
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A promising strategy for improved solubilization of ionic drugs simply by electrostatic pushing†
Mohd Afzal,
Pronab Kundu,
Sinjan Das,
Saptarshi Ghosh and
Nitin Chattopadhyay *
*
Department of Chemistry, Jadavpur University, Kolkata-700032, India. E-mail: nitin.chattopadhyay@yahoo.com; Fax: +91-33-2414-6584
First published on 8th September 2017
Abstract
A simple and prospective strategy has been employed to enhance the solubility of a cationic bioactive photosensitizer, namely, phenosafranin (PSF), within the anionic sodium dodecyl sulfate (SDS) micellar nanocavity using soluble salts. Electrostatic repulsion between the cationic probe trapped at the micelle–water interface and the cation of the added salt plays an important role in effective pushing of probe inside the hydrophobic micellar nanocage. Vivid steady state and time resolved spectroscopic techniques divulge a notable improvement in the fluorescence yield, fluorescence anisotropy as well as the fluorescence lifetime of the SDS-bound probe in the presence of the added salts. Comparative spectral studies using various cations imply that higher charge density on the cation imparts a greater pushing effect on the cationic drug. The present study displays an encouraging demonstration of salt induced increased solubilization of the cationic drugs in the biomimicking target region, providing a promising strategy to a more effective delivery of ionic therapeutics.
Introduction
The therapeutic efficacy of many chemotherapeutic drugs is limited by their poor penetration into the target region and by their adverse side effects on healthy cells.1 To circumvent the issue, development of targeted drug delivery systems (DDS) for improving the therapeutic efficacy of drugs has received much attention owing to the fact that the drugs are selectively delivered to the intended biological target with minimum deleterious side effects.2–5 DDS are designed to improve the efficiency of conventional drugs by altering the pharmacokinetics and biodistribution profile of drugs, reducing systemic toxicity and improving therapeutic index.6–11 To date, various nanoscale delivery systems like liposomes, micelles, carbon nanotubes, dendrimers etc. have been developed that allow more guided delivery of the drugs.2,12–14 These nanocarriers modify the drug release profile, absorption, distribution, and elimination of the drugs for the purpose of improving drug bioavailability. The fundamental aspects of the DDS are their ability to solubilize hydrophilic as well as hydrophobic agents, enhanced bio-distribution and tunable payload release to the specific target domain.12,15,16 The drug efficacy is controlled simply by the use of external stimuli like temperature, electric field, magnetic field, ionic strength, pH, redox potential etc.17–20Micelles, the supramolecular assemblies, formed by the amphiphilic surfactant monomers, are of particular interest as efficient vehicles to serve as drug delivery systems. Micelles can enhance the solubility and hence the bioavailability of a variety of drugs and release the payload preferentially at the target site in response to biological stimuli by means of enhanced permeation and retention effect.8,21–24 The extent of solubilization of the drugs into the micellar nanocavity depends on micellar size, aggregation number and the nature of the solute.25 Partitioning of the drugs between the micelles and the target site controls the sustained release of the drugs from the micellar systems. In micelles, surfactant molecules are held together by weak hydrophobic interactions, which can be modulated in desired fashion by employing an external stimulus.26 Recent studies on a variety of drugs/dyes with various surfactants enlighten our understanding of the probe–micelle binding interaction and the distribution and/or localization of the probes within the micellar environments.22,27–29
There is, however, another possibility of enhancing the drug efficacy: if the drug molecules can be solubilized more in the target site by some means. This is expected to increase the contact area of the drugs with the target system as well as enhance the concentration of the drug in the target region, resulting in an enhanced efficacy. Although, a major effort has been paid on developing and improving the drug carriers, this aspect remains rarely attempted till now. Literature contains only few reports on enhanced penetration of ionic dyes/drugs into the hydrophobic region of bio- or biomimicking targets.29,30 Considering the importance of such enhanced penetration of drugs from the viewpoint of biomedical applications, using a model drug (phenosafranin, PSF) and model target (SDS micelle) we have demonstrated a general strategy for improving the solubilization of the ionic drugs simply by the use of salts.
PSF (Scheme 1), a planar tricyclic cationic phenazinium dye, has significant utility in various photophysical and photobiological applications, including photosensitization.31,32 Phenosafranin functionalized with single-wall carbon nanotubes has been reported to be applied in photodynamic therapy (PDT).33 The polarity sensitive charge transfer emission of PSF enables us to exploit the probe successfully for characterizing various bio- and biomimetic microenvironments.31,32,34–38 In one of our recent studies, we have shown the quantitative release of PSF from an anionic micellar carrier to natural DNA by exogenous means.22
 | ||
| Scheme 1 Schematic structures of (A) phenosafranin (PSF) and (B) sodium dodecyl sulphate (SDS). | ||
In the present work, our objective has been to investigate the effect of different salts on the solubilization of a cationic probe (PSF) within anionic SDS micelle, with a special interest to explore if salts can be exploited for improved cellular internalization of the ionic drugs. To establish the strategy of electrostatic pushing, we have investigated the effect of different salts of varying charge density on the cationic PSF bound to the SDS micelle. Vivid steady state and time resolved spectroscopic techniques have been adopted for this purpose. Differential spectral responses of the SDS-bound probe in various salt environments have been monitored to evaluate the penetration ability of the probe within the micelle. The cations of the salts are prone to be located at the micelle–water interface (the SDS micellar surface being anionic in nature) and electrostatically push the cationic fluorophore inside the micellar nanocavity. The experiments further reveal that cations with higher charge densities impart stronger electrostatic repulsion on PSF to push the cationic probe more inside the SDS micelle.
