residue) after reaction. The hexane was removed via rotary evaporator and the dark viscous liquid was identified as (char + residue). The analyses of carbon balance were conducted by collecting the liquid product every 15 min over a period of 135 min. The changes in the hydrocarbon product and acid values were observed by GC-FID and total acid number (TAN) test.2.5 Optimization study
The optimization studies were carried out via the Box-Behnken design to maximize the hydrocarbon yield from triolein by investigating the effect of 3 combinations of variables. Three independent variables such as (A): reaction temperature (300–390 °C), (B): reaction time (60–120 min) and (C): catalyst loading (1–9 wt%) was applied. All the 17 experiments were augmented with 5 replications and were carried out at the center points to evaluate the pure error. In the present study, the Design Expert 6.0.4 software was used to obtain the regression and graphical analysis of the data. The maximum values of hydrocarbon yield were taken as the response of the design experiment for deoxygenation process. The experimental data obtained from the above procedure was analyzed by the response surface regression using polynomial equation eqn (1), where Y is the predicted response; βo, βj, βij and βjj are constant coefficients; xi and xj are the coded independent variables or factors; ε is random error. The equation was also being validated by carrying out confirmatory experiments.
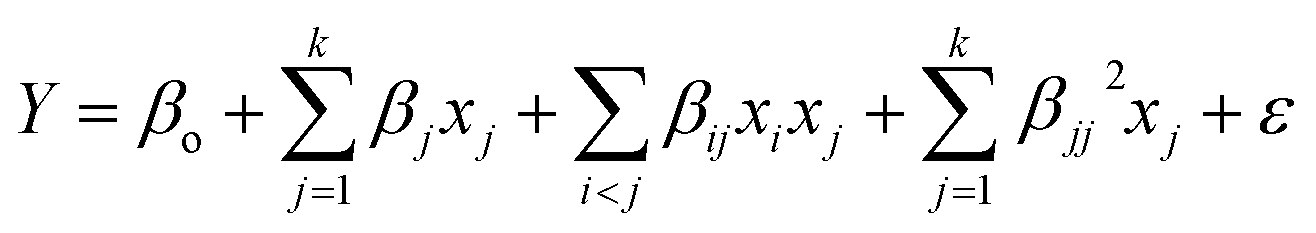 | (1) |
2.6 Product analysis
The deoxygenated liquid products were identified using standard alkane and alkene n-(C8–C20) and were quantitatively analyzed in gas chromatography with flame ionization detector (GC-FID) (Shimadzu GC-14B) equipped with a HP-5 capillary column (length: 30 m × inner diameter: 0.32 mm × film thickness: 0.25 μm) operating at 300 °C. The liquid product was diluted with GC grade n-hexane prior to the yield analysis. 1-Bromo hexane was used as internal standard for quantitative analysis. An aliquot of 1 μL sample was injected with into the GC column. The injection temperature was at 250 °C. Nitrogen gas was served as the carrier gas. The initial temperature of the oven was set at 40 °C and held for 6 min, then ramped up to 270 °C at the heating rate of 7 °C min−1. The hydrocarbon yield and product selectivity were determined by comparing the retention time of standard hydrocarbons (C8–C20) with experimental-based products which shown Fig. S1(see ESI†). In the present study, the catalytic performances of the catalysts was studied by determining the yield of saturated and unsaturated straight-chain hydrocarbons (X) (eqn (2)).14 All analyses were conducted for 3 times to confirm the reproducibility of the results.
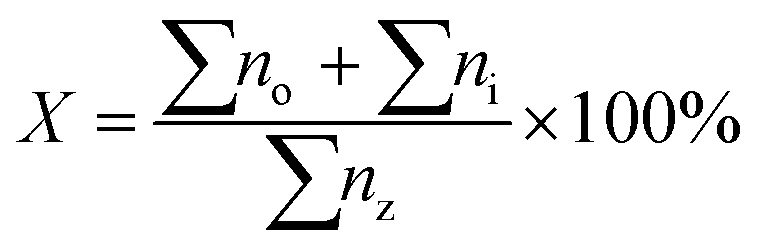 | (2) |
The hydrocarbon selectivity (Sc) or carbon balance were determined by eqn (3)
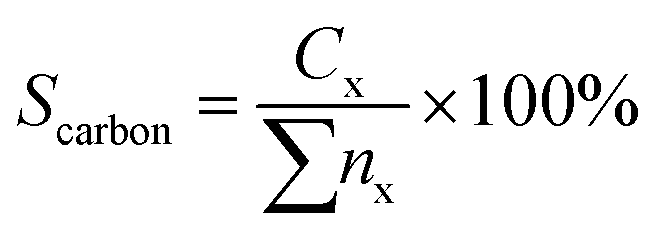 | (3) |
Product distribution of the deoxygenized liquid product were qualitatively characterized using GC-MS (model SHIMADZU QP5050A) equipped with a non-polar DB-5HT column (30 m × 0.32 mm × 0.25 μm) with split-less inlet. The samples were diluted with GC grade n-hexane (purity >98%) to 100 ppm. The generated fraction peaks from the GC-MS spectrum were identified through the National Institute of Standards and Testing library. The compound identification was based on a probability match equal to or higher than 95%. The yield of the organic compound (hydrocarbon fractions, carboxylic acid, alcohol and etc.) was determined using eqn (4):
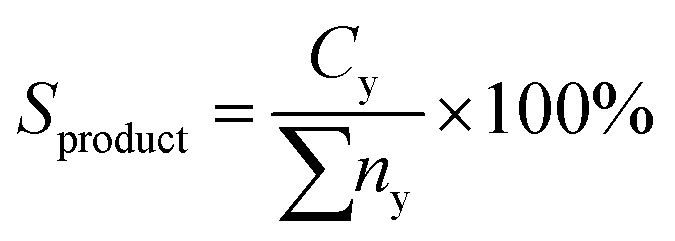 | (4) |
Degree of oxygen to carbon (O/C) atomic ratio and degree of hydrogen to carbon (H/C) atomic ratio were determined by Elemental CHNS/O 2400 analyser. The similar method has been reported by several high inter-disciplinary article focused on deoxygenation reaction.18,19 The results were depicted in terms of H/C and O/C ratio in Van Krevelen diagram. The acid values of the feedstock and liquid products were determined using standard method of AOAS Cd 3d-63. It is important to determine the amount of carboxylic acid/fatty acid in deoxygenated product. The acid value was calculated using equation as shown in eqn (5).
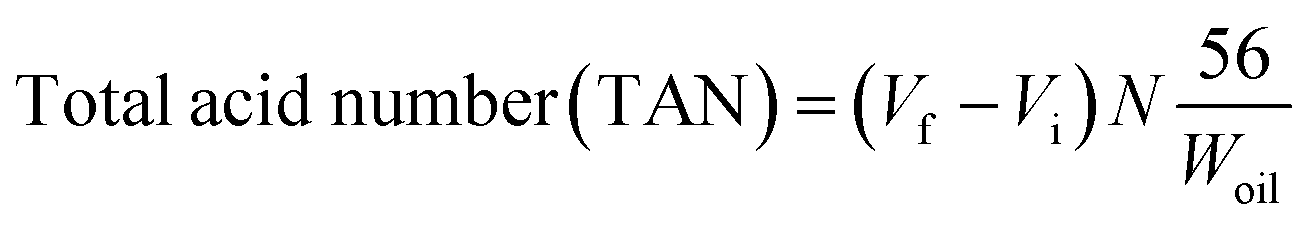 | (5) |
Furthermore, Fourier Transform-Infrared Spectrometer (FT-IR) analysis was performed using PerkinElmer (PC) Spectrum 100 FTIR with a resolution of 4 cm−1 in the IR range of 300–4000 cm−1 which principally followed the concept of Attenuated Total Reflection (ATR) method. This analysis was to determine the chemical functional group contained in the liquid product.
3 Results and discussion
3.1 Physicochemical properties of catalysts
The bulk elemental composition for transition metal oxide (TMO)-doped with CaO catalysts was studied by using XRF analysis (Table 1). The results showed that the presence of transition metal oxides (NiO, ZnO, Fe2O3 and Co2O4) at 15–19 atomic% for each catalyst. Calcium (Ca) content was found as the major element (>80 atomic%) in all catalysts. Thus, experimental ratio (TMO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CaO) of synthesized catalysts (20% of active metal oxide and 80% of CaO) were in agreement with the intended ratio, which is 2:8 atomic ratio. The TMO-doped CaO catalysts were further characterized using FESEM-EDX mapping (Fig. 1a–d) to verify the Ni, Zn, Fe and Co concentration on the surface of support. Ca-rich regions attributed to CaO particles were clearly observed on the catalyst surface. In addition, TMO (Ni, Zn, Fe and Co) was well-dispersed on the surface of CaO. The speckled pattern of transition metal species indicated that the uniform distribution of metallic active sites over the CaO matrix particles contributed to the efficient reactions. Furthermore, the EDX analysis reported that the TMO-doped CaO catalysts rendered the average of 77–87 ± 3.1 wt% of calcium, 14.2 ± 2.1 wt% of Ni for Ni–CaO, 10.4 ± 3.2 wt% of Zn–CaO, 22.9 ± 2.4 wt% of Fe–CaO and 14.9 ± 3.1 wt% of Co–CaO. The high content of CaO in the TMO-doped CaO catalysts acted as effective basic sites which had enhanced the catalysts stability and simultaneously enhanced the occurrence of deCOx reactions.
