
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Versicotides D–F, new cyclopeptides with lipid-lowering activities†
Ran Chen‡
ab,
Zhongbin Cheng‡a,
Jian Huang a,
Dong Liua,
Chongming Wub,
Peng Guo*b and
Wenhan Lin*a
a,
Dong Liua,
Chongming Wub,
Peng Guo*b and
Wenhan Lin*a
aState Key Laboratory of Natural and Biomimetic Drugs, Peking University, Beijing 100191, China. E-mail: whlin@bjmu.edu.cn; Tel: +86-10-8280-6188
bPharmacology and Toxicology Research Center, Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences, Beijing 100193, PR China. E-mail: pguo@imaplad.ac.cn; Tel: +86-10-57833235
First published on 23rd October 2017
Abstract
Lipid-lowering assay guided fractionation of a gorgonian-derived fungus Aspergillus versicolor LZD-14-1 resulted in the isolation of three new cyclopeptides namely versicotides D–F (1–3) and the known analogues versicotides A and B. The new structures were determined by extensive spectroscopic data (1D and 2D NMR, HRESIMS), while the data of the X-ray crystallography and Marfey's method were used for the configurational assignment. Versicotides E–F (2–3) are characterized by the presence of two anthranilic acid residues and a proline that coexisted in the cyclic pentapeptides, which are rarely found from nature. Versicotides D–F (1–3) exerted lipid-lowering effects through the regulation of cholesterol efflux to HDL in RAW264.7 macrophages. The mechanistic study revealed that the cholesterol efflux and influx induced by the peptides are directly related to the promotion of the target genes ABCG1 and LXRα, in addition to the decreasing critical scavenger receptors CD36 and SR-1/SR-2. The present work provided a group of new cyclopeptides which are promising for the development of anti-atherosclerosis leads.
1. Introduction
Atherosclerosis, a multifactorial disease, is one of the major causes of disability of blood vessels induced by the formation and accumulation of lipid plaques in the arteries and by inflammatory responses, which result in insufficient blood supply to organs and tissues and the development of many cardiovascular disorders.1 A main pathological manifestation of atherosclerosis is attributed to the accumulation of excessive cholesterol to form plaques in arterial walls. Reverse cholesterol transport (RCT) can eliminate excessive plasma cholesterol from peripheral tissues and transfer it to the liver for excretion. The current understanding of cellular and molecular mechanisms of atherogenesis is the accumulation of extracellular and intracellular lipids in the arterial intima is caused by low-density lipoprotein (LDL),2,3 thus the enhancement of cholesterol efflux might lead to increased RCT and finally to prevent atherosclerosis development. Oxidized low-density lipoprotein (LDL) induced the foam cell formation, which plays a key role in the step of the pathological process of atherosclerosis.4 Thus, foam cell formation is a main determinant of atherosclerotic lesions, in which macrophages express scavenger receptors on their plasma membranes and uptake oxidized LDL.5 In atherosclerotic lesions, macrophages uptake the oxidized LDL which deposited in the blood vessel walls and developed into foam cells. They secrete various inflammatory cytokines and accelerate the development of atherosclerosis. Scavenger receptors CD36, SR-A1 and SR-A2 bind to and uptake excess oxLDL into macrophages, leading to the accumulation of excess cholesterol and finally inducing toxic cells.6 Besides, ATP-binding cassette transporters (ABCA1 and ABCG1) induce the reverse cholesterol transport (RCT) pathway by the mediation of the translocation of cholesterol across cellular bilayer membranes.7–9 ABCA1 promotes the cholesterol efflux to lipid-poor apolipoproteins, while ABCG1 mediates cholesterol efflux to high density lipoprotein (HDL).10,11 Expression of ABCA1 and ABCG1 is regulated by proliferator-activated receptor gamma (PPARg)-dependent and liver X receptor alpha (LXRa)-dependent pathways, respectively.12 Thus, compounds which are capable of increasing ABCA1 expression and accelerating the reversal of cholesterol transport may lead to a therapeutic benefit for reducing atherosclerosis.Natural products are considered as the continuous source of therapeutic agents and the important pool for the discovery of new anti-atherosclerotic leads.13–15 Statins, the marketed lipid-lowering drugs originated from the fungal Penicillium and Aspergillus species, were the only drugs used for prevention and treatment of atherosclerosis. However, the numerous adverse effects of statin treatment limited the clinic therapy.16 Therefore, discovery of new leads with potent anti-atherosclerotic effects and with no adverse effects from natural products is urgent. With the aim of ongoing discovery of new bioactive natural products with lipid-lowering effects from marine-derived microorganisms, a cell model-based lipid-lowering bioassay was conducted. The results demonstrated that a gorgonian-derived fungus Aspergillus versicolor LZD-14-1 exerted the inhibitory effect to reduce lipid accumulation in RAW264.7 macrophages. Chromatographic separation of the lipid-lowering active ethyl acetate (EtOAc) fraction from the fungus led to the isolation and characterization of five cyclic peptides (1–5), of which 1–3 are the new cyclopeptides (Fig. 1). In this paper, we report the structure elucidation of the new compounds, in addition to the lipid-lowering effects of the analogues and the regulation of target genes.
 | ||
| Fig. 1 Structures of cyclopeptides. | ||
2. Experimental section
2.1. General procedure
Melting points were recorded on an X-5 micromelting point apparatus (Kexian Co.). Specific rotations were measured by an Autopol III automatic polarimeter (Rudolph Research Co., Ltd.). UV spectra were measured on a Cary 300 spectrometer. IR spectra were measured on a Thermo Nicolet Nexus 470 FT-IR spectrometer. The 1H and 13C NMR spectra were recorded on a Bruker Avance-400FT NMR spectrometer. HRESIMS spectra were obtained on a Waters Xevo G2 Q-TOF spectrometer fitted with an ESI source. Sephadex LH-20 (18–110 μm) was provided by Pharmacia. HPLC was performed on Alltech 426 pump employing an UV detector, and the Prevail C18 column (semipreparative, 5 μm) was used for semi-preparative HPLC separation. 25-[N-[(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-methyl]amino]-27-norcholesterol (25-NBD cholesterol), lovastatin, rosiglitazone, oil red O and Dulbecco's modified Eagle's medium (DMEM) were purchased from Sigma-Aldrich, Inc. (St Louis, MO, USA). The kit for intracellular cholesterol was purchased from Jian Cheng Biotechnology Company (Nanjing, China). Human ox-LDL, ApoA1 and HDL were obtained from Yiyuan Biotechnologies (Guangzhou, China). A total RNA extraction reagent RNAiso Plus, a PrimeScript RT reagent kit, and a SYBR-Green PCR kit were purchased from Transgene Biotech, Inc. (Beijing, China). RAW264.7 macrophages originated from the American Type Culture Collection (ATCC: TIB-71™) (Manassas, VA, USA) and obtained from the Peking Union Medical College.2.2. Fungal strain and identification
Fungus Aspergillus versicolor LZD-14-1 was isolated from the gorgonian Pseudopterogorgia sp. (LZD-14), which was collected from the South China Sea in May 2015. The strain was identified by comparing the morphological characteristics and analysis of the ITS region of the rDNA sequence with those of standard records (GeneBank KX254916). The morphological examination was performed by scrutinizing the fungal culture, the mechanism of spore production, and the characteristics of the spores. For inducing sporulation, the fungal strains were separately inoculated onto potato dextrose agar. All experiments and observations were repeated at least twice, leading to the identification of the strain LZD-14-1 as A. versicolor. The strain LZD-14-1 was deposited at the State Key Laboratory of Natural and Biomimetic Drugs, Peking University, China.2.3. Fermentation
The fermentation was carried out in 50 Fernbach flasks (500 mL), each containing 80 g of rice. Distilled H2O (100 mL) was added to each flask, and the contents were soaked overnight before autoclaving at 15 psi for 30 min. After cooling to room temperature, each flask was inoculated with 5.0 mL of the spore inoculum and incubated at 25 °C for 40 days.2.4. Isolation and purification
The fermented material was extracted successively with EtOAc (3 × 500 mL). After evaporation under vacuum, the EtOAc extract (4.0 g) was subjected to a vacuum liquid chromatography (silica gel, 200–300 mesh) with petroleum ether/EtOAc (from 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 0:1, gradient) as an eluent to obtain four fractions (F1 to F4). A bioassay with oil red O staining after elicitation by oxLDL was performed, the bioassay result indicated that fraction F3 (100 μg mL−1) exerted the lipid-lowering effect in oxLDL (50 mg mL−1) induced RAW264.7 macrophages, whereas fractions F1, F2, and F4 showed weak activity in the same dose. Fraction F3 (2.0 g) was chromatographed on a Sephadex LH-20 column eluting with MeOH to collect five subfractions (SF31–SF35). Additional bioassay revealed that SF31 possesses the lipid-lowering effect in RAW264.7 macrophages. SF31 (130 mg) was then subjected to semi-preparative HPLC (ODS) separation using MeCN–H2O (35:65) as a mobile phase to obtain compounds 1 (Rt = 23.7 min, 16 mg), 2 (Rt = 25.2 min, 8.0 mg), 3 (Rt = 25.6 min, 14 mg), and 4/5 (Rt = 27.0 min, 22 mg). After collection of the HPLC pure compounds and dryness in vacuo, compounds 1 and 2 were dissolved in MeOH for crystallization. After one week in 4 °C, the crystals of 1 and 2 were yielded respectively, while the crystals of 4 were obtained from the CH2Cl2/MeOH (1:1) solution of 4/5 mixture.