Experimental section
Materials
The fluorophore, phenosafranin (PSF) was procured from Sigma-Aldrich (USA) and used without further purification. The purity of PSF was verified from its characteristic absorption and emission spectra in standard solvents. The surfactant, sodium dodecyl sulphate (SDS) was used as received from Fluka (USA). Analytical grade salts like, LiCl, NaCl, KCl, NaNO3, Mg(NO3)2 and Al(NO3)3 (Merck, India) were used without further purification. Spectroscopic grade 1,4-dioxane (Spectrochem, India) was used as received. Deionised water from a Milli-Q water purification system (Millipore) was used throughout the experiment. Unless otherwise specified, PSF concentration was maintained at ∼5 μM for the entire study. Freshly prepared micellar solutions were used to avoid aging related problems.Methods
Steady state absorption studies ware carried out using Shimadzu UV-2450 absorption spectrophotometer (Shimadzu Corporation, Kyoto, Japan) after appropriate baseline corrections. Matched quartz cells having 1 cm path length were used in the absorption studies.Steady state fluorescence studies as well as fluorescence anisotropy measurements were carried out using a Horiba Jobin Yvon Fluoromax-4 spectrofluorometer. Measurements were made in a fluorescence quartz cell of 1 cm path length. All measurements were performed keeping both the excitation and the emission slits at 3 nm. Fluorescence anisotropy (r) was determined using the following equation as39
| r = (IVV − GIVH)/(IVV + 2GIVH) | (1) |
| G = IHV/IHH | (2) |
Time resolved fluorescence decay measurements were performed from time resolved intensity decay by the method of time-correlated single photon counting (TCSPC) in a Horiba Jobin Yvon FluoroCube fluorescence lifetime system using diode laser at 490 nm (IBH NanoLED, UK) as the excitation light source with a TBX-04 photon detector. The data stored in a multichannel analyzer were analyzed using IBH DAS-6 decay analysis software. The lamp profile was collected by placing a scatterer (dilute micellar solution of SDS in water) in place of the sample. The acceptability of the fits was evaluated from χ2 criterion and visual inspection of the residuals of the fitted function to the data.
Dynamic light scattering (DLS) measurements were performed using a Nano-ZS90 Malvern instrument (model DLS-nano ZS, Zetasizer, Nano series) equipped with a 4 mW He–Ne laser (λ = 632.8 nm) and a thermostatic sample chamber. The hydrodynamic sizes of the samples were measured using DTS software. The samples were filtered through a 0.11 μm syringe filter before the measurements to remove the dust particles and aggregates, if any. For each sample 20 sets of data were accumulated for 30 s each. The viscosity and refractive index of bulk water was used in the analysis. Final size distributions were obtained by averaging all 20 sets of data for respective samples. All the measurements were carried out at 25 °C.
Results and discussion
Steady state absorption and emission studies
The steady state absorption spectrum of PSF in aqueous medium shows a low energy unstructured broad absorption band with a maximum at 520 nm.22,40 Upon increasing the SDS concentration, the absorbance of PSF slightly increases with a noteworthy red shift (∼10 nm) of the absorption maximum (figure not shown), suggesting the probe–micelle interaction.41,42Upon excited at 520 nm, room temperature emission spectrum of PSF in aqueous solution shows a single and unstructured emission band with a maximum at ∼582 nm, ascribed to the charge transfer (CT) emission.40,41 Gradual addition of the surfactant (SDS) to the aqueous solution of PSF leads to a radical modification of the emission spectrum as depicted in Fig. 1. The fluorescence intensity of PSF drops initially with a progressive hypsochromic shift of ∼11 nm (582 nm to 571 nm) at lower SDS concentrations followed by a remarkable enhancement in the emission intensity with a further blue shift of ∼5 nm (571 nm to 566 nm) as the critical micellar concentration (CMC) is approached. The observed quenching of the PSF emission at lower SDS concentration range had been explained in terms of the formation of dye–surfactant ion pairs which progressively associate to form an aggregated like structure.43,44 The prominent blue shift of the emission maximum of PSF with the addition of surfactant is ascribed to a decrease in the polarity of the microenvironment around the probe compared to the bulk aqueous phase, suggesting the trapping of the cationic probe into the anionic micellar environment. However, the remarkable increase in the emission intensity at higher SDS concentration is attributable to the increased local viscosity around the probe molecule.26 The fluorometric behavior of PSF in anionic SDS micellar atmosphere is quite similar to the observations of the previous studies of the probe in various bio- and biomimicking environments.31,32,36,41,45 Furthermore, the cationic PSF should prefer to reside at the micelle–water interfacial region since the electrostatic force operative between the anionic micelle and the cationic probe is likely to hold the probe in this region rather than allowing the ionic probe to enter the less polar micellar nanocage.
 | ||
| Fig. 1 Emission spectra of PSF in the presence of different SDS concentrations. Inset shows the variation of respective fluorescence maxima against the SDS concentrations. [PSF] = 5 μM, λexc = 520 nm. | ||
Interaction of alkali halide (M+X−) with the SDS-bound PSF leads to the remarkable modification of the fluorometric behavior of the probe. Instead of expected quenching of the fluorescence intensity of PSF by the chloride ion (Fig. 2a), successive addition of potassium chloride (KCl) to the aqueous solution of PSF in 6.64 mM SDS concentration (to be discussed later about the chosen concentration) results in a significant enhancement in the fluorescence intensity (Fig. 2b). Additionally, a further blue shift (although small, ∼1–2 nm) of the emission maximum of PSF in this composite medium (SDS + KCl) is observed relative to its position in the micellar environment in the absence of KCl (Fig. 2b), suggesting that the probe experiences a further less polar environment in the composite medium. The spectral shift and the enhancement in the emission yield of PSF in the composite medium suggest penetration of the probe inside the less polar environment of the SDS micelle. The electrostatic repulsion operative between the cationic probe (PSF) and the solvated cation (K+) of the salt at the micelle–water interface is ascribed to be responsible for this pushing effect.