:CaO) of synthesized catalysts (20% of active metal oxide and 80% of CaO) were in agreement with the intended ratio, which is 2:8 atomic ratio. The TMO-doped CaO catalysts were further characterized using FESEM-EDX mapping (Fig. 1a–d) to verify the Ni, Zn, Fe and Co concentration on the surface of support. Ca-rich regions attributed to CaO particles were clearly observed on the catalyst surface. In addition, TMO (Ni, Zn, Fe and Co) was well-dispersed on the surface of CaO. The speckled pattern of transition metal species indicated that the uniform distribution of metallic active sites over the CaO matrix particles contributed to the efficient reactions. Furthermore, the EDX analysis reported that the TMO-doped CaO catalysts rendered the average of 77–87 ± 3.1 wt% of calcium, 14.2 ± 2.1 wt% of Ni for Ni–CaO, 10.4 ± 3.2 wt% of Zn–CaO, 22.9 ± 2.4 wt% of Fe–CaO and 14.9 ± 3.1 wt% of Co–CaO. The high content of CaO in the TMO-doped CaO catalysts acted as effective basic sites which had enhanced the catalysts stability and simultaneously enhanced the occurrence of deCOx reactions.
| Catalyst | XRF | XRD | BET | TPD | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Elemental compositiona (%) | Crystallite sizeb (nm) | Surface areac (m2 g−1) | Pore size diameter rangec (nm) | CO2 desorption temperatured (°C) | Basic sitesd (μmol g−1) | NH3 desorption temperature (°C) | Acid sitese (μmol g−1) | ||||
| CaO | TMO | Others* | CaO | TMO | |||||||
| a Determined by XRF analysis, *Others minor component of metal oxides: Sc, Sr, Cu, Br, K and Fe.b Determined by using Debye–Scherrer equation. All determinations were carried out twice for each sample.c Determined by BET analysis.d Determined by TPD-CO2 analysis.e Determined by TPD-NH3 analysis. | |||||||||||
| CaO | 98.81 | — | 1.19 | 64.29 ± 0.21 | — | 9.8 | 2.86–78.32 | 636 | 548.52 | — | — |
| Ni–CaO | 81.96 | 15.70 | 0.48 | 50.50 ± 0.18 | 38.97 ± 0.16 | 7.58 | 2.88–121.94 | 650 | 549.71 | 467 | 106.66 |
| Zn–CaO | 84.11 | 17.81 | 0.19 | 55.40 ± 0.11 | 33.32 ± 0.14 | 7.37 | 2.96–152.35 | 664 | 624.57 | 151442 |
73.91100.23 |
| Fe–CaO | 81.75 | 19.37 | 0.44 | 48.02 ± 0.16 | 40.65 ± 0.17 | 7.88 | 2.87–188.68 | 631 | 523.74 | 456 | 32.13 |
| Co–CaO | 80.29 | 17.56 | 0.34 | 53.54 ± 0.13 | 36.71 ± 0.18 | 7.46 | 2.96–116.50 | 603826 |
260.79, 307.77 | 488840 |
257486.91 |
 | ||
| Fig. 1 FESEM-EDX mapping for (a) Ni–CaO, (b) Zn–CaO, (c) Fe–CaO and (d) Co–CaO catalysts. | ||
XRD analysis of CaO, Ni–CaO, Zn–CaO, Fe–CaO and Co–CaO catalysts were shown in Fig. 2a. The TMO-doped CaO showed intense diffraction peaks of CaO phases and low crystallinity peaks of transition metal oxides phases. Based on the XRD results, all the TMO and CaO were segregately presented in individual metal oxide phases. The XRD characteristics of CaO peaks appeared at 2θ = 32.21°, 37.21°, 54.34°, 64.62° and 67.76° that supported by JCPDS card no. 00-037-1497. Ni–CaO catalyst showed the presence of XRD peak at 43.76°, which was attributed to NiO phase (JCPDS card no. 00-047-1049). The Zn–CaO catalyst revealed diffraction peak at 36.76°, which confirmed the presence of ZnO phase composited in CaO (JCPDS card no. 00-036-1451). Besides, Fe2O3 phases from Fe–CaO catalyst exhibited at peaks of 25.8° and 32.61°, respectively (JCPDS card no. 00-021-0917). While Co–CaO catalyst showed the presence of Co2O4 peak at 33.76° (JCPDS card no. 00-042-1467). Moreover, it is observed that a shifting of the diffraction TMO-doped CaO peaks to the higher 2θ values compared to pure CaO. The shifted of peak is well explained with the presumption that Ca2+ lattice parameter (1.06 Å) is partially substituted by the TM lattice (Ni2+: 0.55 Å, Zn2+: 0.6 Å, Fe3+: 0.49 Å and Co4+: 0.40 Å) in TMO-CaO catalysts.20 Result shows that the lattice parameter for pure CaO was a = 2.407 Å, whereas for CaO peak in Ni–CaO, Zn–CaO, Fe–CaO and Co–CaO catalysts were a = 2.39 Å, 2.39 Å, 2.37 Å and 2.38 Å, respectively. These results has suggested that the Ni2+, Zn2+, Fe3+ and Co4+ cations from TMO-doped CaO were replacing the Ca2+cations in the CaO structure. Therefore, minor content of solid (TM(x)Ca(1−x)O) was formed in the solution. As shown in Table 1, the crystallite sizes decreased with the addition of transition metals in CaO catalysts. The crystallite size of cubic phase of CaO catalysts were remarkably decreased from 64 nm to the range of 48–55 nm for TMO-doped CaO catalysts. This indicated that the insertion of the Ni2+, Zn2+, Fe3+ and Co4+ in CaO lattice would reduce the crystallite sizes.
 | ||
| Fig. 2 (a) XRD diffraction patterns and (b) SEM images for CaO and TMO-doped CaO catalysts. | ||
Textural properties of clamshell-derived CaO and TMO-doped CaO catalysts were summarized in Table 1. The surface area of CaO was 9.8 m2 g−1, while Ca-based catalysts (Ni–CaO, Zn–CaO, Fe–CaO and Co–CaO) rendered lower specific surface area, which were 7.58 m2 g−1, 7.37 m2 g−1, 7.88 m2 g−1 and 7.46 m2 g−1, respectively. This finding indicated that the transition metal oxides were successfully impregnated onto the surface of CaO supports and partially covered the CaO active sites.21 It is also observed that the pore diameters of all TMO-doped CaO catalysts were increased in the range of 2–188 nm and varied based on different TMO precursor. The results indicated that the catalysts were mainly consisted of mesoporous (2–50 nm) and macroporous (>50 nm) structure. The significant increase of the pore diameters for TMO-doped CaO catalysts was likely associated with the thermal activation temperature of the synthesized precursor, which collapsed the pore walls.22 This had led to the pitting and erosion on the newly formed TMO-doped CaO catalysts surfaces, which formed a wide channel for diffusion of reactant and product into the TMO-doped CaO pores,23 and thus enhanced the catalytic activity.
The SEM micrographs for all synthesized catalysts were shown in (Fig. 2bi–bv). CaO catalyst prepared from clamshell showed irregular shape with agglomerated structure demonstrated sintering effect of catalyst due to high calcination temperature at 900 °C (Fig. 2b–i).24 However, SEM morphology for all TMO-doped CaO catalysts rendered significant changes of morphology for surface structure. The images for both Ni–CaO and Zn–CaO catalysts revealed that irregular cubic-like structures which were larger in size of particles (Fig. 2b-ii and iii). Interestingly, the particles of Fe–CaO exhibited flat flakes-like structures, which had changed the dominant structure of CaO after the impregnation process (Fig. 2b-iv). In the case of Co–CaO, the aggregated particles consisted of interconnected sheet-like structure with rough surfaces (Fig. 2b-v). The TMO-doped CaO catalysts were found to comprise of large cluster, which had resulted in lower surface area of the catalyst which is shown in Table 1.
The results of XRF and XRD showed that CaO is the dominant metal oxide in the prepared catalysts (Ni–CaO, Zn–CaO, Fe–CaO and Co–CaO). Thus, it was estimated that the catalysts rendered strong base active sties that contributed by Ca2+–O2− phases.25 The basicity study of TMO-doped CaO catalysts was determined by TPD-CO2 analysis (Fig. S2(a)† and Table 1). TMO-doped CaO catalysts showed broad and intense desorption peak at temperature higher than 500 °C. This had proven the presence of stronger Bronsted basic sites.26 Furthermore, basicity trend of synthesized catalysts in the order of Zn–CaO > Co–CaO > Ni–CaO > CaO > Fe–CaO has showed that Zn, Co and Ni-doped CaO were capable of rendering higher amount of basicity than CaO catalyst. This is due the interaction between bi-metal ions, which had promoted synergy effect by enhancing the basicity between active TMO (Zn, Co and Ni) and CaO.27 The acidity profile of synthesized catalysts was studied using TPD-NH3 analysis. TMO-doped CaO catalysts (Ni–CaO, Zn–CaO and Fe–CaO) showed low intensity on NH3 desorption peaks in the range of 151 °C to 450 °C (refer to Fig. S2(b)† and Table 1). This has suggested that the presence of a considerable amount of weak and medium acidic sites on the surfaces of catalysts.10 The presence of acid sites were attributed to Brønsted acid sites associated with the bridging of OH groups and/or Lewis acid sites associated with the presence of transition metal ions [Ni2+, Zn2+, and Fe3+].10,17 From the findings, the acidity of TMO-CaO catalysts were in the order of Co–CaO > Zn–CaO > Ni–CaO > Fe–CaO. Co–CaO catalyst, where Co–CaO rendered the highest acidity (743.91 μmol g−1) with presence of medium and strong acid strength, which had proven the presence of synergy effect between the bonding of Co2O4 and CaO.
