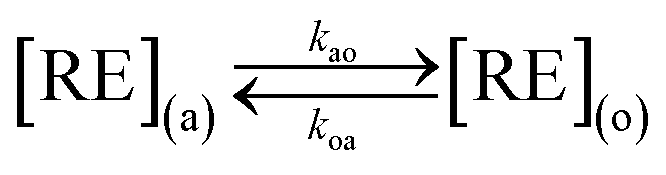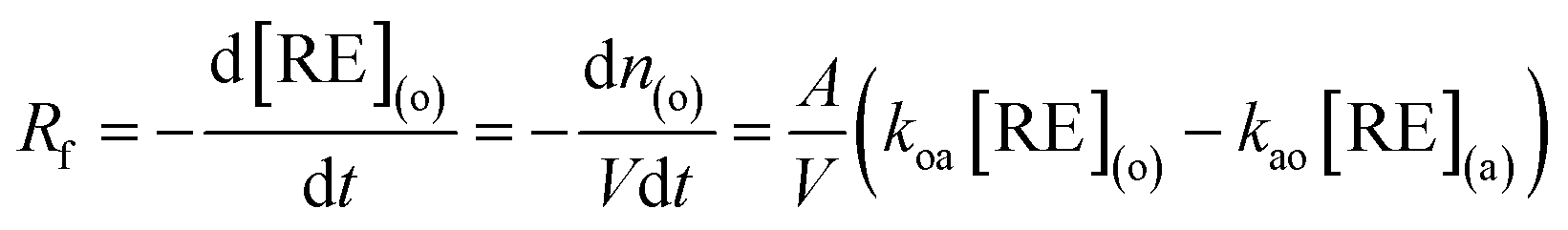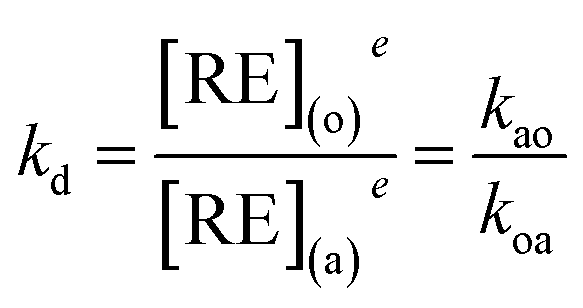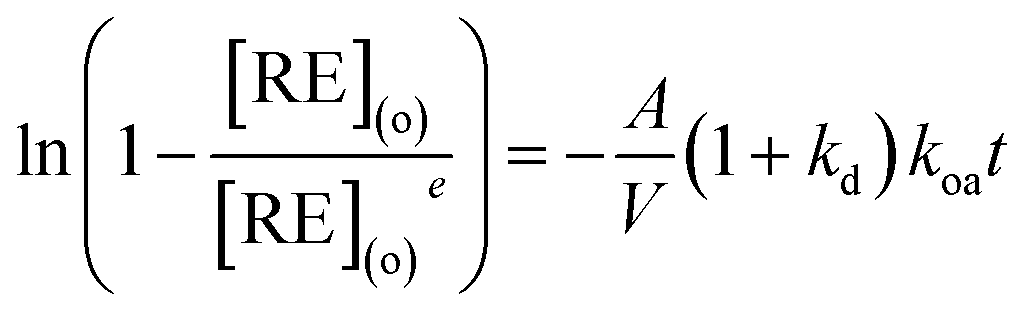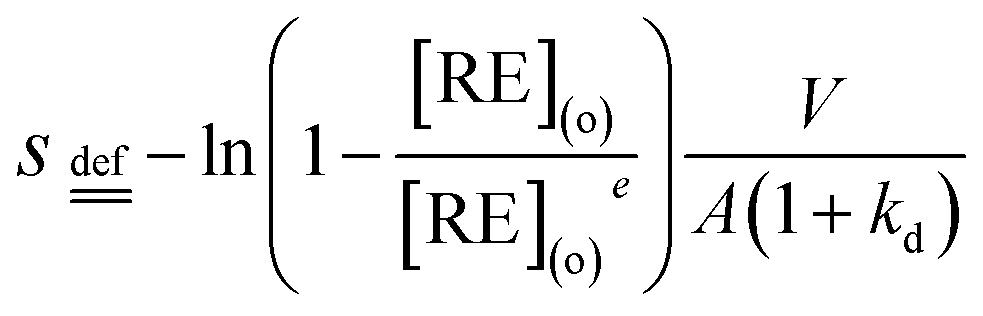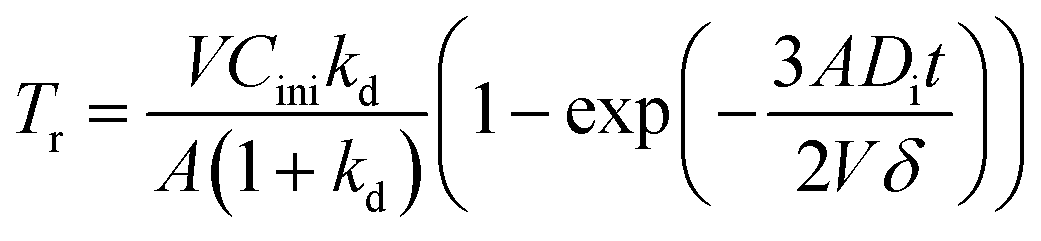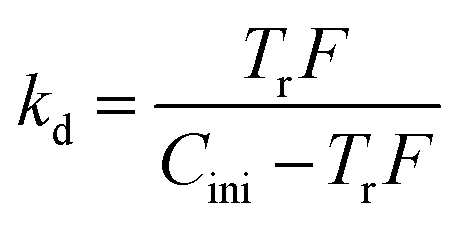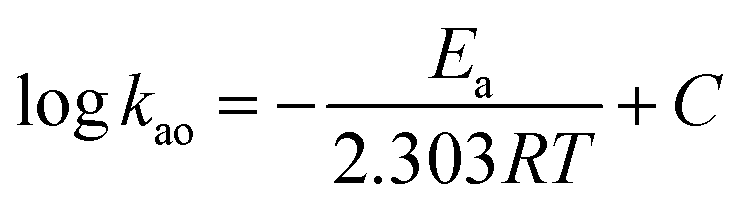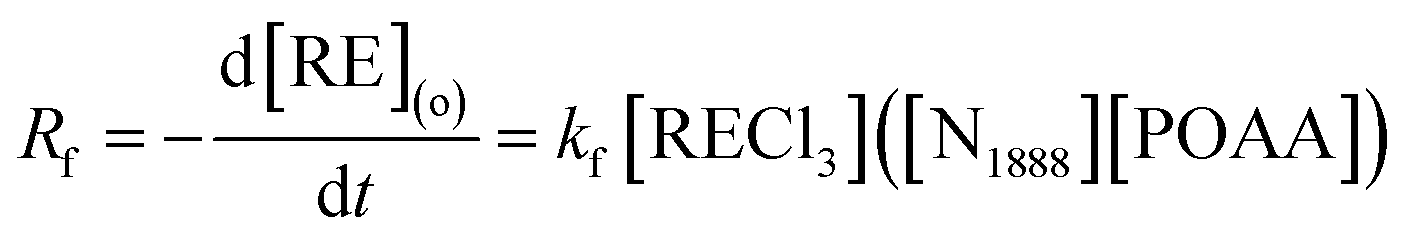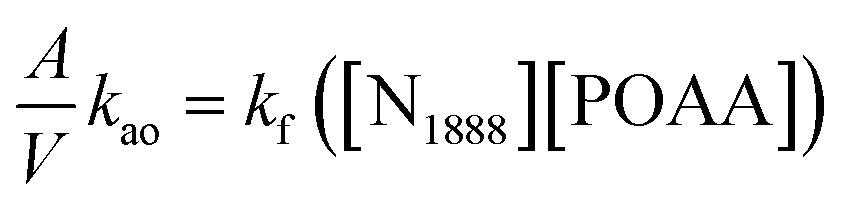DOI:
10.1039/C7RA07851J
(Paper)
RSC Adv., 2017,
7, 39556-39563
Extraction kinetics of mixed rare earth elements with bifunctional ionic liquid using a constant interfacial area cell†
Received
17th July 2017
, Accepted 7th August 2017
First published on 14th August 2017
Abstract
As a bifunctional ionic liquid used for the extraction of yttrium and heavy rare earth elements (REEs), [methyltrioctyl ammonium][(2,6-dimethylheptyl) phenoxy acetic acid] ([N1888][POAA]) has been synthesized in this paper. The extraction kinetics of REEs including La–Lu plus Y except Pm with [N1888][POAA] using a constant interfacial area cell were reported. Comparing with the conventional (2,6-dimethylheptyl) phenoxy acetic acid (HPOAA) extraction system, the forward extraction rate (kao, mm s−1) of heavy REEs in the [N1888][POAA] extraction system increased significantly with an improvement of 8.44 times for yttrium. The effect of stirring speed on the extraction rate of mixed REEs was studied and 300 rpm was suggested to ensure the extraction regimes were chemical