 | ||
| Fig. 2 Fluorescence spectra of PSF in (a) water and (b) 6.64 mM SDS environment with increasing concentration of KCl. Concentrations of added KCl are provided in the legends. λexc = 520 nm. | ||
To find out the effective surfactant concentration for superior drug solubilization inside the micellar nanocavity, we have performed fluorometric experiment to monitor the relative variation of emission intensity of the probe in the presence of definite salt concentration (6 mM) over a broad range of surfactant concentrations.29 At each prefixed SDS concentration, our exhaustive emission studies reveal similar spectral changes, i.e., the fluorescence intensity of PSF increases upon regular addition of KCl, eventually getting saturated at a sufficient concentration of the salt (∼6 mM). Beyond this salt concentration, the solution becomes turbid and precipitation commences. For a better understanding of the effect of addition of salts on the SDS-bound PSF, we have measured the relative changes in the emission intensity (i.e., Fmax/F0) with the addition of KCl over a wide range of predetermined surfactant concentrations as depicted in Fig. 3. F0 and Fmax are the fluorescence intensities of SDS-bound PSF in the absence and in the presence of adequate concentration (6 mM) of KCl. The figure makes a typical bell-like pattern passing through a maximum, i.e., as the SDS concentration increases, Fmax/F0 value increases upto ∼6.3 mM SDS concentration and then it drops with further rise in the SDS concentration. The plot demonstrates that the impact of salt on the SDS-bound PSF is most prominent at around 6.3 mM SDS concentration, close to the CMC value of the SDS micelle in the presence of electrolyte.46 This unique and noteworthy experimental observation can be well explained in terms of our proposed model of electrostatic pushing effect.
 | ||
| Fig. 3 Plot of the maximum enhancement in the relative fluorescence intensity of micelle-bound PSF with the addition of KCl as a function of SDS concentration. | ||
At lower concentration range (i.e., premicellar region), the SDS surfactant stacks parallel most likely forming a hemi-cylindrical structure.47 At any particular time instant due to the chaotic motion of both the probe and the micelle, PSF molecules are exposed to hydrophobic interactions with the alkyl tail of SDS. The positive part of PSF remains closer to the negatively charged sulphate head group of SDS and the long alkyl chain folds back to provide a hydrophobic environment, forming a cluster of SDS around the probe molecule.48,49 Thus, the external salt (KCl) is not allowed to penetrate significantly inside the hydrophobic micellar cluster at lower SDS concentration, resulting in a less prominent effect on the emission intensity of the SDS-bound probe.
Fig. 3 reveals that the relative enhancement of emission intensity of surfactant-bound PSF with the addition of a definite KCl salt (6 mM) reaches its maximum at around the CMC of SDS micelle.46 Because of the hydrophobic environment inside the SDS micellar cavity, the cationic PSF prefers to reside at the micelle–water interface bulging the cationic part toward the aqueous phase.49 Sequential addition of KCl increases the local concentration of K+ ions at the vicinity of micelle–water interface as the K+ ions are electrostatically attracted toward the anionic surface of the SDS micellar units. Additionally, the electrostatic repulsion operates between the solvated K+ ion and cationic PSF molecule at the micelle–water interface effectively pushing the probe more within the micellar assembly. The CT emission band of PSF being sensitive to the solvent polarity (see later), large enhancement in the emission intensity associated with a little blue shift of the emission maximum is observed for the micelle-bound PSF in the presence of the added salt.
At higher SDS concentrations (>CMC), some of the micellar units remain free from the probe. Under this circumstance, increased concentration of K+ ions (from KCl) at the micelle–water interface results in a lesser amount of effective electrostatic interaction with the cationic probe (PSF) relative to the situation at CMC as a part of the K+ ions will be adhered to the micellar units devoid of the probe. Therefore, the relative emission intensity (Fmax/F0) falls down at higher SDS concentrations, finally attaining a plateau (Fig. 3).
Effect of different salts
To decipher the effect of the salts on the micelle-bound probe through the emission study, we strategically varied the cations of the added salts in two ways. First one with the series of salts lithium chloride (LiCl), sodium chloride (NaCl) and potassium chloride (KCl) all having the same valence nature (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and the second one with sodium nitrate (NaNO3), magnesium nitrate (Mg(NO3)2) and aluminium nitrate (Al(NO3)3) differing in valence characteristics (1:1, 2:1 and 3:1). The studied probe (PSF) being cationic in nature, our main emphasis was to change the cations for possible creation of a differential electrostatic repulsion.
:1) and the second one with sodium nitrate (NaNO3), magnesium nitrate (Mg(NO3)2) and aluminium nitrate (Al(NO3)3) differing in valence characteristics (1:1, 2:1 and 3:1). The studied probe (PSF) being cationic in nature, our main emphasis was to change the cations for possible creation of a differential electrostatic repulsion.