3.2 Deoxygenation activity
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching, 1455 cm−1 (–CH2) scissoring, 1398 cm−1 (–CH2), 1285 cm1 (–C–O–C) and 726 cm−1 –(CH)n– bending for alkane.19 FTIR results for triolein and liquid deoxygenated product showed that all spectrum were normalized by the intensity of the absorption band centered at 2858–2950 cm−1(CH stretching, aliphatic). It was noteworthy to mention that the liquid deoxygenated products showed significant shifting of absorption band from 1749 cm−1 that belonged to CO (ester) in molecule triglycerides to the absorption band at 1715 cm−1 which was attributed to CO of carboxylic acid. This implied the occurrence of cracking reaction of ester to intermediates products of carboxylic acid. This fact was in agreement with the acid value test, where the acidity of the triolein was increased drastically from 5 mg KOH per g to >100 mg KOH per g after deoxygenation reaction (Fig. 3a). Besides, deoxygenation reaction also resulted in the absent of C–O–C absorption features at 1285 cm−1 which belonged to carbonyl group in triolein, these was in agreement with the removal of oxygen species via deCOx pathways.
O) stretching, 1455 cm−1 (–CH2) scissoring, 1398 cm−1 (–CH2), 1285 cm1 (–C–O–C) and 726 cm−1 –(CH)n– bending for alkane.19 FTIR results for triolein and liquid deoxygenated product showed that all spectrum were normalized by the intensity of the absorption band centered at 2858–2950 cm−1(CH stretching, aliphatic). It was noteworthy to mention that the liquid deoxygenated products showed significant shifting of absorption band from 1749 cm−1 that belonged to CO (ester) in molecule triglycerides to the absorption band at 1715 cm−1 which was attributed to CO of carboxylic acid. This implied the occurrence of cracking reaction of ester to intermediates products of carboxylic acid. This fact was in agreement with the acid value test, where the acidity of the triolein was increased drastically from 5 mg KOH per g to >100 mg KOH per g after deoxygenation reaction (Fig. 3a). Besides, deoxygenation reaction also resulted in the absent of C–O–C absorption features at 1285 cm−1 which belonged to carbonyl group in triolein, these was in agreement with the removal of oxygen species via deCOx pathways.
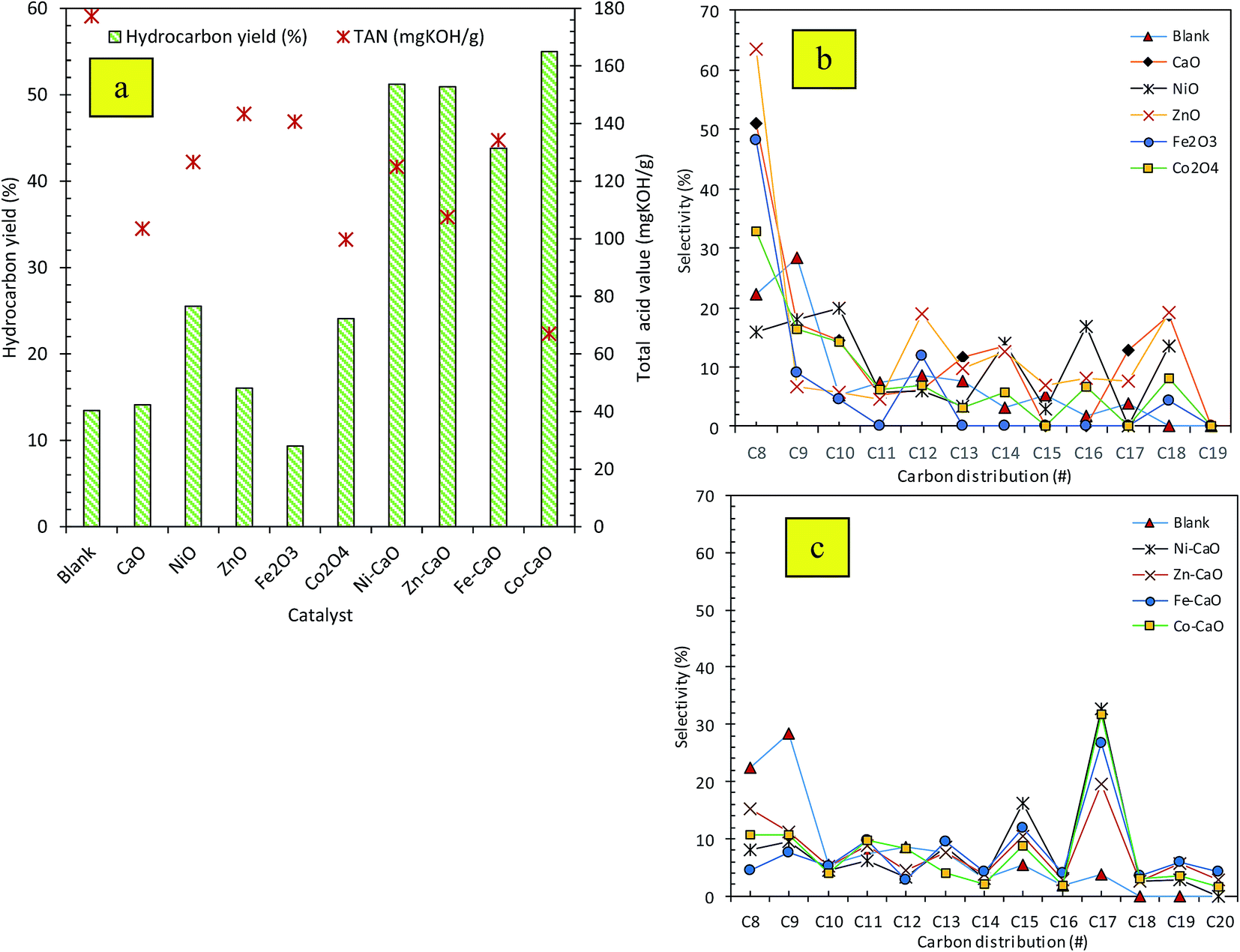 | ||
| Fig. 3 (a) Hydrocarbon yield and (b and c) product selectivity of the liquid deoxygenated product. | ||
As shown in our recent study,15 the GC-MS chromatogram of the trioleins revealed that this feedstock was dominant toward saturated and unsaturated C16 and C18 fatty acids. The product of the subsequent fatty acid deCOx process are predicted to be resulted in formation of the long-chain hydrocarbon of n-C15 and n-C17 fractions. Based on the carbon distribution profile of the product Fig. 3b and c, all the catalyst-free and TMO catalyzed reaction showed wide distribution of hydrocarbon within the range of n-(C8–C20). Our observations also verified that majority of the hydrocarbons fractions tended to form light hydrocarbon n-C8. Nevertheless, the product distribution from TMO-doped CaO catalyzed deoxygenation of triolein was improved compared to deoxygenation reaction without catalyst and with TMO catalysts. TMO-doped CaO catalysts were more selective to form desired deCOx product (n-C15, n-C17) and there were only minor formation of n-(C8–C12). Co–CaO and Ni–CaO catalysts showed the highest (n-C15 + n-C17) selectivity at 41% and 48%, respectively. Thus, it can be suggested that the use Co- and Ni-doped CaO catalyzed deoxygenation was in favor to the formation of Cn−1 hydrocarbons, which has implied higher deCOx activity. Formation of n-(C8–C12) hydrocarbon fractions were detected in the reaction which indicated that cracking reaction via C–C scission had also occurred, this was then proved that TMO-doped CaO had catalyzed simultaneous cracking-deCOx reaction. Co–CaO is more favored toward deoxygenation reactivity with the highest hydrocarbon yield (54%) and high production of desired deCOx product (n-C15 + n-C17). The efficiency of Co–CaO catalyst in deoxygenizing triolein was confirmed by producing the lowest acid value (67 mg KOH per g) in deoxygenated liquid product (Fig. 3a).
Fig. 4a showed the chemical composition of deoxygenated liquid products catalyzed by TMO-doped CaO catalysts. Results had showed that 91% of the total unsaturated and saturated C18 + C16 fatty acids in triolein had gone through remarkable changes after catalyzed by TMO-doped CaO catalysts. All the catalyst rendered desired reaction with saturated and unsaturated hydrocarbon fractions n-(C8–C20) in the range of 60% to 76%, minor coupling products (ketone), alcohol, cyclic, heavy hydrocarbon n-(C21–C24) fractions and acid compounds. The unfavourable reaction towards the formation of ketones rich compound was dominated by the high numbers of basic sites (Fe–CaO). It was in agreement with Parida's group, which high ketonization degree (86.5%) of acetic acid to acetone catalyzed by basic catalyst derived Mg/Al hydrotalcite at 350 °C.28 However, ketonization rate reactions could be inhibited with the increase of acid sites follow order Co–CaO – Ni–CaO > Zn–CaO > Fe–CaO (Fig. S2(b)† and Table 1). This implied that the relative acidity in catalyst is important in inhibiting the coupling process.29 We also found that the Fe–CaO promoted significant formation of heavy hydrocarbon n-(C21–C24). It is possible to infer that the polymerization reaction in deoxygenation of triolein was expected to occur due to poor acidic properties of Fe2O3 and CaO metal oxides. Both Co–CaO and Ni–CaO rendered similar trend of product selectivity, which is in the range of 29–35% for gasoline and 38–44% for diesel based on the gasoline and diesel profile (Fig. 4b). As a summary, Co–CaO showed the highest hydrocarbon yield compared to other catalysts, thus it is selected for further optimization study to improvise on the catalytic activity and product selectivity.