reaction-controlled. The effect of temperature was studied and the extraction reaction was controlled by chemical reaction when the temperature was below 298 K. The effect of specific interfacial area was also evaluated which indicated that the bulk phase was the reaction zone. The average extraction rate equation of REEs has also been obtained as: −d[RE](o)/dt = 0.141[RECl3]([N1888][POAA]).
1 Introduction
Rare earth elements (REEs), including La–Lu, Sc and Y, have become critical materials in various fields, such as in catalysts, alloys, magnets, lasers, batteries, electronics, lighting, and telecommunications.1 Heavy REEs consisting of Y, Gd, Tb, Dy, Ho, Er, Tm, Yb and Lu exhibit irreplaceable specific performance, therefore, they have become important elements in exploring high technology materials.2 Solvent extraction processes have been well established and widely applied in the REEs separation industry of China with organic phosphonic/phosphoric acids and carboxylic acids.3,4 However, the saponification of the acidic extractants results in the release of NH4+, Na+, Ca2+ or Mg2+ to wastewaters and serious pollution. There is a growing interest in developing new extractants and corresponding materials to solve these problems.5
Ionic liquids are a series of organic salts which have received much attention as green solvents in many fields.6,7 Bifunctional ionic liquids (BILs) composed of quaternary ammonium/phosphonium cation and monobasic acidic extractant, have become the focus of research for the separation of yttrium and heavy REEs in recent years.8 The inner synergistic effects of BILs for solvent extraction of metal ions using [tricaprylmethyl ammonium][di-2-ethylhexyl phosphinate] as extractant have been reported.9–11 The results show that the extraction efficiency of REEs has been enhanced and the selectivity has been changed in the BIL system. Other advantages, such as avoiding saponification wastewater from the application of acidic extractants, low acidity for extraction and good interfacial phenomena, have emerged.12,13 (2,6-Dimethylheptyl) phenoxy acetic acid (HPOAA) and [methyltrioctyl ammonium (N1888)][POAA] have been synthesized in this lab recently. The former was used for the separation of high-purity yttrium from ion-absorbed rare earth ore.14–16 The later was used for the development of sustainable yttrium separation process from heavy rare earth enrichments.17 Equilibrium data have been collected and the separation process for industrial application may contribute to eliminating the saponification process and avoiding the acid or base consumption. However, there still remains considerable uncertainty about the kinetics involved in the extraction of REEs, such as extraction regimes (diffusion, chemical reaction or mixed controlled) and chemical reaction zone which control the rate of extraction in the bulk phases or at the liquid–liquid interface.