The steady state fluorescence spectra of SDS-bound PSF in the presence of 1:1 salts (LiCl, NaCl and KCl) are provided in Fig. S1 in the ESI.† The figure reveals a remarkable increase in the emission intensity with increasing salt concentration, eventually getting saturated at around 6 mM salt concentration. Beside this intensity enhancement, a slight blue shift of the emission maximum was also observed in the presence of the salts. The shift indicates that the probe experiences even less polar environment relative to that in the micellar environment in the absence of the salts. In order to scrutinize the variation of fluorescence intensity of SDS-bound PSF in the presence of these salts, we have plotted the relative changes in the fluorescence intensities (F/F0) against the salt concentration as depicted in Fig. 4a, where F and F0 are the fluorescence intensities of SDS-bound PSF in the presence and absence of the added salts respectively. Though the spectral observation is qualitatively similar in nature for all the 1:1 salts studied, the extent of enhancement in the fluorescence intensity of the SDS-bound PSF follows the order LiCl < NaCl < KCl. This relative enhancement can be explained by considering the charge density of the cations since all the salts contain same anion. It is known that the ionic radii of the alkali metal ions in gaseous phase follows the order Li+ < Na+ < K+. In aqueous solution, however, sizes of the hydrated ions follow the trend Li+ (aq.) > Na+ (aq.) > K+ (aq.) and hence the charge densities of these cations in aqueous medium follow the order Li+ (aq.) < Na+ (aq.) < K+ (aq.), all being univalent. The experimental observations (Fig. 4a), thus, reveals that the degree of penetration of the cationic probe inside the SDS micellar nanocavity increases with increasing charge density of the cation of the salt, i.e., the electrostatic pushing effect increases gradually from LiCl to KCl through NaCl.
 | ||
| Fig. 4 Variation of the relative fluorescence intensities (F/F0) of SDS-bound PSF as a function of concentrations of (a) chloride salts with same valencies of the cations and (b) nitrate salts with variable valencies of the cations. The added salts are mentioned in the legends. | ||
The effect of charge density on the electrostatic pushing effect has also been established from the fluorometric observations in the presence of added cations differing in valency state, namely Na+, Mg2+ and Al3+ (all in nitrate salts). The corresponding fluorescence spectra are given in Fig. S2 in the ESI.† The appreciable enhancement in the fluorescence intensity as well as slight blue shift of the emission maxima of SDS-bound probe was observed upon gradual addition of these salts, suggesting interaction with the SDS-bound probe. Plot of F/F0 against the salt concentrations reveal that the extent of enhancement in the emission intensity of the SDS-bound probe follows the order NaNO3 < Mg(NO3)2 < Al(NO3)3 as evident from Fig. 4b. This implies that the salt with trivalent cation (Al3+) having higher charge density results in a greater pushing effect on the cationic probe followed gradually by the salts with bivalent (Mg2+) and then monovalent (Na+) cations with lower charge densities. Thus, salts having higher charge densities help in solubilizing the probe/drug more into the micelles promising a more efficient delivery of the drug in less polar targeted bio-environment. It is pertinent to mention here that the apparent mismatch of fluorescence enhancement for NaCl and NaNO3 (although both bear same cation) is due to the difference in their concentration. A careful look at Fig. 4 reveals that at same concentration, both these salts display similar fluorescence yield suggesting comparable effect of these salts on the pushing phenomenon. We have adopted other experimental tools as well, to be described in the forthcoming sections, to further establish the extent of pushing effect.
Fluorescence anisotropy measurements
Fluorescence anisotropy measurement provides important information regarding the physical characteristics and nature of the microenvironment around a fluorophore.39–41,50,51 It reveals the extent of rigidity imposed by the microenvironment on the dynamic properties of the probe and thereby is capable of assessing the precise location of the probe in a rather complex microheterogeneous assembly.39 An increase in the rigidity around the fluorophore causes an enhancement in the fluorescence anisotropy. In fluid medium devoid of any specific interaction with the probe, the fluorophore rotates very fast within its fluorescence lifetime giving rise to very low fluorescence anisotropy. However, the anisotropy is enhanced in confined media like biomacromolecules, micelle, cyclodextrins etc. where the tumbling motion of the embedded fluorophore is restricted because of the interactions with the microheterogeneous environments.39,40 In the present study, fluorescence anisotropy demonstrates a picture of the electrostatic pushing effect together with the differential impact of varying cationic charge density. For the purpose, we have monitored the fluorescence anisotropy of PSF in 6.64 mM SDS environment in the absence and presence of different salts to judge the location of the probe from the micelle–water interface to the interior of the micelle in the presence of the salts.Fig. 5a shows the variation of fluorescence anisotropy of the probe in aqueous SDS medium as a function of increasing concentration of the aforesaid 1:1 salts and the significant data are summarised in Table 1. Very low anisotropy value (r = 0.03) of PSF in aqueous solution indicates that the probe molecule rotates almost freely in water. Upon gradual addition of SDS, the fluorescence anisotropy of PSF rises and at 6.64 mM SDS concentration it was found to be 0.054 (figure not shown), suggesting the binding interaction between the probe and the micelle. Measurements of fluorescence anisotropy reveal that with the addition of the salts on the micelle-bound probe, there is further increase in the fluorescence anisotropy (r) of PSF, eventually attaining plateaus with r ∼0.080, 0.082 and 0.087 for the addition of LiCl, NaCl and KCl respectively (Table 1). The enhancement of the anisotropy values (from the value 0.054) reveals the electrostatic pushing of the cationic probe more into the micelle by the solvated cations of the added salts leading to imposition of further motional restriction on the probe as compared to the situation in the micellar medium in the absence of the salts. However, the relative enhancement of the anisotropy was found to be LiCl < NaCl < KCl, in line with the increase in the charge densities from Li+ (aq.) to K+ (aq.) through Na+ (aq.) (already discussed).