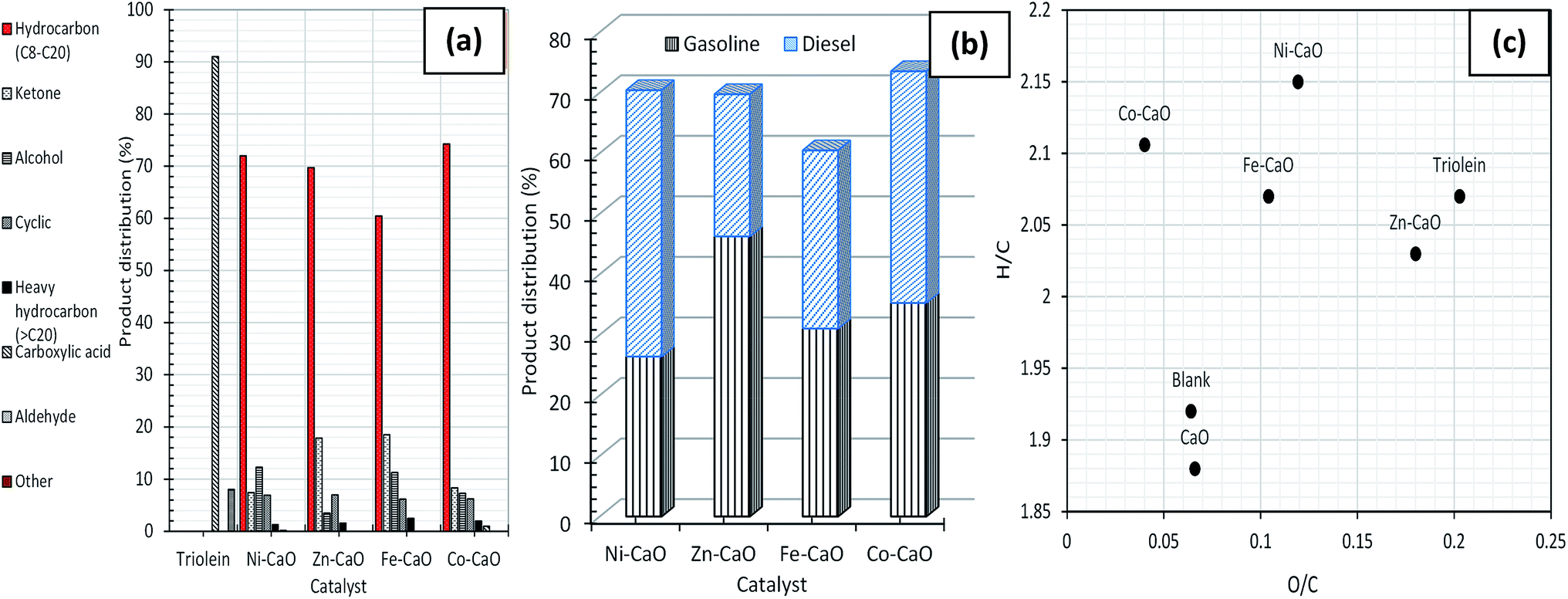 | ||
| Fig. 4 (a) Product distribution of deoxygenated liquid product and (b) gasoline and diesel distribution catalysed by TMO-doped CaO catalysts and (c) H/C and O/C ratio for deoxygenized liquid product (Van Krevelen diagram). | ||
Generally, the desired catalytic deoxygenation pathway is the capability of the catalyst to efficiently remove oxygen molecules while reducing the carbon loss to maintain the quality of final fuel product. Based on the catalytic activity study, Co–CaO catalyst showed the highest extent of deoxygenation efficiency and high selectivity towards formation of n-C15 + n-C17 products. The presence of bifunctional acidity-basicity in Co–CaO catalyst is the key factor to influence the deoxygenation pathway (refer to TPD profile, Table 1). The presence of acid sites facilitated oxygen removal via hydrolysis of carboxylic ester to acid (eqn (6)) and follow by dehydration pathway (eqn (7)).34 Both oxygen and hydrogen content can be removed effectively in reaction pathway. On the other hand, active basic sites generated from CaO are able to promote synergic effect towards the deCOx pathways of carboxylic acid chain to form hydrocarbon products (eqn (8) and (9)).19,35 It was speculated that the strong basic site derived from oxygen on the metal oxides able to create a forces by abstracting an alpha hydrogen in carbonyl compound, followed by C–O scission to form hydrocarbon.36 Thus, deoxygenation processes via hydrolysis, dehydration, decarboxylation and decarbonylation can perform effectively with the existence of both strong acid–base catalyst. This is in agreement with the GC-MS results (Fig. 4a), where lowest oxygenates compound (<15%) was detected with the use of strong acid–base Co–CaO catalyst. In contrast, other catalysts with strong basic sites medium acidic sites characteristic (Ni–CaO, Fe–CaO and Zn–CaO) showed formation of high oxygenates compound (19–30%). In addition, side reaction such as cracking of C–C bond pathway occurred during deoxygenation process. The cracking pathway is unavoidable in deoxygenation reaction as this reaction predominantly occur at high reaction temperature (350–500 °C).37 The deoxygenized liquid product will be thermally decomposed to form shorter hydrocarbon.35,38 It is foreseeable that the presence of strong acidic sites on the catalysts will induce cracking of fatty acid (eqn (10)) or deoxygenated product via C–C scission to form light hydrocarbon fractions (n-(C8–C12)) (eqn (11)). It was summarized that during catalysis, acidic sites facilitated dehydration and cracking, while the basic sites mainly promoted deCOx and partially removing oxygen from the triolein simultaneously.
Hydrolysis of carboxylic ester to acid
| RCOOR + H2O → RCOOH + ROH | (6) |
Dehydration of fatty acid
| RCOOH → RCO + H2O | (7) |
Decarboxylation of fatty acid
| RCOOH → CO2 + RH | (8) |
Decarbonylation of fatty acid
| RCOOH → CO + H2O + RH | (9) |
Cracking
| RxCOOH → Rx−1H + RxCOOH | (10) |
|
Rx–Ry → RxCH + RyCH
| (11) |
| Theoretical deCOx | Triolein liquid (oil) + 3mol CO2/CO (g) + 3mol H2O (aq) + by product (12) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Reactiona | Feedstock | Liq-productb | Gasc | Waterd | Char + residuee | ||||
| (g) | (g) | (wt%) | (g) | (wt%) | (g) | (wt%) | (g) | (wt%) | |
| a Deoxygenation condition: reaction temperature of 350 °C, 60 min reaction time, 5 wt% of catalyst, under vacuum condition 10 mbar pressure and stirring 400 ppm.b Mass fraction for liq-product = [(mass of liq-product/mass of feedstock) × 100].c Material fraction for gas = [(mass of feedstock − mass of liq-product − mass of (char + residue) − mass of water)/mass of feedstock × 100].d Material fraction for water = [(mass of water/mass of feedstock) × 100].e Material fraction for (char + residue) = [mass of (char + residue)/mass of feedstock) × 100)]. | |||||||||
| Theoretical data (deCOx) | 10.00 | 6.89 | 68.90 | 2.49 | 24.90 | 0.62 | 6.20 | — | — |
| CaO | 10.05 | 3.25 | 32.33 | 2.62 | 26.06 | 0.55 | 5.23 | 3.63 | 35.76 |
| Ni–CaO | 10.01 | 2.20 | 21.97 | 2.65 | 26.47 | 0.52 | 5.19 | 4.64 | 46.35 |
| Zn–CaO | 10.01 | 2.36 | 23.57 | 2.88 | 28.87 | 0.61 | 6.09 | 4.16 | 41.55 |
| Fe–CaO | 10.02 | 3.03 | 30.23 | 2.54 | 25.34 | 0.55 | 5.48 | 3.90 | 38.92 |
| Co–CaO | 10.09 | 3.80 | 37.66 | 3.20 | 31.71 | 0.62 | 6.14 | 2.67 | 26.46 |
3.3 Optimization studies via Box-Behnken design
| Hydrocarbon yield = 54.38 + 3.62A + 1.66B − 3.63C −5.53A2 − 8.41B2 − 6.90C2 − 8.57AB − 0.050AC − 8.61BC | (13) |
| Run | A: reaction temperature (°C) | B: reaction time (min) | C: catalyst loading (wt%) | Hydrocarbon yield (%) | |
|---|---|---|---|---|---|
| Experimental response | Predicted response | ||||
| 1 | 300 | 60 | 5 | 25.73 | 26.59 |
| 2 | 360 | 60 | 5 | 52.26 | 50.97 |
| 3 | 300 | 120 | 5 | 45.75 | 47.04 |
| 4 | 360 | 120 | 5 | 38.00 | 37.14 |
| 5 | 300 | 90 | 3 | 42.80 | 41.91 |
| 6 | 360 | 90 | 3 | 48.00 | 49.25 |
| 7 | 300 | 90 | 7 | 36.00 | 34.75 |
| 8 | 360 | 90 | 7 | 41.00 | 41.89 |
| 9 | 330 | 60 | 3 | 32.40 | 32.44 |
| 10 | 330 | 120 | 3 | 53.36 | 52.97 |
| 11 | 330 | 60 | 7 | 42.00 | 42.40 |
| 12 | 330 | 120 | 7 | 28.53 | 28.49 |
| 13 | 330 | 90 | 5 | 53.52 | 54.38 |
| 14 | 330 | 90 | 5 | 54.42 | 54.38 |
| 15 | 330 | 90 | 5 | 55.00 | 54.38 |
| 16 | 330 | 90 | 5 | 55.26 | 54.38 |
| 17 | 330 | 90 | 5 | 53.69 | 54.38 |
Positive sign in front of the terms indicates synergistic effect, while negative sign indicates antagonistic effect.40 The generated model equation implied that positive coefficient of A and B rendered linear effect toward the response. Meanwhile, the quadratic term of C, A2, B2, C2, AB, AC, and BC showed negative effect, which decreased the hydrocarbon yield. Table 3 showed the percentage of hydrocarbon yield was varied between 26–54%. The highest hydrocarbon yield was at 330 °C, 90 min, 5 wt% catalyst loading and the lowest yield was at 300 °C, 60 min, 5 wt% catalyst loading. It was proven that the modeled and experimental values were similar, hence, validating the reliability of the model developed for establishing a correlation between the process variables and the hydrocarbon yield.