Although there were various instruments and methods to determine how the extraction rates depended on the time, such as single drop technique,18,19 hollow fiber membrane method,20,21 centrifugal partition chromatography,22 constant interfacial area cell23,24 and microfluidic method,25 the cell among which was used wildly for its stability and reproducibility data. Literature related to this research was shown in Table 1. The main problem of these conventional extraction systems was that, the interfacial transfer limited by diffusion of molecules to the interface at the relatively slow mixing and small specific interfacial areas (1–100 m2 m−3) rather than chemical reaction, and reaction zone which controlled the rate of extraction usually at the liquid–liquid interface.
Table 1 The kinetic mechanism including extraction regime and reaction zone of metal ions with extractants using constant interfacial area cell
| Extractants |
Metal |
Kinetic mechanism |
Ref. |
| Extraction regime |
Reaction zone |
HDEHP is di(2-ethylhexyl)phosphoric acid. EHEHPA is (2-ethylhexyl)phosphoric acid mono-ethylhexyl ester. C272 is Cyanex 272, bis(2,4,4-trimethylpentyl)phosphinic acid. DEHEHP is di-(2-ethylhexyl)-2-ethylhexyl phosphonate. CA12 is sec-octylphenoxy acetic acid. CA100 is sec-nonylphenoxy acetic acid. C923 is Cyanex 923 (R3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), a mixture of four trialkylphosphine oxides in which each R group is either hexyl or octyl. TODGA is N,N,N′,N′-tetraoctyl diglycolamide. O), a mixture of four trialkylphosphine oxides in which each R group is either hexyl or octyl. TODGA is N,N,N′,N′-tetraoctyl diglycolamide. |
| HDEHPa |
Pr3+ |
Diffusion |
The interface |
26 |
| EHEHPAb |
Co3+ |
Chemical reaction |
Liquid–liquid interface |
27 |
| C272c |
Er3+ |
Mixed controlled |
Liquid–liquid interface |
28 |
| DEHEHPd |
Th4+ |
Mixed controlled |
The bulk phase |
29 |
| CA12e |
RE3+ |
Mixed controlled |
Two-phase interface |
30 |
| CA100f |
Y3+ |
Mixed controlled |
Liquid–liquid interface |
31 |
| C923g |
Ce4+ |
Mixed controlled |
Liquid–liquid interface |
32 |
| N1923 |
Ce4+ |
Mixed controlled |
Liquid–liquid interface |
23 |
| TODGAh |
La3+ |
Diffusion and chemical reaction |
N/A |
33 |
| [A336][CA12] |
La3+ |
Mixed controlled |
N/A |
34 |
| [A336][P227] |
Lu3+ |
Chemical reaction |
Liquid–liquid interface |
35 |
Task-specific ionic liquids [tricaprylmethylammonium][sec-octylphenoxy acetic acid] ([A336][CA12]) and [tricaprylmethyl ammonium][di-(2-ethylhexyl) phosphinic acid] ([A336][P227]) were synthesized and the extraction kinetics of La3+ and Lu3+ from aqueous chloride solutions into n-heptane solutions were discussed, respectively.34,35 However, less reports to our knowledge concern the extraction kinetics of mixed rare earth elements using a constant interfacial area cell. The main idea of this paper is to elaborate the extraction rates of 15 kinds of REEs from aqueous solution into bifunctional ionic liquid [N1888][POAA] which closer to the industrial conditions. The effects of stirring speed, temperature, specific interfacial area and extractant concentration on the extraction rate were examined. The kinetic mechanism concerns whether the extraction process was controlled by diffusion or chemical reaction, and whether the reaction zone was in the bulk phases or at the interface were involved.
2 Experimental
2.1 Materials and methods
Methyltrioctyl ammonium chloride ([N1888]Cl, purity > 99%) was purchased from Anhui Benma Pioneer Technology Co., Ltd., China. (2,6-Dimethylheptyl) phenol and sodium chloroacetate were purchased from Chendu Xiya Reagent Chemical Technology Co., Ltd. Individual REE stock solutions, including La–Lu plus Y, were prepared by dissolving the corresponding oxide (>99.99%, Ganzhou Rare Earth Group Co., Ltd., China) with hydrochloric acid and diluting with deionized water. All the chemicals were used without further purification.