:1 to 0:1, gradient) as an eluent to obtain four fractions (F1 to F4). A bioassay with oil red O staining after elicitation by oxLDL was performed, the bioassay result indicated that fraction F3 (100 μg mL−1) exerted the lipid-lowering effect in oxLDL (50 mg mL−1) induced RAW264.7 macrophages, whereas fractions F1, F2, and F4 showed weak activity in the same dose. Fraction F3 (2.0 g) was chromatographed on a Sephadex LH-20 column eluting with MeOH to collect five subfractions (SF31–SF35). Additional bioassay revealed that SF31 possesses the lipid-lowering effect in RAW264.7 macrophages. SF31 (130 mg) was then subjected to semi-preparative HPLC (ODS) separation using MeCN–H2O (35:65) as a mobile phase to obtain compounds 1 (Rt = 23.7 min, 16 mg), 2 (Rt = 25.2 min, 8.0 mg), 3 (Rt = 25.6 min, 14 mg), and 4/5 (Rt = 27.0 min, 22 mg). After collection of the HPLC pure compounds and dryness in vacuo, compounds 1 and 2 were dissolved in MeOH for crystallization. After one week in 4 °C, the crystals of 1 and 2 were yielded respectively, while the crystals of 4 were obtained from the CH2Cl2/MeOH (1:1) solution of 4/5 mixture.
| Ala | 1 | No. | 2 | Ala | 3 | |||
|---|---|---|---|---|---|---|---|---|
| δH | δC | δH | δCb | δH | δC | |||
| Ala | ||||||||
| a 1H NMR (400 MHz), 13C NMR (100 MHz), J in Hz, δ in ppm. | ||||||||
| 1 | 171.0, C | 1 | 170.5, C | 1 | 167.8, C | |||
| 2 | 4.16, dq (8.0, 6.9) | 47.2, CH | 2 | 4.20, dq (6.6, 6.4) | 48.0, CH | 2 | 4.90, q (6.6) | 59.1, CH |
| 3 | 0.78, d (6.9) | 17.0, CH3 | 3 | 1.36, d (6.6) | 16.9, CH3 | 3 | 1.57, d (6.6) | 14.4, CH |
| NH | 7.59, d (8.0) | NH | 9.42, d (6.4) | |||||
| Phe | ATA | ATA | ||||||
| 4 | 169.7, C | 4 | 168.1, C | 4 | 168.5, C | |||
| 5 | 5.08, t (7.0) | 53.6, CH | 5 | 117.7, C | 5 | 120.1, C | ||
| 6 | 2.97, dd (13.9, 7.0) | 33.8, CH2 | 6 | 7.72, d (8.0) | 128.4, CH | 6 | 7.45, d (7.6) | 127.7, CH |
| 2.65, dd (13.9, 7.0) | ||||||||
| 7 | 138.0, C | 7 | 7.11, dd (8.0, 7.8) | 122.6, CH | 7 | 7.18, dd (7.6, 7.5) | 122.6, CH | |
| 8 | 7.17, m | 129.4, CH | 8 | 7.39, dd (8.4, 7.8) | 132.8, CH | 8 | 7.50, dd (8.4, 7.5) | 131.3, CH |
| 9 | 7.17, m | 127.9, CH | 9 | 8.30, d (8.4) | 118.7, C | 9 | 8.70, d (8.4) | 120.2, CH |
| 10 | 7.13, t (7.9) | 126.0, CH | 10 | 132.8, C | 10 | 131.3, C | ||
| 11 | 7.17, m | 127.9, CH | NH | 12.0, s | NH | 10.1, s | ||
| 12 | 7.17, m | 129.4, CH | Pro | Pro | ||||
| Gly | 11 | 169.6, C | 11 | 169.8, C | ||||
| 13 | 168.3, C | 12 | 4.78, dd (8.1, 4.5) | 63.7, CH | 12 | 4.55, d (8.3) | 61.7, CH | |
| 14 | 3.87, dd (17.6, 8.6) | 42.4, CH2 | 13 | 2.43, m; 2.03, m | 32.2, CH2 | 13 | 2.19, m; 2.30, m | 32.1, CH2 |
| 3.50, dd (17.6, 6.3) | ||||||||
| NH | 7.82, dd (8.6, 6.3) | 14 | 1.98, m; 1.89, m | 22.3, CH2 | 14 | 1.93, m; 1.84, m | 21.8, CH2 | |
| Phe | 15 | 3.86, m; 3.70, m | 47.5, CH2 | 15 | 3.82, m | 46.4 | ||
| 3.45, td (10.7, 7.7) | ||||||||
| 15 | 171.3, C | ATA | Ala | |||||
| 16 | 5.41, dd (9.9, 5.4) | 53.5, CH | 16 | 166.5, C | 16 | 171.7, C | ||
| 17 | 2.88, dd (14.1, 5.4) | 33.6, CH2 | 17 | 123.6, C | 17 | 4.15, dq (7.0, 4.5) | 49.1, CH | |
| 2.79, dd (14.1, 9.9) | ||||||||
| 18 | 137.5, C | 18 | 7.61, d (7.6) | 126.7, CH | 18 | 1.38, d (7.0) | 15.2, CH3 | |
| 19 | 7.17, m | 129.5, CH | 19 | 7.06, dd (7.8, 7.6) | 122.8, CH | NH | 9.17, d (4.5) | |
| 20 | 7.17, m | 127.7, CH | 20 | 7.28, dd (8.3, 7.8) | 130.4, CH | ATA | ||
| 21 | 7.13, t (7.9) | 126.0, CH | 21 | 8.24, d (8.3) | 119.5, CH | 19 | 168.8, C | |
| 22 | 7.17, m | 127.7, CH | 22 | 135.9, C | 20 | 116.0, C | ||
| 23 | 7.17, m | 129.5, CH | NH | 9.20, s | 21 | 8.02, d (7.9) | 128.8, C | |
| Ala | 22 | 7.14, dd (7.9, 7.6) | 122.3, CH | |||||
| 23 | 169.6, C | 23 | 7.52, dd (8.4, 7.6) | 131.3, CH | ||||
| 24 | 4.84, q (6.9) | 56.6, CH | 24 | 8.56, d (8.4) | 118.8, CH | |||
| 25 | 1.40, d (6.9) | 15.2, CH3 | 25 | 140.2, C | ||||
| NH | 12.68, s | |||||||
| N–Me | 2.61, s | 28.5, CH3 | N–Me | 2.90, s | 30.0, CH3 | N–Me | 2.74, s | 29.7, CH3 |
| N–Me | 2.69, s | 29.7, CH3 | ||||||
2.5. X-ray crystallographic analysis
Crystal data were obtained on a Rigaku MicroMax 002+ single-crystal X-ray diffractometer. Cell parameter measurements and data collection were performed with a Bruker APEX2 CCD diffractometer using the wavelength for CuKα (λ = 1.5418 Å) radiation. Compounds were crystallized in polar space groups. The crystal structures were solved by direct methods (SHELXS-97), and subsequent Fourier difference techniques (SHELEX-97, version 6.10, Bruker AXS Inc.). The Crystallographic data for the structures have been deposited in Cambridge Crystallographic Data Center (CCDC numbers: 1491883, 1491882, and 1491884†).2.6. Marfey's method