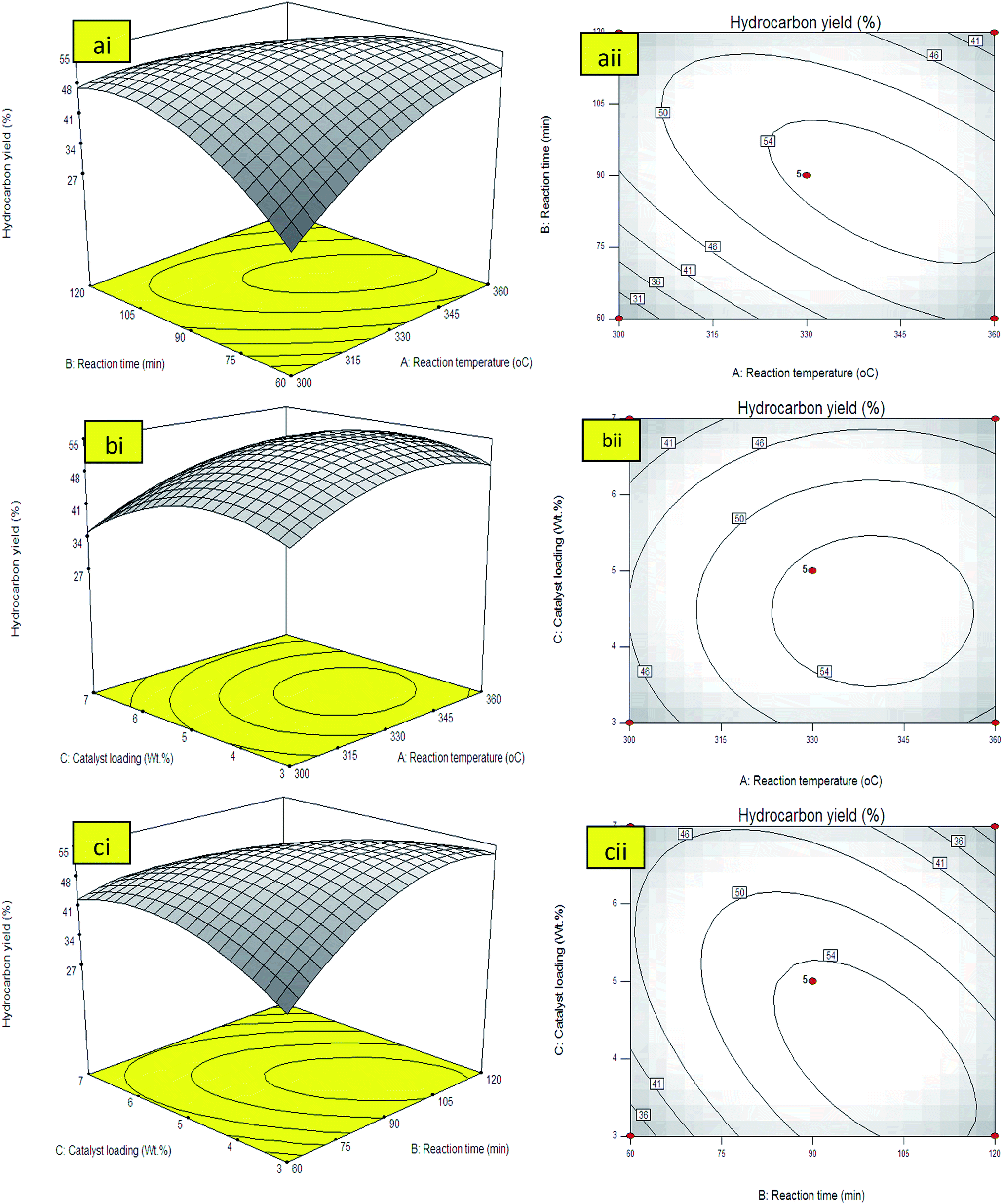 | ||
| Fig. 5 3D-response surface plot and 2D-contour plot for (a) interaction of reaction temperature and reaction time (AB), (b) interaction of reaction temperature and catalyst loading (AC), and (c) interaction of reaction time and catalyst loading (BC). | ||
The simultaneous interaction effects towards hydrocarbons yield by varying reaction temperature (A) and the catalyst loading (C) at constant reaction time of 90 min and 400 rpm was presented by 3D and contour lines plot in Fig. 5(bi and bii). The results showed that the catalyst loading rendered low impact on the hydrocarbon yield. The hydrocarbon yield increased with the increment of catalyst loading within the range of 3–5 wt% at reaction temperature up to 330 °C. The trend was reversed when catalyst loading was further increased to >5 wt% and reaction temperature exceeded 330 °C. Theoretically, higher catalyst loading would increase the availability of catalyst sites for deoxygenation reaction, thus increase the hydrocarbon yield. However, the usage of excess of catalyst would increase the possibilities of secondary reactions such as polymerization and cracking reactions. Polymerization reaction will lead to the formation of heavy product such as large aromatic-by product and asphaltenes which is unhealthy for the catalyst surface and lead to deactivation of catalyst.42 Moreover, further increment of the catalyst loading had resulted in high thermal cracking efficiency towards the formation of shorter hydrocarbon fraction like gaseous product.43 The occurrence of secondary reactions due to thermal cracking at high temperature will eventually resulted in production of more volatile species and caused reduction in the yield.
The effect of reaction time (B) and catalyst loading (C) on the hydrocarbon yield under constant reaction temperature at 330 °C were shown in both surface and contour plots in Fig. 5(ci and cii). Analysis of these two effects showed that the increment in both reaction time and catalyst loading increased the hydrocarbon yield up to a certain point. It has clearly shown that hydrocarbon yield was increased with the increment of reaction time from 60 to 105 min. Similar trend was also observed thru the increment of catalyst loading from 3 to 5 wt%. The contour plot Fig. 5(cii) indicated similar results with 3D-surface plot. It is obvious that the maximum hydrocarbon yield region can be obtained around ∼55% within 105 min using 5 wt% catalyst loading. It can be concluded that reaction temperature and reaction time shall have significant effect to deoxygenation of triglycerides into hydrocarbon fractions. The effect of increasing catalyst loading towards hydrocarbon yield is unsubstantial compared to reaction temperature and reaction time. It was suggested that increasing of all parameters above the optimum level will reduce the product yield. This was in agreement with eqn (13) when all the interaction of AB, BC and AC showed negative interaction. The model predicted that the maximum yield can be obtained at 340 °C, reaction time, 5 wt% catalyst loading within 105 reaction time. The optimum response variables were then tested following suggested conditions to verify the model prediction. The experimental results indicated that the hydrocarbons yield (56%) was reasonably close to the predicted value (55%) generated from the model.