 | ||
| Fig. 5 Variation of the fluorescence anisotropy (r) of SDS-bound PSF as a function of concentrations of (a) chloride salts with same valencies of the cations and (b) nitrate salts with variable valencies of the cations. The added salts are mentioned in the legends. | ||
| Environment | Anisotropy (r) (±0.004) |
|---|---|
| Water | 0.030 |
| SDS (6.64 mM) | 0.054 |
| SDS (6.64 mM) + LiCl | 0.080 |
| SDS (6.64 mM) + NaCl | 0.082 |
| SDS (6.64 mM) + KCl | 0.087 |
| SDS (6.64 mM) + NaNO3 | 0.076 |
| SDS (6.64 mM) + Mg(NO3)2 | 0.091 |
| SDS (6.64 mM) + Al(NO3)3 | 0.096 |
Variations of the fluorescence anisotropy of PSF in 6.64 mM SDS upon addition of increasing salt concentrations with varying cationic charge (NaNO3, Mg(NO3)2 and Al(NO3)3) are depicted in Fig. 5b and the anisotropy values are tabulated in Table 1. The experimental observations are qualitatively similar to that for 1:1 type salts and the anisotropies finally reach their saturation levels at r ∼ 0.076, 0.091 and 0.096 for Na+, Mg2+ and Al3+ salt respectively, relative to its value in 6.64 mM SDS (r = 0.054). The enhancement of the anisotropy values in the presence of the added salts is attributed to the restriction of movement of the probe within micellar environment. The study implies that the extent of increase in the fluorescence anisotropy value follows the order NaNO3 < Mg(NO3)2 < Al(NO3)3 that goes parallel to the increase in the fluorescence intensity in the presence of the respective salts. This observation substantiates the aforesaid rationale that introduction of the cations of higher charge density has a greater pushing effect than one of lower charge density.
Time resolved fluorescence decay measurements
Being sensitive to the environment and excited state interactions, fluorescence lifetime serves as an important parameter to identify precisely the residence of a probe in a microheterogeneous assembly.22,32,36,40,41,50–53 The fluorescence decay profiles of PSF with increasing concentration of SDS are provided in Fig. S3 in the ESI† and the corresponding deconvoluted lifetime data are tabulated in Table S1.† In aqueous as well as in SDS micellar environment, the single exponential decay pattern of PSF indicates the residence of the probe in a single environment.41 Addition of SDS to the aqueous solution of PSF results in a significant enhancement in the fluorescence lifetime (0.88 ns to 2.30 ns, Table S1†) suggesting a binding interaction of the probe with the micelle; substantiating the steady state fluorometric results. The increment in the fluorescence lifetime of PSF in micellar environment implies a reduction in the polarity at the local environment around the probe molecule, whereby there is a decrease in the rate of nonradiative transitions.41,54,55The fluorescence decay profiles of PSF in 6.64 mM SDS with increasing concentrations of the aforesaid 1:1 salts are depicted in Fig. 6a–c and the deconvoluted lifetime values are tabulated in Tables S2–S4.† With increasing salt concentrations to the solution of SDS-bound PSF, the fluorescence lifetime of PSF increases significantly relative to its value in 6.64 mM SDS environment (Tables S2–S4†). This enhancement in the fluorescence lifetime is assigned to a reduction in the polarity of the microenvironment around the probe because of the electrostatic pushing of PSF into the less polar micellar environment as induced by the added salts. For a better understanding of the impact of different salts, we have plotted the variation of the fluorescence lifetime (τ) of SDS-bound PSF against concentration of the added salts as shown in Fig. 7a. The extent of enhancement of fluorescence lifetime is found to be highest in case of KCl followed by NaCl and LiCl (although the differences are not so big). This is attributed to the fact that higher charge density of the solvated cation leads to a greater electrostatic pushing effect on the probe forcing it to penetrate deeper into the micellar interior as compared to the effect of the cation of lower charge density. Similar enhancements in the fluorescence lifetime (τ/τ0) and fluorescence intensity (F/F0) of PSF in 6.64 mM SDS upon gradual addition of the said salts (Tables S2–S4†) strengthen our proposition of salt-induced electrostatic pushing of the probe inside the micellar nanocavity as framed from the results of the steady state emission and anisotropy measurements.