Inductively coupled plasma optical emission spectroscopy (ICP-OES) Horiba Ultima 2 was used to determine the concentrations of REEs. 1H and 13C nuclear magnetic resonance (NMR) spectra were obtained in CDCl3 with an AV III-500 BRUKER spectrometer.
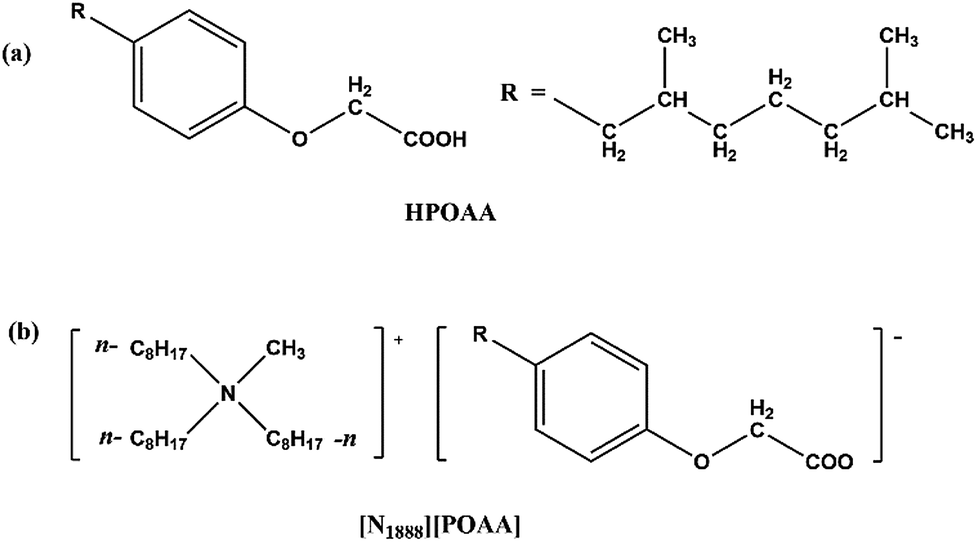
The molecular structure of (2,6-dimethylheptyl) phenoxy acetic acid (HPOAA) and [N1888][POAA] are shown in Fig. 1. The former was prepared in laboratory scale via Williamson reaction and the synthetic routes were described.16 The later was prepared by acid–base neutralization method.17 The amount of residual Cl− in the product of [N1888][POAA] determined was lower than 0.07 wt%.
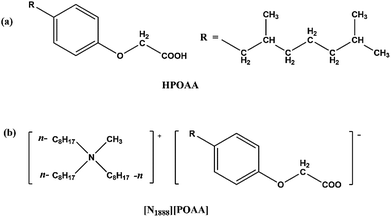 |
| | Fig. 1 The molecular structures of HPOAA (a) and [N1888][POAA] (b). | |
2.2 Experimental procedures
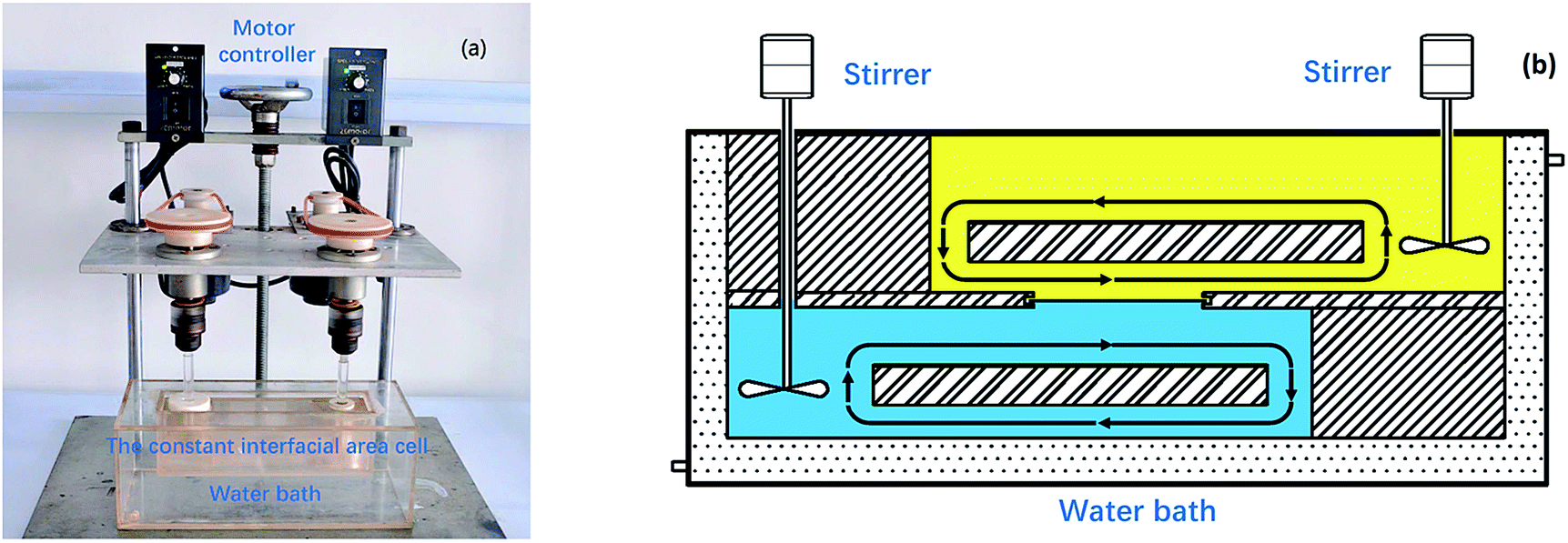
A constant interfacial area cell which improved from Nitsch-type stirred cell36 was adopted to investigate the extraction kinetics. As shown in Fig. 2(a), the cell whose phase was independently agitated with a stirring paddle was made of transparent polymethyl methacrylate. Under the combined action of stirring paddles and flow deflectors, the lighter phase and the heavier phase circulate in each chamber and move near the interface in lamina, respectively (Fig. 2(b)). The interfacial plate was a rectangle