3.4 Effect of catalyst/oil weight ratio
In order to investigate the deoxygenation activity of the Co–CaO with different catalyst/oil weight ratio, deoxygenation reactions of triolein were carried out at the catalyst/oil weight ratio of 0.01, 0.05 and 0.09 at the reaction conditions of 340 °C, 5 wt% catalyst loading within 105 min of reaction time under inert N2 flow. The results are shown in Table 4. Catalytic deoxygenation of triolein into hydrocarbons was found to be greatly affected by the catalyst/oil weight ratio. The hydrocarbon yield was found to increase in the order of 0.05 (56%) > 0.01 (50%) > 0.09 (43%). Similar trend was observed on the n-C15 + n-C17 and diesel selectivity of the deoxygenated liquid products. Lower rate of deoxygenation activity via deCOx reaction was observed with high catalyst/oil weight ratio tests, the result was further endorsed with the presence of high acid value (72 mg KOH per g) of the product. In contrast, the acid value for catalyst/oil weight ratio at 0.01 and 0.05 was very low (38–42 mg KOH per g), which indicated that conversion of carboxylic acid group occurred efficiently and the liquid product could be regarded as high quality hydrocarbon mixtures. Overall, it was implied that high catalyst/oil weight ratio favored C–C scissions of triglycerides and decomposed into intermediates product especially carboxylic acid groups with less occurrence of C–O scission via deCOx reaction.44 This indicates that high catalyst/oil weight ratio condition is not preferable in deCOx reaction.| Catalyst/oil weight ratioa (wt%) | Hydrocarbon yield (%) | n-C15 + n-C17 (%) | Diesel (C13–C20) | Total acid value (TAN) (mg KOH per g) |
|---|---|---|---|---|
| a Reaction condition: 340 °C reaction temperature, 105 reaction time, 5wt% catalyst loading within 105 reaction time. | ||||
| 0.01 | 50 | 52 | 63 | 42 ± 1.2 |
| 0.05 | 56 | 58 | 65 | 38 ± 0.8 |
| 0.09 | 43 | 44 | 58 | 72 ± 0.3 |
3.5 Co–CaO derived waste clamshell versus commercial CaO
The catalytic deoxygenation of triolein at 340 °C, 5wt% catalyst loading within 15–135 min reaction time using Co-CaOclamshell and Co-CaOcommercial under inert N2 flow condition was shown in Fig. 6a–d. The total amount of hydrocarbon fractions for Co-CaOclamshell and Co-CaOcommercial achieved 77% and 72%, respectively at the reaction time of ≥105 min (Fig. 6(a and b)). This finding indicated that the deoxygenation activity of Co-CaOclamshell is comparable to Co-CaOcommercial catalyst. Further study on carbon distribution profile was shown in Fig. 6(c and d). The results showed similar trend of carbon distribution curve for both catalysts. The desired product for n-C17 fraction showed maximum content at 60 min of reaction time. The product in carbon range of n-(C13–C16) increase after 60 min, this suggested that mild C–C dissociation of fatty acid has occurred. Meanwhile, reduction of product range of n-(C8–C12) after 60 min indicated that the cracking reaction has occurred mildly. The efficiency of Co-CaOclamshell and Co-CaOcommercial catalysts in converting the free fatty acids into hydrocarbon were in agreement with changes in acid values, FTIR analysis and GC-MS analysis. It can be seen that both Co-CaOclamshell and Co-CaOcommercial catalysts resulted in drastic changes in the acid values of liquid product from high-acid-value (145–135 mg KOH per g) in the first 15 min of reaction time to low-acid value–value (<2 mg KOH per g) after 105 min of reaction time. This is equivalent to acid value conversion of ∼98% (Fig. 6(c and d)). The elimination of intermediates acid species were further confirmed by FTIR analysis (Fig. 7a) with a significant reduction of CO stretch at 1713–1705 cm−1 which is belonged to carboxylic acid and elimination of C–O stretch at 1285 cm−1 which is belonged to carbonyl. Meanwhile, GC-MS analysis showed that zero detection of carboxylic acid compound (Fig. 7b). These observations had shown that the gradual formation of the hydrocarbon-based liquid fuel along with the elimination of intermediate acid compound during the deoxygenation reaction. Moreover, GC-MS also showed a trace amount of oxygenates and non-oxygenates compound which consisted of ketone, alcoholic, cyclic and heavy hydrocarbons n-(C21–C25). In contrast, straight chains alkanes and alkenes compounds with carbon number n-(C8–C20) was the most predominant in its concentration (68–83%). This had shown that the product selectivity is leaning towards diesel-range hydrocarbon product. These findings suggested that both Co-CaOclamshell and Co-CaOcommercial catalyzed the reaction toward diesel-range hydrocarbon formation via cracking-deCOx reaction and retarded the side reactions (polymerization, cyclization, ketonization and etc.) simultaneously.
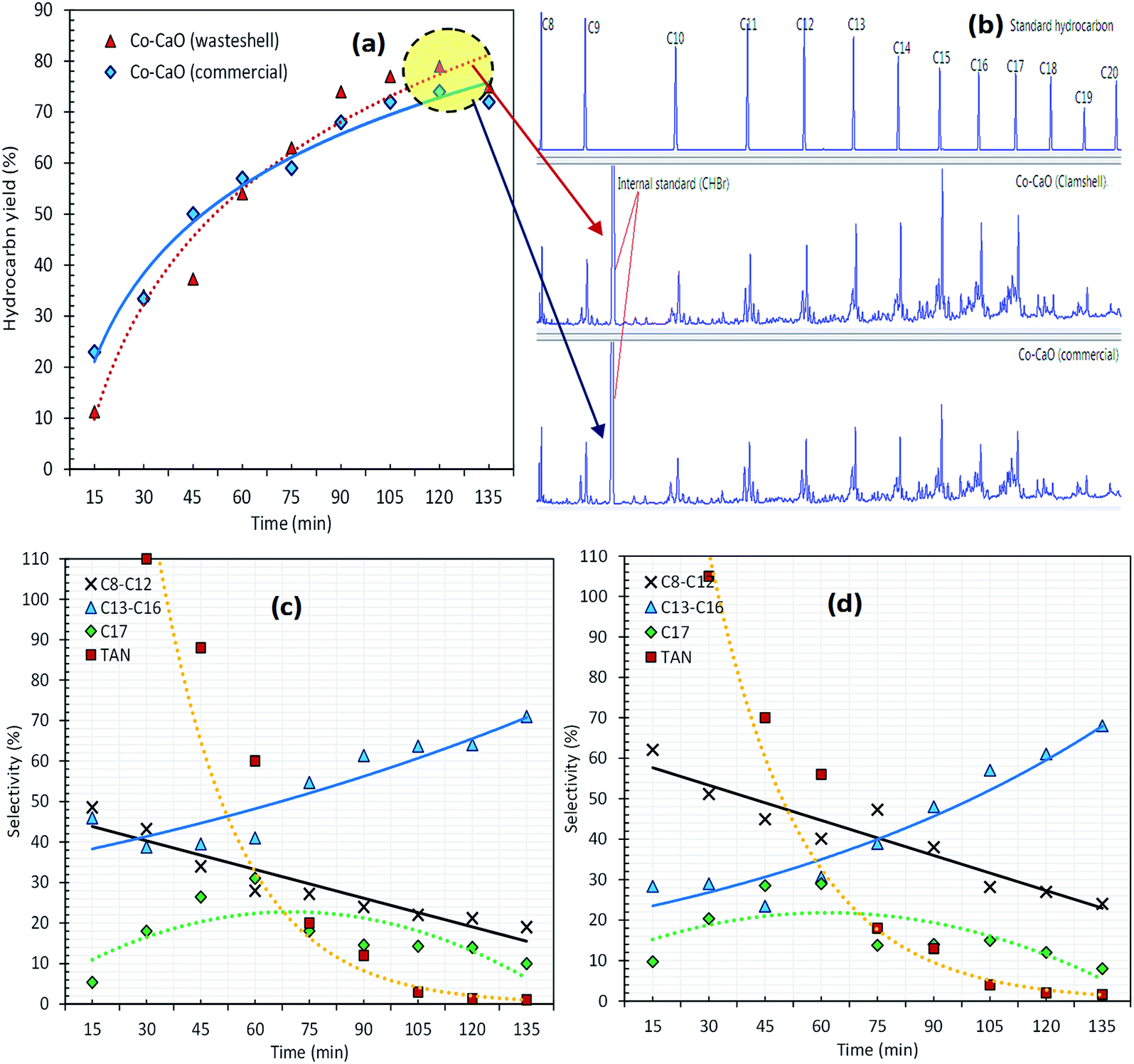 | ||
| Fig. 6 (a) Hydrocarbon yield, (b) GC chromatogram of the optimize sample and (c, d) carbon balance for deoxygenation of triolein via Co–CaO (wasteshell) and Co–CaO (commercial). | ||
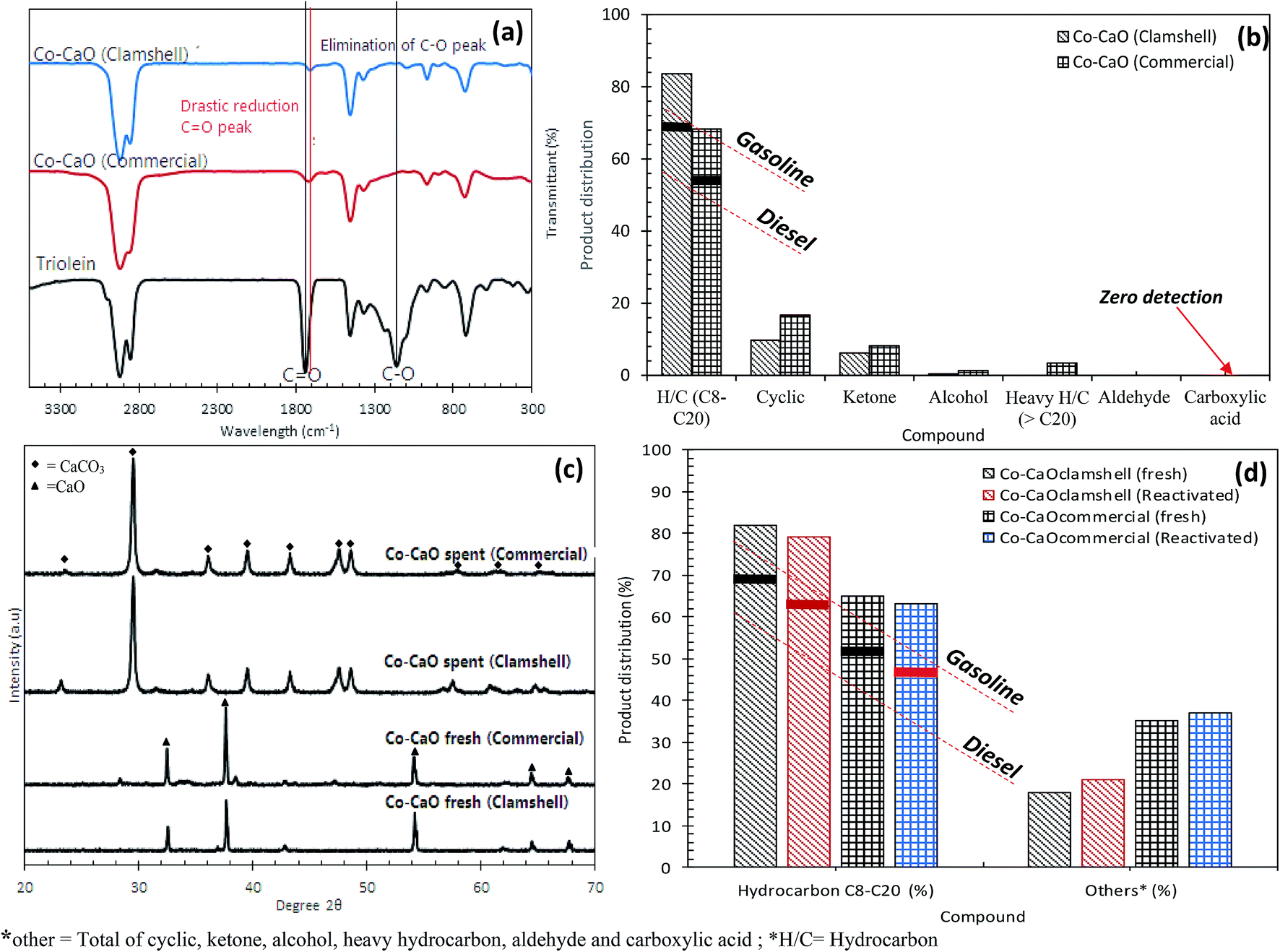 | ||
| Fig. 7 (a) FTIR spectra for triolein and deoxygenated liquid products, (b) product distribution for deoxygenated liquid product, (c) XRD diffraction for the as-synthesized catalysts of Co–CaO and Co–CaO after deoxygenation reaction and (d) product distribution for deoxygenated liquid product using fresh and re-activated Co–CaO catalysts. *other = total of cyclic, ketone, alcohol, heavy hydrocarbon, aldehyde and carboxylic acid; *H/C= hydrocarbon. | ||