Cyclopeptide (0.5 mg) was placed in a 5 mL conical vial containing HCl (6 M, 1 mL), and the sealed vials were heated at 110 °C for 20 h. After evaporation of the solvent, H2O (100 μL) was added. Then NaHCO3 (1 M, 50 μL) and 1-fluoro-2,4-dinitrophenyl-5-L-alanine amide (L-FDAA, 1%, 100 μL) in acetone were added to the hydrolysis solution, and the sealed vial was heated at 40 °C for 2 h. Then HCl (2 M, 20 μL) was added to the reaction mixture to stop the reaction. After evaporation, the reacted products were dissolved in MeOH for HPLC analysis in Grace Allsphere ODS column (250 mm × 4.6 mm, 25 μm) and Thermo BDS Hypersil C18 column (150 mm × 4.6 mm, 5 μm) using MeOH–H2O (H3PO4) as a mobile phase (gradient: 0 min: 30% MeOH–H2O; 40 min: 70% MeOH–H2O) with a flow rate of 1 mL min−1. UV detection was performed at a wavelength of 340 nm.2.7. Lipid-lowering assay and cell-based total cholesterol (TC) accumulation assay
RAW264.7 macrophages were cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS, Gibco) at 37 °C and 5% CO2 for 12 h. Subsequently, fractions F1–F5 (each 100 μg mL−1), respective compound in the doses of 1, 10, and 50 μM, and positive control simvastatin (10 μM) were added in DMEM containing oxLDL (50 μg mL−1) to incubate for 12 h. The blank control was attributed to FBS-free DMEM solo. Cells were then fixed with 4% w/v paraformaldehyde (30 min, rt) and stained with filtered oil red O solution (60 min, room temperature). The oil red O staining was evaluated by both microscopic examination (Olympus, Tokyo, Japan) and spectrophotometry at 358 nm. The concentrations of the total cholesterol were detected by special kits according to the operating directions. Subsequently, the cells were subjected to TC determination by special kits according to the operating directions, while the doses of each compound was selected as 10 and 50 μM. Each experiment (n = 8) was repetitive with three times.2.8. Cholesterol influx assay
Cholesterol influx assays were carried out with 25-NBD cholesterol in RAW264.7 macrophages. The cells were plated equably in 96 well clear-bottom black plates at 5 × 105 cells per well. In the next 4 hours, the medium was removed and the cells were labeled with 25-NBD-cholesterol (5 μg mL−1) in serum-free DMEM containing 10 μM of respective compounds or an equal volume of serum-free DMEM. Then the cells were washed three times with phosphate buffered saline (PBS), and cholesterol contents in the cells were lysated by 0.5% Triton-X100. The lysis solution was measured using a Tecan Infinite M1000Pro Microplate Reader (TECAN Group Ltd. Shanghai, China; excitation 485 nm, emission 535 nm). Rosiglitazone is used as a positive control. Each uptake assay (n = 8) was tested repetitively in three experiments.2.9. Cholesterol efflux assay
RAW264.7 macrophages were equilibrated with 25-NBD cholesterol (5 μg mL−1) for 24 h. NBD-cholesterol-labeled cells were washed with PBS three times and incubated in serum-free DMEM containing 50 μg mL−1 HDL and 10 μM respective compounds for 6 h. The fluorescence-labeled cholesterol that released from the cells into the medium was measured with a Tecan Infinite M1000Pro Microplate Reader (TECAN Group Ltd. Shanghai, China). Cholesterol efflux was expressed as a percentage of fluorescence in the medium relative to the total amount of fluorescence detected in the cells and the medium. Rosiglitazone is used as a positive control. Each efflux assay (n = 8) was repetitive with three experiments.2.10. Quantitative real-time PCR
Total RNA extraction, cDNA synthesis, and quantitative PCR assays were performed as described previously.17 Total RNA was extracted from RAW264.7 macrophages using the EasyPure RNA kit (ER101; TransGen Biotech) on the basis of the manufacturer's instructions. cDNA was synthesized using TransScript All-in-One First-Strand synthesis SuperMix for qPCR system (AT341; TransGen Biotech). Samples were adopted 40 cycles using a Fast ABI-7500 sequence detector (Applied Biosystems, Foster City, USA). ABI Prism7500 fluorescent signal acquisition conditions were conducted for 30 s at 95 °C, and were followed by 40 cycles of 5 s at 95 °C, 15 s at 60 °C, and 10 s at 72 °C. The cycle threshold (Ct) was calculated under default settings for real-time sequence detection software (Applied Biosystems). At least three independent biological replicates were performed to verify the repeat ability of the data. The gene-specific primers used for real-time quantitative PCR are tabulated in Table 2.| Name | Forward (5′–3′) | Reverse (5′–3′) |
|---|---|---|
| LXRα | AGGAGTGTCGACTTCGCAAA | CTCTTCTTGCCGCTTCAGTTT |
| ABCA1 | CCCAGAGCAAAAAGGGACTC | GGTCATCATCACTTTGGTCCTTG |
| ABCG1 | CAAGACCCTTTTGAAAGGGATCTC | GCCAGAATATTCATGAGTGTGGAC |
| CD36 | CAAGCTCCTTGGCATGGTAGA | TGGATTTGCAAGCACAATATGAA |
| SR-1 | TTAAAGGTGATCGGGGACAAA | CAACCAGTCGAACTGTCTTAAG |
| β-actin | CCTGGCACCCAGCACAAT | GCCGATCCACACACGGAGTACT |
2.11. Statistical analysis
The data were expressed as the mean ± SEM. Statistical significance were determined by one-way analysis of variance (ANOVA) followed by Dunnett's post hoc test. SPSS 17.0 software (SPSS, Chicago, IL, USA) was used for statistical analysis. A probability level (p) of 0.05 was considered as significant.2.12. Cytotoxicity assays
Cytotoxicity of the compounds against RAW264.7 macrophages using the MTT method was evaluated.2.13. Evaluation of the TrxR inhibitory activities