 | ||
| Fig. 6 Time resolved fluorescence decay profiles of PSF in 6.64 mM SDS in the presence of different concentrations of (a) LiCl (b) NaCl, (c) KCl, (d) NaNO3, (e) Mg(NO3)2 and (f) Al(NO3)3. Legends give concentrations of the added salts. The sharp black profiles on the left represent the instrument response function (IRF). λexc = 490 nm and λem = λmaxem. | ||
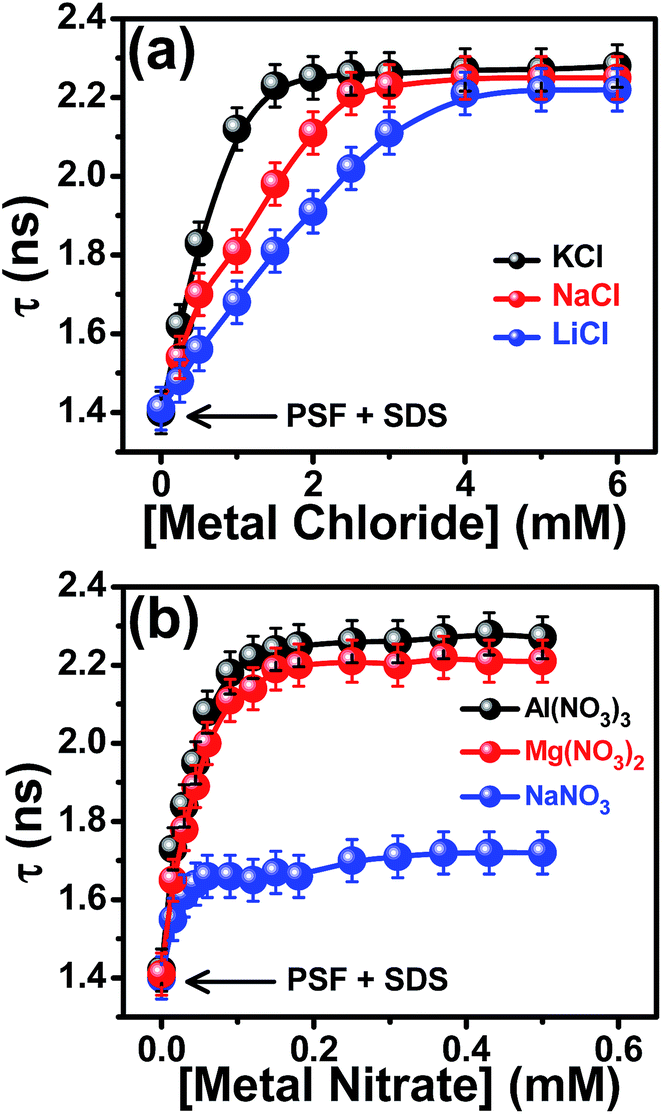 | ||
| Fig. 7 Variation of the fluorescence lifetime (τ) of SDS-bound PSF as a function of concentrations of (a) chloride salts with same valencies of the cations and (b) nitrate salts with variable valencies of the cations. The added salts are mentioned in the legends. [SDS] = 6.64 mM. For details see text. | ||
The fluorescence decay measurements of the probe in 6.64 mM SDS are also carried out in the presence of cations of varying valency (Fig. 6d–f) and the analyzed lifetime data are collected in Tables S5–S7.† It is evident from the figure that with increasing concentration of the salts, the lifetime of PSF increases and gets saturated at a sufficiently high salt concentration. The increase in the fluorescence lifetime is ascribed to be associated with a decrease in the polarity of the microenvironment of the probe, as already discussed. Fig. 7b demonstrates that the extent of increase in the lifetime of SDS-bound probe is largest in case of Al(NO3)3 as compared to those observed with Mg(NO3)2 and NaNO3 at the same concentration. This implies that the cation of higher valency (Al3+) helps in solubilizing the cationic probe more into the micellar environment as compared to bivalent (Mg2+) and monovalent cations (Na+), consistent with the steady state emission and anisotropy experiments. The outcomes of all these experiments confirm that the cation of higher charge density imparts a greater electrostatic pushing effect on the cationic probe. The similarity in the relative enhancements of fluorescence lifetime (τ/τ0) and the emission intensity (F/F0) in the presence of the above mentioned salts run parallel (Tables S5–S7†) and suggests that the increment is due to the reduction in polarity at the local environment around the probe.
We have also performed the steady state and time resolved fluorometric studies of PSF in water–dioxane mixtures of varying polarity to get an idea about the reduction in the polarity of the microenvironment around the probe in the micellar medium with the addition of the salts and hence, to justify the electrostatic pushing effect. Fig. S4 in the ESI† depicts an increase in both fluorescence intensity and fluorescence lifetime of PSF with an increase in the dioxane proportion in the solvent mixture. The fluorometric observations, thus, infer that with a reduction in the polarity of the environment, fluorescence intensity as well as lifetime of the probe increases. Similar enhancements of these two fluorometric parameters of the micelle-bound probe (SDS–PSF) with the addition of the salts are ascribed to result from a reduction in the micropolarity around the fluorophore which refers to a penetration of the probe within SDS micelles with the addition of the salts; substantiating the salt-induced electrostatic pushing of the cationic probe by the cations of the added salts.
To have a qualitative idea about the micropolarities around the micelle-bound probe in the absence and in the presence of added KCl, the fluorometric response of the probe in these two situations have been compared to those in a series of dioxane–water solvent mixtures of known polarity equivalent ET(30)29,50,55,56 values. Fig. 8 presents the calibration plot of relative fluorescence intensity of PSF in various dioxane–water mixtures with the interpolation of the same variant in 6.64 mM SDS micellar solution in the absence and in the presence of 6 mM KCl. It is evident from the figure that the added salt leads to a reduction in the micropolarity (ET(30)) around the cationic fluorophore (PSF) from 55.6 kcal mol−1 to 51.4 kcal mol−1; supporting that the added cation of the salt exerts an electrostatic pushing effect resulting in a greater penetration and hence solubilization of the cationic probe.