plate with a hole of 8–28 cm2. Temperature was controlled with a circulating water bath at constant temperature (298 K) except for temperature experiments. Equal volumes of 90 mL each of aqueous and organic phases were individually added to the cell chamber using a syringe. After stirring was started, 0.2 mL of sample was collected at 10 min intervals from the aqueous phase to monitor the extraction of REEs with [N1888][POAA] during the kinetic process. The experiments were carried out at a constant stirring rate of 300 rpm except for stirring rate experiments.
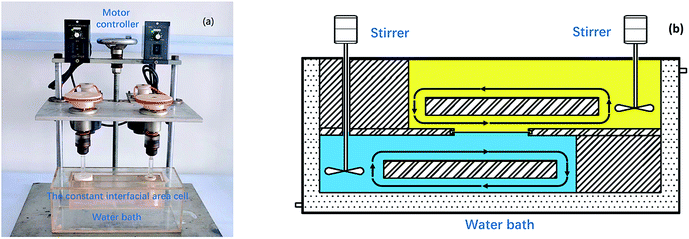 |
| | Fig. 2 The experimental equipment of kinetics (a) and detailed structure of the constant interfacial area cell (b) used to study the extraction kinetics. | |
2.3 Data treatment
This paper discussed the extraction of the mixed rare earth ions with bifunctional ionic liquid and time was the new parameter introduced into the kinetic processes which differ from equilibrium thermodynamics. Because the extraction reactions occurred simultaneously, the extraction model can be formulated as parallel reactions.| |
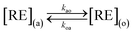 | (1) |
where (a), (o), kao (cm s−1) and koa (cm s−1) represent the aqueous phase, organic phase, forward extraction rate and backward extraction rate, respectively. RE equals La–Lu plus Y except Pm.