For determining the TrxR inhibitory activities of the compounds, the DTNB (5,5′-dithiobis 2-nitrobenzoic acid) reduction assay was employed. All assays were conducted at 25 °C in a total volume of 40 μL. In each measurement, 0.3 μL of TrxR (from rat liver, Sigma-Aldrich) (0.04 μM) was added to an assay buffer containing 1 M potassium phosphate (pH 7.0), 500 mM EDTA (pH 7.4), NADPH (0.48 mM) and 1 μL of inhibitor at various concentrations. After 5 min pre-incubation, the reaction was initiated with the addition of 3.2 μL of DTNB (final concentration of 5.0 mM). The positive control curcumin was incubated with the same amount of 2.5% DMSO (v/v), while the compounds were tested by the same protocol as for positive control. The increasing absorbance at 412 nm (Δε 412 = 13.6 mM−1 cm−1) was monitored in the initial 120 s. The IC50 values were calculated to represent the TrxR inhibitory effects of compounds.3. Result and discussion
3.1. Structure elucidation of new cyclopeptides
Lipid-lowering assay in association with chromatographic separation of a gorgonian (Pseudopterogorgia sp.) associated fungus A. versicolor LZD-14-1 resulted in the isolation of three new cyclopeptides namely versicotides D–F (1–3) and two known analogues (4–5) (Fig. 1).Versicotide D (1) has a molecular formula of C25H30N4O4 as determined by the HRESIMS and NMR data. The 13C NMR and DEPT spectra exhibited 12 aromatic carbons for two phenyl groups, four carbonyl resonances, three methylene, four methine, and two methyl groups. In association with the 1H NMR data for two NH and five α-protons, compound 1 was characteristic of a peptidic derivative. The 2D NMR data (COSY, HMQC, HMBC, and NOESY) established the segments for two phenylalanines, an alanine, and a glycine. The HMBC data such as the correlations from NH (δH 7.59, Ala) and α-H (δH 5.08, Phe-1) to the carbonyl carbon C-4 (δC 169.7), α-H (Phe-1) and NH (δH 7.82, Gly) to C-13 (δC 168.3), NH (Gly) and α-H (δH 5.41, Phe-2) to C-15 (δC 171.3), as well as α-H (Phe-2) and α-H (Ala) to C-1 (δC 171.0), linked the amino acid residues in a sequence of Ala–Phe(1)-Gly–Phe(2), while Phe-2 connected to Ala to cyclize the amino acid residues was assigned on the basis of the NOE relationships between NH (Ala)/α-H (Phe-2) and NH (Gly)/α-H (Phe-1). In addition, the HMBC correlations from the methyl protons at δH 2.61 (s) to C-13 (δC 168.3) and C-5, and from the methyl protons at 2.69 (s) to C-1 (171.0) and C-16, located the methyl groups at the nitrogen atoms of Phe-2 and Phe-1, respectively. The data of Cu/Kα X-ray diffraction18 using flack parameter of −0.02(6) (Fig. 2) unambiguously confirmed the structure and determined the absolute configurations of the amino acid residues to be L-Ala, L-Phe(1), and D-Phe(2). It is noted that this is the first sample of cyclic tetracyclopeptides containing both D- and L-phenylalanine.
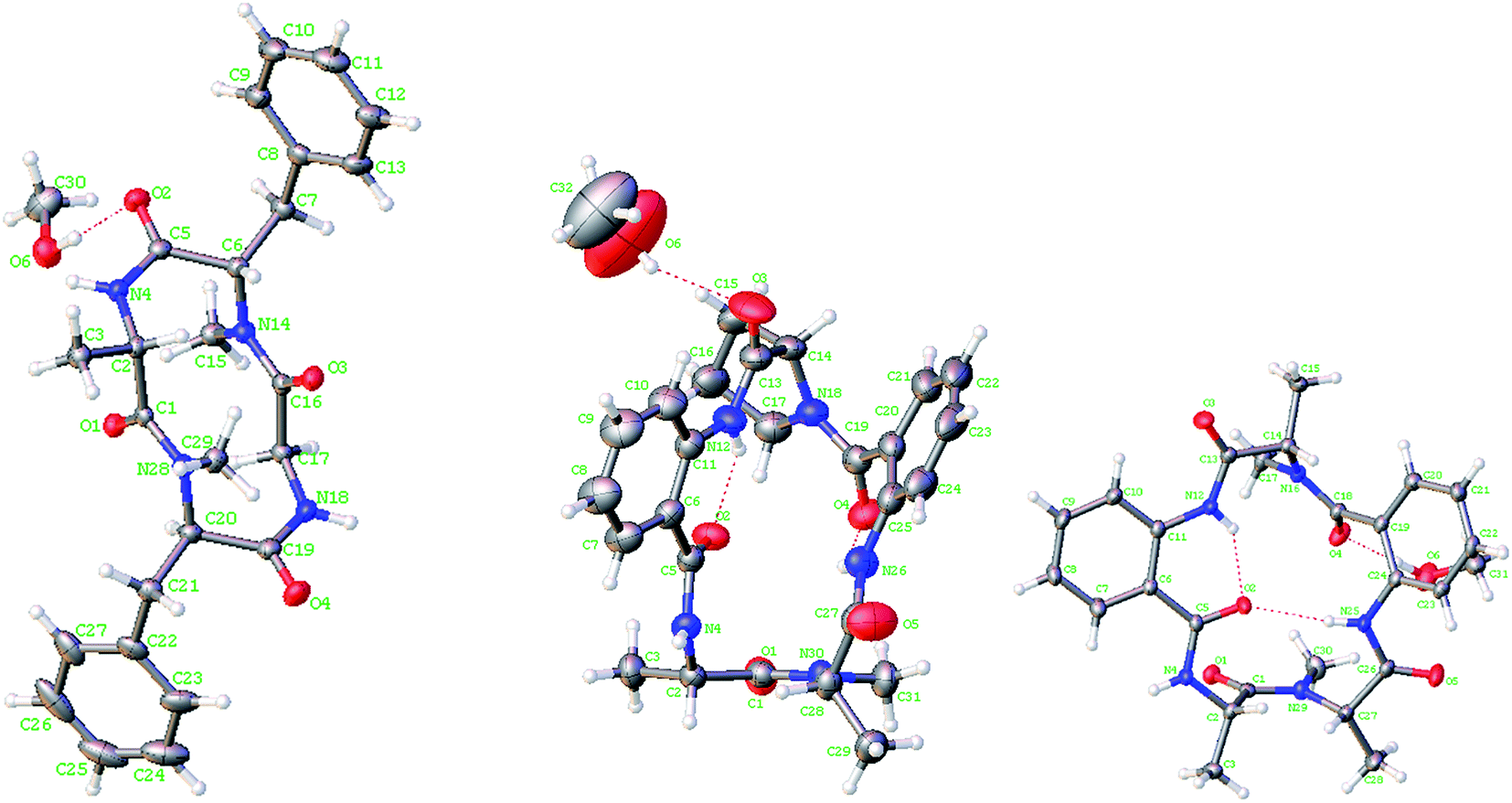 | ||
| Fig. 2 X-ray crystallographic structures of 1, 2 and 4. | ||
Versicotide E (2) has a molecular formula of C26H29N5O5 as established by the HRESIMS and NMR data, requiring 15 degrees of unsaturation. The 13C NMR spectrum (Table 1) exhibited the resonances for five carbonyl carbons (δC 170.5, 166.5, 169.6, 168.1, and 169.6), whereas the 1H NMR data showed three α-H protons for the amino residues (δH 4.20, 4.78, 4.84), in addition to three methyl groups, eight aromatic protons and a number of alkyl protons. These data were characteristic of a peptidic structure. Analyses of the 2D NMR data (COSY, HMQC, and HMBC) uncovered three amino acid resides for two alanine (Ala) and a proline (Pro), while two anthranilic acid residues (ATA) were recognized. The sequence assignment was based on the HMBC and NOESY data initiating from ATA-1. The presence of an ABCD aromatic spin system from H-6 (δH 7.72, d, J = 8.0 Hz) to H-9 (δH 8.30, d, J = 8.4 Hz), in addition to the HMBC correlations from H-9 and NH (δH 12.0, ATA-1) to the aromatic carbons C-5 (δC 117.7) and C-10 (δC 132.8) and from NH (ATA-1) and α-H (Pro) (δH 4.78, dd, J = 4.5, 8.1 Hz) to the carbonyl carbon C-11 (δC 169.6), clarified the linkage of ATA-1 and Pro unit. Subsequently, the HMBC correlations from α-H (Pro) and the aromatic proton H-17 (δH 7.61, d, J = 7.6 Hz) (ATA-2) to the carbonyl carbon C-16 (δC 166.5), NH (δH 9.20, s, ATA-2) and α-H (δH 4.84, Ala-2) to the carbonyl carbon C-23 (δC 169.6), in association with the HMBC correlations from α-H (Ala-2) and α-H (Ala-1) to the carbonyl carbon C-1 (δC 170.5), established a linear sequence of ATA(1)-Pro-ATA(2)-Ala (2)-Ala (1). Additional HMBC correlations from the aromatic proton H-6 (ATA-1) and NH (δH 9.42) (Ala-1) to the carbonyl carbon C-4 (δC 168.1) cyclized the amino sequence. Moreover, a methyl singlet at δH 2.90 (s) showed the HMBC correlations with the carbonyl carbon C-1 and the α-carbon of Ala-2 (C-24), clarified the substitution of a methyl group at the nitrogen atom of Ala-2. These assignments were further supported by the data of the X-ray diffraction of 2 with the flack parameter of 0.03 (5) (Fig. 2),18 which enabled to assign the absolute configurations of each amino acid residue to be L-form.