 | ||
| Fig. 8 Plot of variation of the relative fluorescence enhancement of PSF as a function of ET(30) in dioxane–water solvent mixtures. F0 represents the fluorescence intensity in pure water. The magenta and the wine points are the interpolated relative fluorescence intensities in 6.64 mM SDS solution in the absence and in the presence of 6 mM KCl salt respectively. | ||
Dynamic light scattering (DLS) study
To see whether or not there is a change in the size of the micelle in the presence of the added salts, we have measured the hydrodynamic size of the micelle in different situations through DLS study. The hydrodynamic diameter of SDS micellar units are obtained from the monomodal distribution and the value is found to be 4.3 ± 0.2 nm (Fig. 9) that agrees well with the literature report.57 DLS measurements reveal that the presence of the probe does not affect the size of the micelle. That the addition of the individual salts (NaCl, NaNO3 or Al(NO3)3) to the SDS-bound probe do not bring any significant change (within error limit of ±5%) in the size of the micelle is evident from Fig. 9. Therefore, the fluorometric enhancement of the probe in SDS micelle in the presence of added salts is not due to the salt-induced perturbation of the micellar size, but because of the electrostatic pushing effect.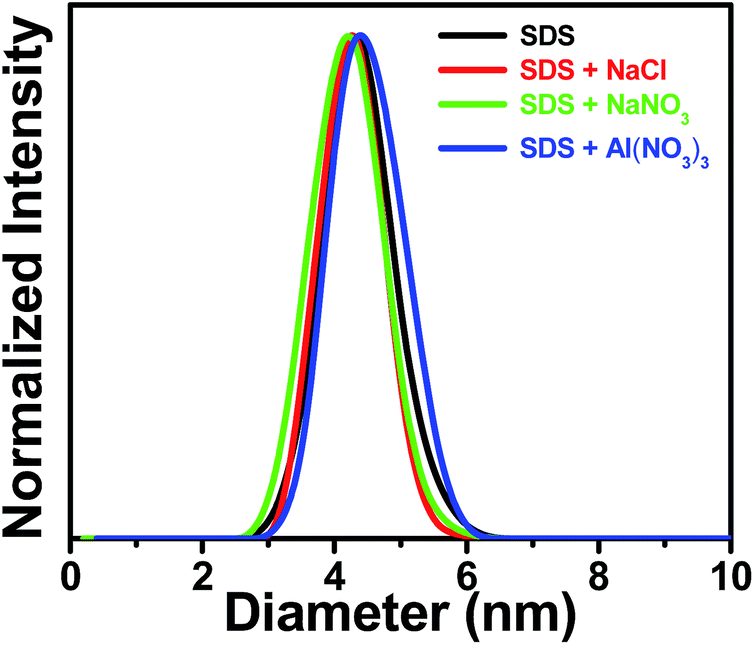 | ||
| Fig. 9 Normalized size distribution of SDS micelle in water and in the presence of different salts. Added salts are indicated in the legends. Concentrations of the salts are 6 mM for NaCl and 0.5 mM for both NaNO3 and Al(NO3)3. | ||
Conclusion
The present work articulates the electrostatic pushing of a bioactive photosensitizer (PSF) more inside the anionic micelle (SDS), by the use of different salts. Vivid steady state fluorescence studies, anisotropy measurements and time resolved fluorescence decay experiments unambiguously reveal that the added cations induce pushing of the probe electrostatically (both being positively charged) within the micellar cavity from its initial location at the micelle–water interfacial region. The bunch of experiments with different salts concludes that the extent of penetration increases with an increase in the effective charge density of the cation. The simple strategy projects enhanced solubilization of the ionic drugs in less polar regions of the bio-environments by the application of permissible salts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support received from Department of Science and Technology, Govt. of India (Project no. EMR/2016/001087) is gratefully acknowledged. M. A. thanks U.G.C. for Dr D. S. Kothari Postdoctoral Fellowship. P. K. thanks C.S.I.R. and S. D. and S. G. thank U.G.C. for their research fellowships.References
- A. I. Minchinton and I. F. Tannock, Nat. Rev. Cancer, 2006, 6, 583–592 CrossRef CAS PubMed.
- V. V. Ranade, M. A. Hollinger and J. B. Cannon, Drug Delivery Systems, CRC Press, Boca Raton, 2nd edn, 2004 Search PubMed.
- V. Sanna, N. Pala and M. Sechi, Int. J. Nanomed., 2014, 9, 467–483 CAS.
- S. Y. Qin, A. Q. Zhang, S. X. Cheng, L. Rong and X. Z. Zhang, Biomaterials, 2017, 112, 234–247 CrossRef CAS PubMed.
- Y. H. Bae and K. Park, J. Controlled Release, 2011, 153, 198–205 CrossRef CAS PubMed.
- A. Bochot and E. Fattal, J. Controlled Release, 2012, 161, 628–634 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS PubMed.
- K. Park, Controlled Drug Delivery, Challenges and Strategies, American Chemical Society, Washington, DC, 1997 Search PubMed.
- R. Langer, Nature, 1998, 392, 5–10 CAS.
- E. Soussan, S. Cassel, M. Blanzat and I. Rico-Lattes, Angew. Chem., Int. Ed., 2009, 48, 274–288 CrossRef CAS PubMed.
- V. P. Torchilin, Pharm. Res., 2007, 24, 1–16 CAS.
- C. K. Kim, P. Ghosh, C. Pagliuca, Z.-J. Zhu, S. Menichetti and V. M. Rotello, J. Am. Chem. Soc., 2009, 131, 1360–1361 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- A. Bianco, K. Kostarelos and M. Prato, Curr. Opin. Chem. Biol., 2005, 9, 674–679 CrossRef CAS PubMed.
- V. P. Torchilin, Adv. Drug Delivery Rev., 2006, 58, 1532–1555 CrossRef CAS PubMed.
- R. Duncan and R. Gaspar, Mol. Pharm., 2011, 8, 2101–2141 CrossRef CAS PubMed.
- H. Suzuki and Y. H. Bae, Biomaterials, 2016, 98, 120–130 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- G. D. Sagar, M. A. Arunagirinathan and J. R. Bellare, Indian J. Exp. Biol., 2007, 45, 133–159 CAS.
- J. Zhou, M. V. Pishko and J. L. Lutkenhaus, Langmuir, 2014, 30, 5903–5910 CrossRef CAS PubMed.
- H. Maeda, Bioconjugate Chem., 2010, 21, 797–802 CrossRef CAS PubMed.
- P. Kundu, S. Ghosh, S. Das and N. Chattopadhyay, Phys. Chem. Chem. Phys., 2016, 18, 3685–3693 RSC.
- E. Soussan, S. Cassel, M. Blanzat and I. Rico-Lattes, Angew. Chem., Int. Ed., 2009, 48, 274–288 CrossRef CAS PubMed.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS PubMed.
- M. Malmsten, Surfactants and Polymers in Drug Delivery, Marcel Dekker, New York, NY, 2002 Search PubMed.
- P. K. Singh and S. Nath, J. Phys. Chem. B, 2013, 117, 10370–10375 CrossRef CAS PubMed.