The stability of Co–CaO catalyst for deoxygenation reaction under optimum condition (340 °C reaction temperature, 5 wt% catalyst loading, 105 reaction time under inert N2 flow) was further studied by investigation of the chemical characteristic of spent catalysts (Co-CaOclamshell and Co-CaOcommercial). The XRD spectra of the fresh and spent Co-CaOclamshell and Co-CaOcommercial catalysts (i.e., catalyst with highest hydrocarbon yield) are displayed in Fig. 7c. The XRD pattern of the spent Co–CaO catalyst showed that the presence of intense XRD peaks that is ascribed to inactive carbonate phases at 2θ: 23.30°, 29.64°, 36.11°, 39.66°, 43.38°, 47.73°, 48.77°, 57.66°, 60.80°, 64.93° (JCPDS Card no. 00.005-0585). This finding was in agreement with our previous study.29 Both clamshell-derived CaO support and commercial CaO support were reacted with carboxylic acid during deoxygenation process, and it was further deactivated into non-reactive carbonate phases in order to enhance the occurrence of deCOx pathways by absorbing the CO2/CO gas from the acid.45,46 It is also noteworthy to mention that the inactive carbonate phases of spent Co–CaO catalyst can be re-converted into active CaO phase through thermal activation. The recoverability of the spent Co-CaOclamshell and Co-CaOcommercial catalysts were performed via hydrothermal treatment with hexane to remove oil coating, followed by thermal activation at 800 °C for 1 h to convert inactive CaCO3 phases into active CaO phases. Results indicated that the deoxygenation activity of reactivated catalysts under N2 flow was comparable to fresh catalyst. Similar trend was reported by our former study in deoxygenation of triolein by using CaO-based catalyst under vacuum condition.29 The total hydrocarbon fraction (C8–C20) obtained from reactivated Co-CaOclamshell and Co-CaOcommercial catalysts was slightly reduced from 82% to 79% and 68% to 63%, respectively (Fig. 7d). Meanwhile, a trace amount of oxygenates and non-oxygenates compound were present in the deoxygenated liquid product catalyzed by reactivated Co–CaO catalyst. This implied that the active sites on the treated catalyst remained active after reactivation for further reaction. Utilization of Co-CaOclamshell as a catalyst has more benefits than Co-CaOcommercial catalyst as it was derived from natural sources which are abundant in nature, economically feasible and environment friendly.
3.6 Comparison studies and the reaction pathway of deoxygenation of triolein
The catalytic activity of Co–CaO at optimum conditions was further compared to other Ca-based catalysts which tabulated in Table 5.29,35,47,48 The summary showed that Co–CaO (present study) rendered better hydrocarbon yield (56%) and selectivity toward diesel range (65%) at optimized conditions (reaction time: 105 min, temperature: 340 °C and 5 wt% catalyst loading) under in N2 flow condition via deoxygenation reaction compared to mesoporous CaO/AC, MgO–SiO2, Co- and W- supported catalysts. Our former study shown that Co-supported catalyst rendered higher yield in gasoline-range product via cracking reaction in a shorter reaction time (45 min) under partial vacuum condition.29 Meanwhile, in the present study, Co–CaO rendered higher selectivity towards diesel-range within 105 min via cracking-deCOx reaction under inert N2 flow condition. Therefore, other than the synergy effect between TMO and CaO with improved acidity-basicity profile, the reaction parameters especially time and reaction atmospheres were also some of the crucial criteria to look into to enhance the product selectivity toward diesel-range hydrocarbon. This was in agreement with former studies, where deoxygenation under inert N2 or He flow condition rendered hydrocarbon within diesel-range fractions.35,47,48 Continuous flow of N2 and He during the reaction has helped in maintaining a high catalytic activity by avoiding CO2/CO gases produced by the deCOx reaction from poisoning the active catalyst surface.49 Furthermore, flowing N2 and He has the ability to promote desorption of organic contaminants at the active surfaces of the catalyst, therefore conserving its activity for longer reaction time.| Catalyst | Reaction condition | Feeds | H/C yield (%) | Reaction mode | Product selectivity (%) | References |
|---|---|---|---|---|---|---|
| a deCOx: decarboxylation/decarbonylation.b Decarboxy: decarboxylation. | ||||||
| CaO/AC | Time = 60 min, temperature = 350 °C, catalyst loading = 5 wt%, under inert N2 flow | WCO | ∼50 | deCOxa | Diesel | 47 |
| Cox–CaO, Wx–CaO | Time = 45 min, temperature = 350 °C, catalyst loading = 5 wt%, under partial vacuum condition | Triolein | ∼32, ∼22 | Cracking | Gasoline | 29 |
| CaO | Time = 360 min, temperature = 400 °C, catalyst loading = 3 wt%, under inert N2 flow | WCO, JCO | 70, <75 | Cracking-deCOxa | Diesel | 48 |
| MgO–SiO2 | Oil feed rate: 15 mL h−1, catalyst: 50 mL, LHSV: 0.3 h−1, temperature = 430 °C, under inert He flow | Palm oil | 50 | Cracking-decarboxyb | Diesel | 35 |
| Co–CaO | Time = 105 min, temperature = 340 °C, catalyst loading = 5 wt%, under inert N2 flow | Triolein | 56 | Cracking-deCOxa | Diesel | Present study |
4 Conclusion
The present study has investigated the performance of clamshell-derived catalysts (Ni–CaO, Zn–CaO, Fe–CaO and Co–CaO) for deoxygenation reaction of triolein. The binary metal oxide system between TMO promoters (NiO, ZnO, Co2O4 and Fe2O3) and CaO support has created both acidic-basic active sites that synergically contributed to the production of diesel-range product mainly composed of desired deCOx product. It was summarized that during catalysis, the oxygen removal from the triolein facilitated by the existence of strong acid–base sites. Based on the deoxygenation profile, the liquid hydrocarbon yield increased in the order of Co–CaO > Ni–CaO > Zn–CaO > Fe–CaO. Co–CaO catalyst rendered the highest yield (55%) with product selective toward deCOx product (n-C15 + n-C17), which due to the presence of excellent acidity/basicity profile (density, strength). The optimization study via RSM Box-Behnken suggested that the optimal value of ∼56% hydrocarbon yield were achieved under reaction conditions of 5 wt% of catalyst, 340 °C within 105 min of reaction time with product selectivity towards diesel-range. The optimization study also proposed that the interaction effect between reaction temperature–time would greatly influence the product yield. The application of clamshell derived CaO-based support in catalytic deoxygenation to produce green diesel is proven effective by comparison study between Co-CaOclamshell and Co-CaOcommercial, which found both catalysts rendered comparable deoxygenation activity with Co-CaOclamshell having an edge over Co-CaOcommercial as it was derived from natural clamshells which were abundant in nature, economical and environment friendly.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express great appreciation to financial support from University of Malaya's Research Grant (UMRG, RP025A/B/C-14AET), SATU Joint Research Scheme (ST014-2017) and Grand Challenge Grant (GC, Project number: GC001B-14AET).References
- H. Song, N. Wang, H.-L. Song and F. Li, Catal. Commun., 2015, 59, 61–64 CrossRef CAS.