The HRESIMS and NMR data provided the molecular formula of versicotide F (3) to be the same as that of 2. The 1H and 13C NMR data in association with the COSY and HMBC correlations assigned 3 also possessing the amino acid residues with two ATA residues, a Pro and two Ala, while the similar HMBC correlations indicated that 3 has a segment of Pro-ATA(1), the same as that of 2. However, the HMBC data revealed the correlation of α-H (δH 4.55, Pro) and α-H (δH 4.15, Ala-2) with a carbonyl carbon C-16 the (δC 171.7), indicating the presence of a Pro–Ala(2) residue. The connection of Ala-2 with ATA-2 was demonstrated by the HMBC correlations from NH (δH 9.17, Ala-2) and the aromatic proton H-21 (δH 8.02, ATA-2) to the carbonyl carbon C-19 (δC 168.8). Additional HMBC correlations from NH (δH 12.68, ATA-2) and α-H (δH 4.90, Ala-1) to the carbonyl carbon C-1 (δC 167.8) established the linear sequence of 2 to be ATA(1)-Pro–Ala(2)-ATA(2)-Ala(1). The cyclization between Ala-1 and ATA-1 was evident from the HMBC correlations between the aromatic proton H-6 (δH 7.45) and C-4 (δC 168.5) and from the methyl singlet (δH 2.74, s) to C-4 and C-2 (δC 59.1, α-carbon of Ala-1) (Fig. 3), which also deduced the location of a methyl group at the nitrogen atom of Ala-1. It is noted that both 2 and 3 exhibited the ΔδCβ–Cγ value to be 9.9 and 10.3 ppm, respectively, which was in accordance with a cis configuration instead of trans-form.19 Based on the advanced Marfey's method,20 the HPLC retention times of the hydrolyzed derivatives of 3 in comparison with the those of the derivatives of the authentic samples, assigned the configurations of the chiral amino acid residues to be L-alanine and L-proline.
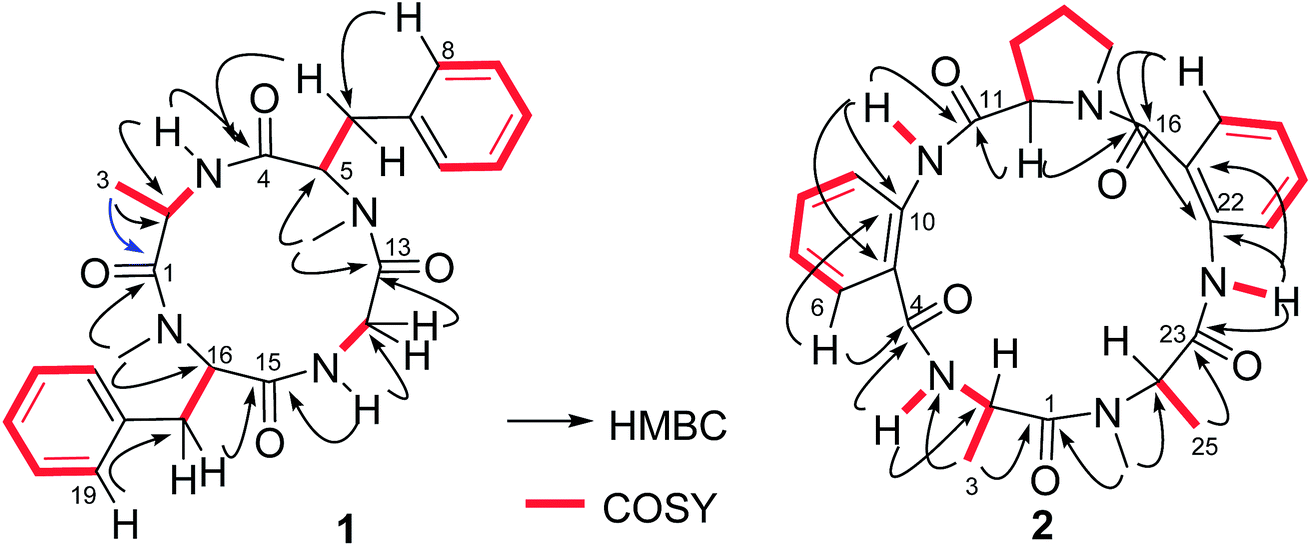 | ||
| Fig. 3 Key COSY and HMBC correlations of 1 and 2. | ||
In addition, compounds 4 and 5, a pair of inseparable isomers with a ratio of 2.5:1, were identical to versicotides A and B respectively,21 based on the comparison of the NMR and MS data with those reported in the literature in addition to the 2D NMR data. Fortunately, versicotide A (4) was able to be crystalized in CH2Cl2/MeOH (1:1) solution. The X-ray single crystal diffraction using a flack parameter of 0.04(5) led to the assignment of the absolute configuration of 4 (Fig. 2). Therefore, the absolute configurations of 5 were determined to be the same as those of 4 biogenetically.