- M. Afzal, S. Ghosh, S. Das and N. Chattopadhyay, J. Phys. Chem. B, 2016, 120, 11492–11501 CrossRef CAS PubMed.
- M. Afzal, S. Ghosh and N. Chattopadhyay, Biophys. Chem., 2016, 219, 75–81 CrossRef CAS PubMed.
- D. Sarkar, D. Ghosh, P. Das and N. Chattopadhyay, J. Phys. Chem. B, 2010, 114, 12541–12548 CrossRef CAS PubMed.
- S. Ghosh, P. Kundu, B. K. Paul and N. Chattopadhyay, RSC Adv., 2014, 4, 63549–63558 RSC.
- D. Sarkar, P. Das, S. Basak and N. Chattopadhyay, J. Phys. Chem. B, 2008, 112, 9243–9249 CrossRef CAS PubMed.
- D. Bose, D. Ghosh, P. Das, A. Girigoswami, D. Sarkar and N. Chattopadhyay, Chem. Phys. Lipids, 2010, 163, 94–101 CrossRef CAS PubMed.
- G. M. do Nascimento, R. C. de Oliveira, N. A. Pradie, P. R. G. Lins, P. R. Worfel, G. R. Martinez, P. D. Mascio, M. S. Dresselhaus and P. Corio, J. Photochem. Photobiol., A, 2010, 211, 99–107 CrossRef CAS.
- M. F. Broglia, M. L. Gomez, S. G. Bertolotti, H. A. Montejano and C. M. Previtali, J. Photochem. Photobiol., A, 2005, 173, 115–120 CrossRef CAS.
- S. Jockush, H. J. Timpe, W. Schnabel and N. J. Turro, J. Phys. Chem. A, 1997, 101, 440–445 CrossRef.
- S. Ghosh, P. Kundu and N. Chattopadhyay, J. Photochem. Photobiol., B, 2016, 154, 118–125 CrossRef CAS PubMed.
- K. R. Gopidas and P. V. Kamat, J. Phys. Chem., 1990, 94, 4723–4727 CrossRef CAS.
- D. Bose, D. Sarkar and N. Chattopadhyay, Photochem. Photobiol., 2010, 86, 538–544 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- R. Chaudhury, J. Guharay and P. K. Sengupta, J. Photochem. Photobiol., A, 1996, 101, 241–244 CrossRef.
- P. Das, A. Chakrabarty, A. Mallick and N. Chattopadhyay, J. Phys. Chem. B, 2007, 111, 11169–11176 CrossRef CAS PubMed.
- R. K. Dutta and S. N. Bhat, Colloids Surf., A, 1996, 106, 127–134 CrossRef CAS.
- P. Pal, H. Zeng, G. Durocher, D. Girard, R. Giasson, L. Blanchard, L. Gaboury and L. Villeneuve, J. Photochem. Photobiol., A, 1996, 98, 65–72 CrossRef CAS.
- M. Sarkar and S. Poddar, J. Colloid Interface Sci., 2000, 221, 181–185 CrossRef CAS PubMed.
- A. Mahata, D. Bose, D. Ghosh, B. Jana, B. Bhattacharya, D. Sarkar and N. Chattopadhyay, J. Colloid Interface Sci., 2010, 347, 252–259 CrossRef CAS PubMed.
- E. Dutkiewicz and A. Jakubowska, Colloid Polym. Sci., 2002, 280, 1009–1014 CAS.
- V. K. Paruchuri, J. Nalaskowski, D. O. Shah and J. D. Miller, Colloids Surf., A, 2006, 272, 157–163 CrossRef CAS.
- S. Niu, K. R. Gopidas, N. J. Turro and G. Gabor, Langmuir, 1992, 8, 1271–1277 CrossRef CAS.
- B. J. Kim, S. G. Oh, M. G. Han and S. S. Im, Synth. Met., 2001, 122, 297–304 CrossRef CAS.
- P. Kundu, S. Ghosh and N. Chattopadhyay, Phys. Chem. Chem. Phys., 2015, 17, 17699–17709 RSC.
- A. Mahata, D. Sarkar, D. Bose, D. Ghosh, A. Girigoswami, P. Das and N. Chattopadhyay, J. Phys. Chem. B, 2009, 113, 7517–7526 CrossRef CAS PubMed.
- J. Oshima, S. Shiobara, H. Naoumi, S. Kaneko, T. Yoshihara, A. K. Mishra and S. Tobita, J. Phys. Chem. A, 2006, 110, 4629–4637 CrossRef CAS PubMed.
- P. Das, A. Chakrabarty, B. Haldar, A. Mallick and N. Chattopadhyay, J. Phys. Chem. B, 2007, 111, 7401–7408 CrossRef CAS PubMed.
- E. M. Kosower, H. Dodiuk, K. Tanizawa, M. Ottolenghi and N. Orbach, J. Am. Chem. Soc., 1975, 97, 2167–2178 CrossRef CAS.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- C. Reichardt, H. Ratajazak and W. J. Orville-Thomus, Molecular Interactions, Wiley, New York, 1982, vol. 3, p. 255 Search PubMed.
- N. C. Maiti, M. M. G. Krishna, P. J. Britto and N. Periasamy, J. Phys. Chem. B, 1997, 101, 11051–11060 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Emission spectra of PSF bound SDS in the presence of different salts, time resolved fluorescence decay profiles of PSF with the addition of SDS, emission and decay profiles of PSF in varying composition of dioxane–water mixtures and tables collecting the fluorescence intensity and deconvoluted fluorescence lifetime data of SDS-bound PSF in the absence and presence of different salts. See DOI: 10.1039/c7ra08056e |
| This journal is © The Royal Society of Chemistry 2017 |