- S. A. W. Hollak, J. H. Bitter, J. Van Haveren, K. P. De Jong and D. S. Van Es, RSC Adv., 2012, 2, 9387 RSC.
- W. Wang, Z. Qiao, K. Zhang, P. Liu, Y. Yang and K. Wu, RSC Adv., 2014, 4, 37288 RSC.
- L. T. Chiam and C. T. Tye, Malays. J. Anal. Sci., 2013, 17, 129–138 Search PubMed.
- Y. Lin, C. Zhang, M. Zhang and J. Zhang, Energy Fuels, 2010, 24, 5686–5695 CrossRef CAS.
- L. E. Oi, M.-Y. Choo, H. V. Lee, H. C. Ong, S. B. A. Hamid and J. C. Juan, RSC Adv., 2016, 6, 108741–108754 RSC.
- L. Chen, Y. Zhu, H. Zheng, C. Zhang and Y. Li, Appl. Catal., A, 2012, 411–412, 95–104 CrossRef CAS.
- X. Zhang, Q. Zhang, T. Wang, L. Ma, Y. Yu and L. Chen, Bioresour. Technol., 2013, 134, 73–80 CrossRef PubMed.
- P. J. Dietrich, F. G. Sollberger, M. C. Akatay, E. A. Stach, W. N. Delgass, J. T. Miller and F. H. Ribeiro, Appl. Catal., B, 2014, 156–157, 236–248 CrossRef CAS.
- K. Kandel, J. W. Anderegg, N. C. Nelson, U. Chaudhary and I. I. Slowing, J. Catal., 2014, 314, 142–148 CrossRef CAS.
- Z. He and X. Wang, Catal. Today, 2014, 240, 220–228 CrossRef.
- E. Meller, U. Green, Z. Aizenshtat and Y. Sasson, Fuel, 2014, 133, 89–95 CrossRef CAS.
- S. K. Kim, J. Y. Han, H. S. Lee, T. Yum, Y. Kim and J. Kim, Appl. Energy, 2014, 116, 199–205 CrossRef CAS.
- N. Asikin-mijan, H. V Lee, G. Abdulkareem-alsultan and A. Afandi, J. Cleaner Prod., 2016, 1–12 Search PubMed.
- N. Asikin-Mijan, H. V. Lee, Y. H. Taufiq-Yap, G. Abdulkrem-Alsultan, M. S. Mastuli and H. C. Ong, Energy Convers. Manage., 2016, 141, 325–338 CrossRef.
- N. Asikin-Mijan, Y. H. Taufiq-Yap and H. V. Lee, Chem. Eng. J., 2015, 262, 1043–1051 CrossRef CAS.
- Y. H. Taufiq-Yap, S. Sivasangar and a. Salmiaton, Energy, 2012, 47, 158–165 CrossRef CAS.
- J. Payormhorm, K. Kangvansaichol, P. Reubroycharoen, P. Kuchonthara and N. Hinchiranan, Bioresour. Technol., 2013, 139, 128–135 CrossRef CAS PubMed.
- N. Asikin-Mijan, H. V. Lee, Y. H. Taufiq-Yap, J. C. Juan and N. A. Rahman, J. Anal. Appl. Pyrolysis, 2015, 117, 46–55 CrossRef.
- J. O. Shim, D. W. Jeong, W. J. Jang, K. W. Jeon, B. H. Jeon, S. Y. Cho, H. S. Roh, J. G. Na, C. H. Ko, Y. K. Oh and S. S. Han, Renewable Energy, 2014, 65, 36–40 CrossRef CAS.
- A. Siahvashi and A. A. Adesina, Chem. Eng. Sci., 2013, 93, 313–325 CrossRef CAS.
- H. V. Lee, J. C. Juan, N. F. Binti Abdullah, R. Nizah Mf and Y. H. Taufiq-Yap, Chem. Cent. J., 2014, 8, 30 CrossRef PubMed.
- K. Chandra Mouli, K. Soni, A. Dalai and J. Adjaye, Appl. Catal., A, 2011, 404, 21–29 CrossRef CAS.
- S. Sirisomboonchai, M. Abuduwayiti, G. Guan, C. Samart, S. Abliz, X. Hao, K. Kusakabe and A. Abudula, Energy Convers. Manage., 2015, 95, 242–247 CrossRef CAS.
- N. Asikin-Mijan, H. V. Lee and Y. H. Taufiq-Yap, Chem. Eng. Res. Des., 2015, 102, 368–377 CrossRef CAS.
- J. Wu, Q. Xia, H. Wang and Z. Li, Appl. Catal., B, 2014, 156–157, 265–272 CrossRef CAS.
- P. Nair, B. Singh, S. N. Upadhyay and Y. C. Sharma, J. Cleaner Prod., 2012, 29–30, 82–90 CrossRef CAS.
- K. Parida and J. Das, J. Mol. Catal. A: Chem., 2000, 151, 185–192 CrossRef CAS.
- N. Asikin-Mijan, H. V. Lee, J. C. Juan, A. R. Noorsaadah, G. Abdulkareem-Alsultan, M. Arumugam and Y. H. Taufiq-Yap, J. Anal. Appl. Pyrolysis, 2016, 120, 110–120 CrossRef CAS.
- Y. Lou, P. He, L. Zhao and H. Song, Fuel, 2016, 183, 396–404 CrossRef CAS.
- S.-S. Kee, A. Mohammadi, Y. Kidoguchi and K. Miwa, SAE Tech. Pap. Ser., 2005 DOI:10.4271/2005-01-2086.
- J.-S. Chang, J.-C. Cheng, T.-R. Ling, J.-M. Chern, G.-B. Wang, T.-C. Chou and C.-T. Kuo, J. Taiwan Inst. Chem. Eng., 2016, 1–11 Search PubMed.
- X. Dupain, D. J. Costa, C. J. Schaverien, M. Makkee and J. A. Moulijn, Appl. Catal., B, 2007, 72, 44–61 CrossRef CAS.
- J. Zhang, Y. S. Choi and B. H. Shanks, Catal. Sci. Technol., 2016, 6, 7468–7476 CAS.
- H. Tani, T. Hasegawa, M. Shimouchi, K. Asami and K. Fujimoto, Catal. Today, 2011, 164, 410–414 CrossRef CAS.
- J. Zhang, K. Wang, M. W. Nolte, Y. S. Choi, R. C. Brown and B. H. Shanks, ACS Catal., 2016, 6, 2608–2621 CrossRef CAS.
- T. J. Benson, R. Hernandez, W. T. French, E. G. Alley and W. E. Holmes, J. Mol. Catal. A: Chem., 2009, 303, 117–123 CrossRef CAS.
- R. Parvizsedghy, S. M. Sadrameli and J. Towfighi Darian, Energy Fuels, 2015, 30, 326–333 CrossRef.
- S. Vitolo, M. Seggiani, P. Frediani, G. Ambrosini and L. Politi, Fuel, 1999, 78, 1147–1159 CrossRef CAS.
- S. F. A. Halim, A. H. Kamaruddin and W. J. N. Fernando, Bioresour. Technol., 2009, 100, 710–716 CrossRef CAS PubMed.
- J. Gan and W. Yuan, Appl. Energy, 2013, 103, 350–357 CrossRef CAS.
- K. C. Kwon, H. Mayfield, T. Marolla, B. Nichols and M. Mashburn, Renewable Energy, 2011, 36, 907–915 CrossRef CAS.
- K. Hengst, M. Arend, R. Pfützenreuter and W. F. Hoelderich, Appl. Catal., B, 2015, 174–175, 383–394 CrossRef CAS.
- K. Murata, Y. Liu, M. Inaba and I. Takahara, Energy Fuels, 2010, 24, 2404–2409 CrossRef CAS.
- M. Tymchyshyn, Z. Yuan and C. Xu, Fuel, 2013, 112, 193–202 CrossRef CAS.
- L. Han, Q. Wang, Q. Ma, C. Yu, Z. Luo and K. Cen, J. Anal. Appl. Pyrolysis, 2010, 88, 199–206 CrossRef CAS.
- G. Abdulkareem-Alsultan, N. Asikin-Mijan and Y. H. Taufiq-Yap, Key Eng. Mater., 2016, 707, 175–181 CrossRef.
- M. J. A. Romero, A. Pizzi, G. Toscano, G. Busca, B. Bosio and E. Arato, Waste Manag., 2015, 47, 62–68 CrossRef PubMed.
- H. Bernas, K. Eränen, I. Simakova, A. R. Leino, K. Kordás, J. Myllyoja, P. Mäki-Arvela, T. Salmi and D. Y. Murzin, Fuel, 2010, 89, 2033–2039 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08061a |
| This journal is © The Royal Society of Chemistry 2017 |