3.2. Bioassay
The cellular model based bioassay indicated that the cyclopeptide containing fraction (F3) possessed lipid-lowering effect in oxLDL induced RAW264.7 macrophage cells based on the oil red O staining method. The same experiment was conducted to test whether the isolated cyclopeptides possess the relative activities. As shown in Fig. 4A, compounds 1–3 significantly reduced the lipid accumulation with the reduction of 11%, 13%, and 12%, respectively in a dose of 10 μM, whereas compounds 4/5 showed weak activity in comparison with the positive control simvastatin which showed the reduction of 22% in the same dose. Increasing the dose to 50 μM, compounds 1–3 exerted the activities similar to that of simvastatin in 10 μM. Macrophages uptake oxidized LDL (oxLDL) to accumulate lipids and differentiate into foam cells to form the early lesions that mature into atherosclerotic plaques, thus macrophage foam cell formation is a crucial determinant of atherosclerotic lesion occurrence and is associated with abnormalities in cellular cholesterol homeostasis in macrophages.22 The bioassay results indicated that compounds 1–3 play important role to prevent the formation of foam cells. In addition, compounds 1–3 decreased the intracellular total cholesterol levels (Fig. 4B), of which compounds 2 and 3 exhibited the effects with the decreasing ratio of 8.7% and 9.2% that were comparable to the positive control simvastatin (9.3%) at the same dose (10 μM). In a dose of 50 μM, compounds 2 and 3 decreased the intracellular total cholesterol in the ratio of 14.6% and 18.7%, respectively, whereas compound 1 showed the decreasing data of 11.3%. As determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, these compounds did not decrease cell viability of RAW264.7 macrophages up to a dose of 100 μM (Fig. 5), indicating that their inhibiting effect on cellular lipid accumulation was not due to cytotoxicity. These findings revealed that compounds 1–3 were potent to inhibit the cholesterol accumulation and to delay the transformation of macrophages into foam cells under oxLDL overloading.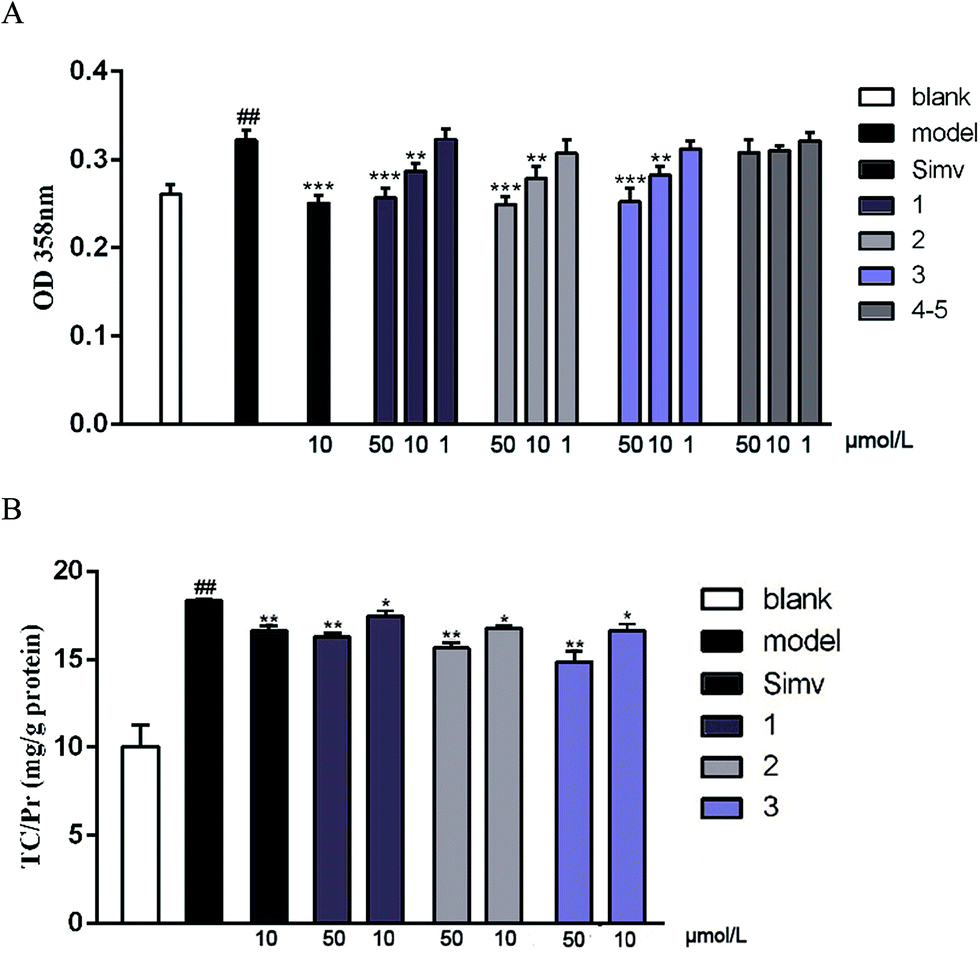 | ||
| Fig. 4 Compounds suppress oxLDL-induced foam cell formation and intracellular total cholesterol (TC) accumulation in RAW264.7 macrophages. (A) Spectrophotometry at 358 nm after oil red O staining; (B) intracellular TC levels. The blank group was given DMEM only, while the other groups were given 50 μg mL−1 of oxLDL to induce foam cell formation. Bars depict the means ± SEM in triplicate. ##p < 0.01 blank group vs. model group; *p < 0.05, **p < 0.01, test group vs. model group. Simv: simvastatin. | ||
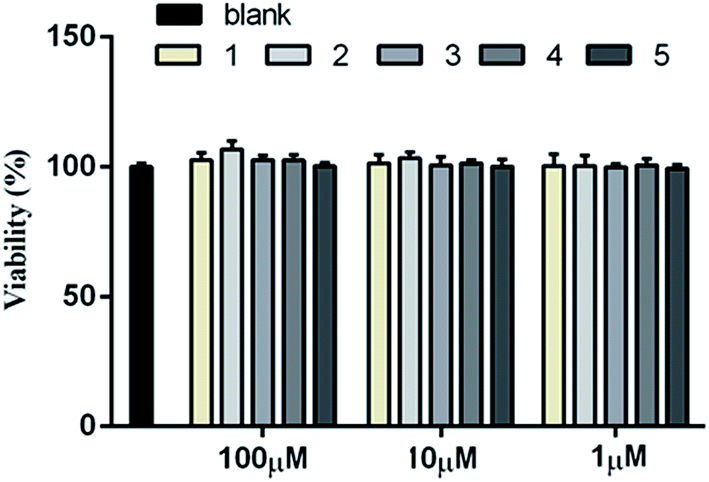 | ||
| Fig. 5 Cytotoxicity of 1–5 by MTT assay. The blank group was given DMEM only, while the other groups were given respective compound at the indicated concentration. Bars depict the means ± SEM in triplicate. | ||
It is noted that the imbalance of cholesterol influx and efflux caused the cholesterol accumulation, which could induce the development of atherosclerosis. Removal of excessive cholesterol from macrophages (macrophage cholesterol efflux), plays a protective role during the development of atherosclerosis.23 Subsequently, cholesterol influx experiment was performed to evaluate the capability of the active compounds 1–3 for the regulation of cholesterol influx. As shown in Fig. 6A, an experiment using 25-NBD-labeled cholesterol as a fluorescence marker revealed that compounds 1–3 at a dose of 10 μM significantly inhibited cholesterol influx. In parallel, three compounds (1–3) effectively promoted cholesterol efflux to HDL in RAW264.7 macrophages at a dose of 10 μM. However, they showed slight weaker activities than that of rosiglitazone, an agent used for the treatment of atherogenesis through the stimulation of HDL-induced cholesterol efflux (Fig. 6B). These results demonstrated that compounds 1–3 can inhibit macrophage-derived foam cells formation via inhibiting cholesterol influx and promoting efflux. Preliminarily analyses of the structure–activity relationship indicated that proline unit such as 2 and 3 is requirement to induce the reduction of lipid accumulation among the ATA-bearing cyclic peptides. However, compound 1 with the inhibitory effect is an exception due to the absence of ATA and proline units in the structure.
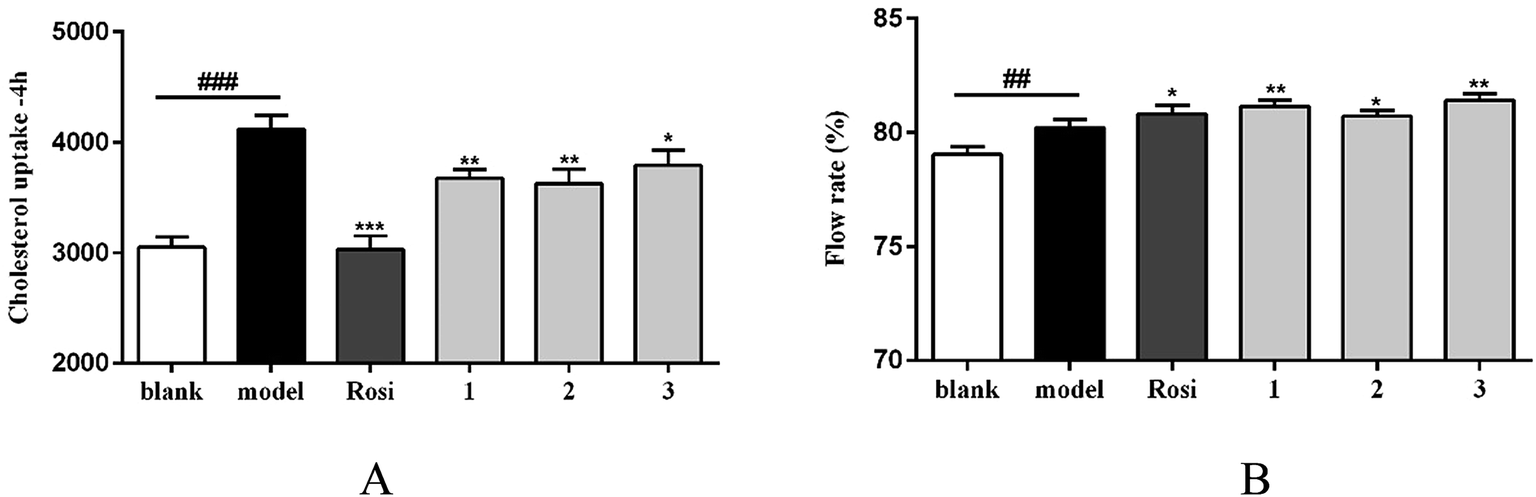 | ||
| Fig. 6 Compounds regulate cholesterol influx/efflux in RAW264.7 macrophages. (A) Cholesterol uptake data indicated by 25-NBD cholesterol at 4 h; (B) 25-NBD cholesterol efflux to HDL. Bars depict the means ± SEM in triplicate.##p < 0.01, ###p < 0.001,blank group vs. model group; *p < 0.05, **p < 0.01, test group vs. model group. Rosi: rosiglitazone. | ||
Mechanistically, the critical scavenger receptors including CD36 and SR-1 are the main targets to regulate cholesterol dynamics such as cholesterol uptake, while liver X receptor a (LXRa) and ATP-binding cassette transporters A1 and G1 (ABCA1 and ABCG1) play key roles in stimulating cholesterol efflux.24 Scavenger receptors cluster of differentiation 36 (CD36) and class A scavenge receptors (SR-1 and SR-2) bind to and mediate excess oxLDL into macrophages, resulting in excess cholesterol accumulation and pernicious cells. High expression of scavenger receptors led to foam cell formation and early atherosclerotic lesions. Removing excess intracellular cholesterol from macrophages mainly depends on the cholesterol efflux pathway. The main transporters of cholesterol efflux include the ATP binding cassette transporters A1 and G1 (ABCA1 and ABCG1) and the scavenger receptor B1 (SR-B1). Liver X receptor alpha (LXRα), as cholesterol sensors, plays vital role in stimulating the efflux of cholesterol from cells to HDL through ABCA1, ABCG1 and SR-B1. In order to clarify the targets of the active cyclopeptides related to the cholesterol efflux or influx, real-time quantitative PCR was performed to test the expression of cholesterol flux modulating genes. The amplification of β-actin was served as an internal control. As shown in Fig. 7A, compounds 1–3 significantly restrained the expression of CD36 and SR-II transcription at a dose of 10 μM in RAW264.7 cells. Moreover, compounds 2 and 3 exhibited the inhibitory effects against the SR-I expression, whereas compound 1 showed no regulation of SR-I. Subsequently, the real-time quantitative PCR data revealed that compounds 1–3 significantly promoted the expression of target genes ABCG1 and LXRα (Fig. 7B), and compound 1 induced the up-regulation of ABCA1, while no induction was observed in compounds 2 and 3. These findings suggested the effects of compounds 1–3 against the oxLDL-induced foam cell formation and the regulation of the cholesterol influx and efflux to be induced by the down-regulation of CD36, SR-II and/or SR-I and the up-regulation of ABCG1, LXRα and/or ABCA1. Comparison of the data of the target gene regulation induced by 1–3 revealed the analogues with the sequence differentiation and the amino acid variation exerted different mechanism for gene regulation.
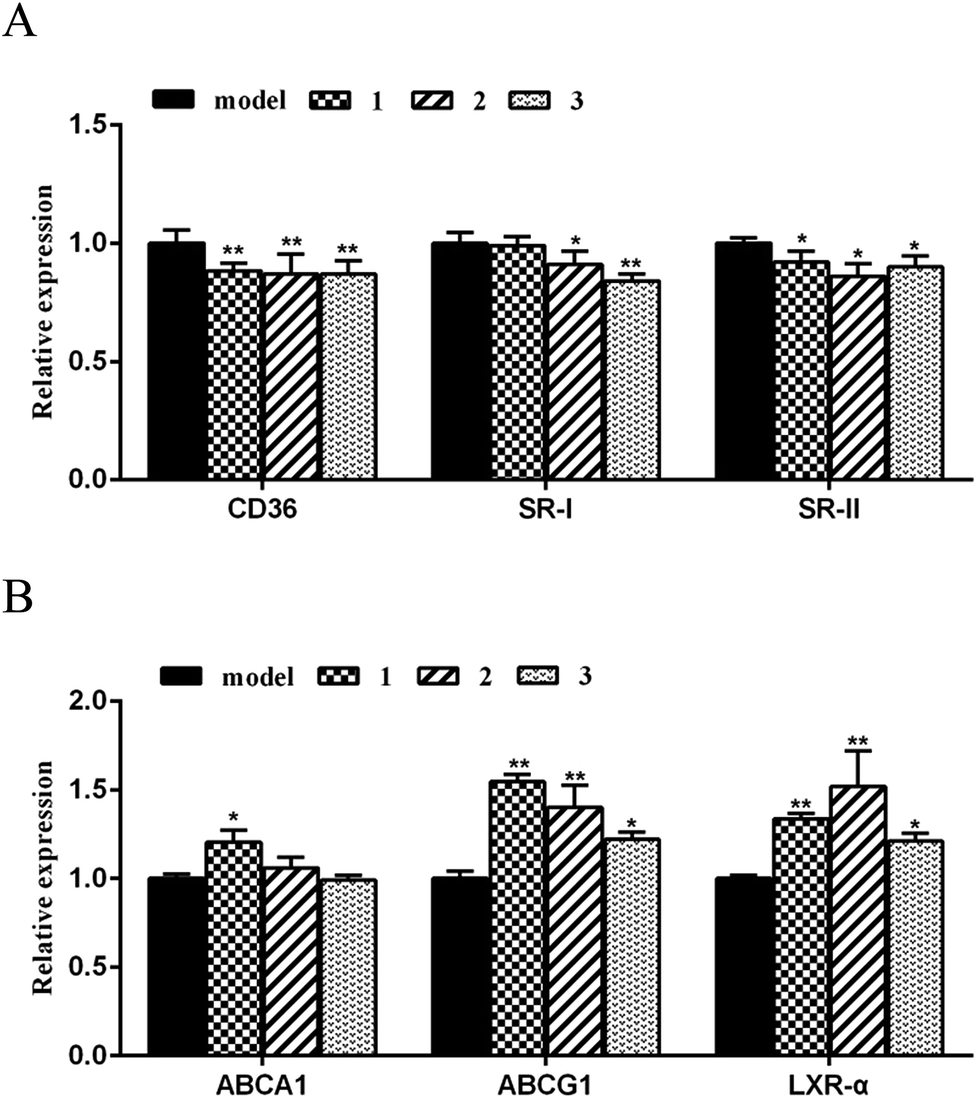 | ||
| Fig. 7 Compounds on the mRNA levels of CD36 and SR-I,ABCA1, ABCG1, LXRα in RAW264.7 cells. (A) Relative genes expression on cholesterol influx; (B) relative genes expression on cholesterol efflux; real-time PCR was conducted with gene-specific oligonucleotide primers. The amplification of β-actin served as an internal control. The values shown are the means ± SEM at least three experiments. *p < 0.05, **p < 0.01. | ||
In addition, thioredoxin reductase (TrxR) is a key enzyme for cellular redox control and antioxidant defense, and which was upregulated in human atherosclerotic plaques and was expressed in foam cells.24 oxLDLs dose-dependently increased TrxR1 mRNA in human monocyte derived macrophages. Compounds 2 and 3 exhibited inhibitory activity against TrxR with IC50 values of 28.8 and 38.4 μM, respectively (Table 3).
4. Conclusion
In conclusion, the present work reported the isolation and the structural characterization of three new cyclic peptides derived from a gorgonian associated fungus A. versicolor LZD-14-1, while two of them are characterized by the naturally unique ATA-bearing pentacyclopeptides. Compounds 1–3 prevented foam cells formation in RAW264.7 cells by the reduction of lipid accumulation through the regulation of cholesterol efflux and influx. The mechanistic study revealed that the cyclopeptides induced cholesterol influx or efflux was related to the promotion of the target genes ABCG1 and LXRα expression, in addition to the down-regulation of critical scavenger receptors CD36 and SR-1/SR-2. Thus, this work provided the new natural scaffolds which may be developed as the anti-atherosclerosis leads through structural optimization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the grants of National High Technology and Science 973 program (2015CB755906), and NSFC (81630089, 81573436, 41376127).References
- L. Fernandez-Friera, J. L. Penalvo, A. Fernandez-Ortiz, B. Ibanez, B. Lopez-Melgar, M. Laclaustra, B. Oliva, A. Mocoroa, J. Mendiguren, V. Martinez de Vega, L. Garcia, J. Molina, J. Sanchez-Gonzalez, G. Guzman, J. C. Alonso-Farto, E. Guallar, F. Civeira, H. Sillesen, S. Pocock, J. M. Ordovas, G. Sanz, L. J. Jimenez-Borreguero and V. Fuster, Circulation, 2015, 131, 2104–2113 CrossRef PubMed.
- K. M. Patel, A. Strong, J. Tohyama, X. Jin, C. R. Morales, J. Billheimer, J. Millar, H. Kruth and D. J. Rader, Circ. Res., 2015, 116, 789–796 CrossRef CAS PubMed.
- L. N. Rao, T. Ponnusamy, S. Philip, R. Mukhopadhyay, V. V. Kakkar and L. Mundkur, Lipids, 2015, 50, 785–797 CrossRef CAS PubMed.
- H. Y. Lee, S. D. Kim, S. H. Baek, J. H. Choi and Y. S. Bae, Biochem. Biophys. Res. Commun., 2013, 433, 255–259 CrossRef CAS PubMed.
- X. H. Yu, Y. C. Fu, D. W. Zhang, K. Yin and C. K. Tang, Clin. Chim. Acta, 2013, 424, 245–252 CrossRef CAS PubMed.
- S. O. Rahaman, W. Swat, M. Febbraio and R. L. Silverstein, J. Biol. Chem., 2011, 286, 7010–7017 CrossRef CAS PubMed.
- G. J. Zhao, K. Yin, Y. C. Fu and C. K. Tang, Mol. Med., 2012, 18, 149–158 CAS.
- J. Y. Lee, J. Karwatsky, L. Ma and X. Zha, Am. J. Physiol.: Cell Physiol., 2011, 301, C886–C894 CrossRef CAS PubMed.
- M. A. Kennedy, G. C. Barrera, K. Nakamura, A. Baldan, P. Tarr, M. C. Fishbein, J. Frank, O. L. Francone and P. A. Edwards, Cell Metab., 2005, 1, 121–131 CrossRef CAS PubMed.
- A. Ji, J. M. Wroblewski, L. Cai, M. C. de Beer, N. R. Webb and D. R. van der Westhuyzen, J. Lipid Res., 2012, 53, 446–455 CrossRef CAS PubMed.
- L. Yvan-Charvet, N. Wang and A. R. Tall, Arterioscler., Thromb., Vasc. Biol., 2010, 30, 139–143 CrossRef CAS PubMed.
- A. Chawla, W. A. Boisvert, C. H. Lee, B. A. Laffitte, Y. Barak, S. B. Joseph, D. Liao, L. Nagy, P. A. Edwards, L. K. Curtiss, R. M. Evans and P. Tontonoz, Mol. Cell, 2001, 7, 161–171 CrossRef CAS PubMed.
- C. Wu, R. Chen, M. Liu, D. Liu, X. Li, S. Wang, S. Niu, P. Guo and W. Lin, Mar. Drugs, 2015, 13, 6352–6365 CrossRef CAS PubMed.
- S. Wang, X. Zhang, M. Liu, H. Luan, Y. Ji, P. Guo and C. Wu, Pharm. Microbiol., 2015, 53, 1481–1487 CAS.
- C. Wu, H. Luan, X. Zhang, S. Wang, X. Zhang, X. Sun and P. Guo, PLoS One, 2014, 9, e95452 Search PubMed.
- P. Amarenco, J. Labreuche, P. Lavallee and P. J. Touboul, Stroke, 2004, 35, 2902–2909 CrossRef CAS PubMed.
- K. J. Livak and T. D. Schmittgen, Methods, 2001, 25, 402–408 CrossRef CAS PubMed.
- T. Nakano, H. Kakuda, Y. Mori and M. Shiro, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, o331–332 Search PubMed.
- I. Z. Siemion, T. Wieland and K. H. Pook, Angew. Chem., Int. Ed. Engl., 1975, 14, 702–703 CrossRef CAS PubMed.
- Y. Nozawa, A. Kawashima, E. H. Hashimoto, H. Kato and K. Harada, J. Chromatogr. A, 2009, 1216, 3807–3811 CrossRef CAS PubMed.
- L. Zhou, H. Gao, S. Cai, T. Zhu, Q. Gu and D. Li, Helv. Chim. Acta, 2011, 94, 1065–1070 CrossRef CAS.
- A. N. Orekhov and E. A. Ivanova, Phytomedicine, 2016, 23, 1190–1197 CrossRef CAS PubMed.
- A. V. Khera, M. Cuchel, M. de la Llera-Moya, A. Rodrigues, M. F. Burke, K. Jafri, B. C. French, J. A. Phillips, M. L. Mucksavage, R. L. Wilensky, E. R. Mohler, G. H. Rothblat and D. J. Rader, N. Engl. J. Med., 2011, 364, 127–135 CrossRef CAS PubMed.
- G. H. Hwang, J. M. Ryu, Y. J. Jeon, J. Choi, H. J. Han, Y. M. Lee, S. Lee, J. S. Bae, J. W. Jung, W. Chang, L. K. Kim, J. G. Jee and M. Y. Lee, Eur. J. Pharmacol., 2015, 765, 384–393 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1491882–1491884. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra07940k |
| ‡ Equal contribution to this work. |
| This journal is © The Royal Society of Chemistry 2017 |