The extraction rate is given by the following equation.37
| |
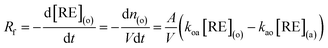 | (2) |
where
n(o) is the total number of moles of REEs in the organic phase,
V (mL) is the volume of either aqueous or organic phase, and
A (cm
2) is the interfacial area.
At equilibrium time, eqn (2) is equal to zero. Thus,
| |
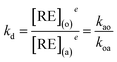 | (3) |
where
kd refers to the distribution ratio, and
e is the concentration at equilibrium. From the mass balance,
eqn (2) can be integrated and rewritten as,
| |
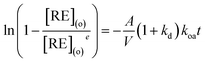 | (4) |
kd was determined by collecting samples from the aqueous phase at equilibrium.
| |
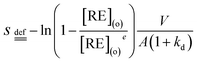 | (5) |
koa and
kao can be obtained by curve fitting the plots of
s versus time (
t).
3 Results and discussion
3.1 Characterization of the experimental equipment
To verify the accuracy of mass transfer of REEs, theoretical calculation and practical kinetic experiments using the constant interfacial area cell were completed. According to mass transfer model, mass transfer per square centimetre, Tr, can be expressed as follows,38| |
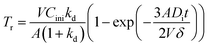 | (6) |
where V is the volume of aqueous phase (mL), A is interfacial area (cm2), Cini is initial concentration of RE ion in aqueous phase (mol L−1), kd is distribution ratio, t is time (second).
kd can be deduced from Tr as the following equation,
| |
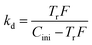 | (7) |
where
F is the conversion factor.
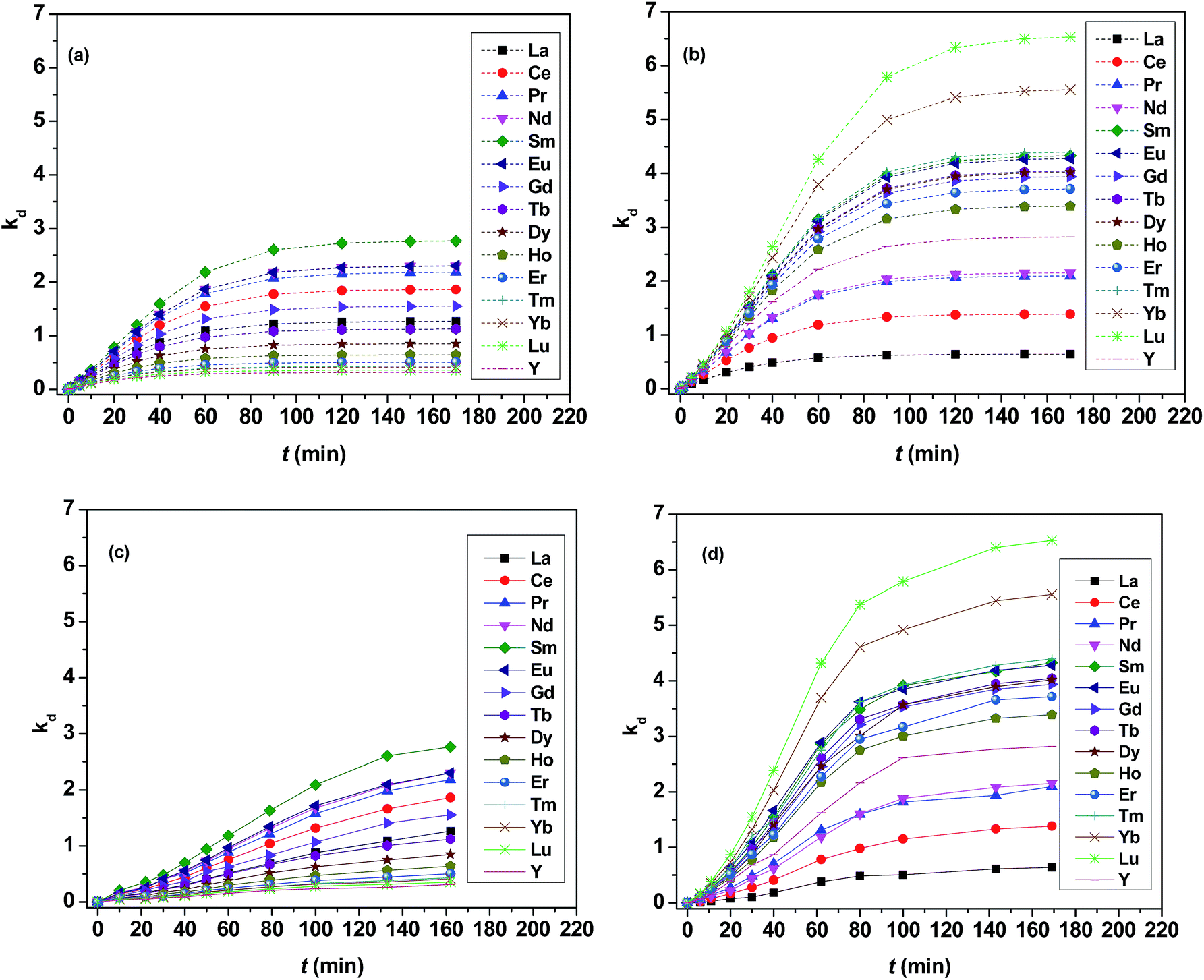
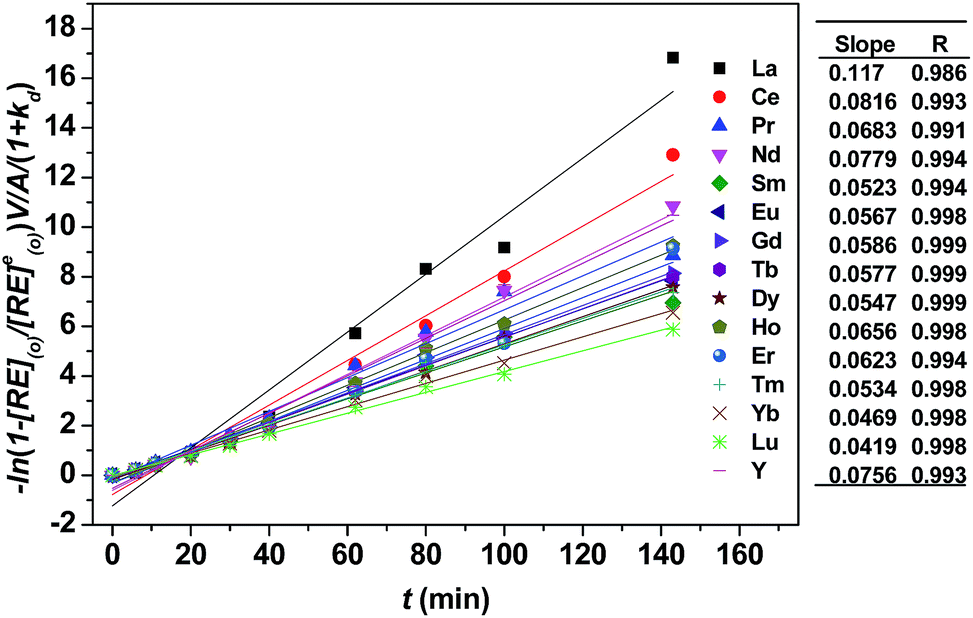
As shown in Fig. 3, kd values predicted by theoretical calculations (Fig. 3(a) and (b)) are close to practical kinetic values obtained from kinetic experiments (Fig. 3(c) and (d)) using the constant interfacial area cell. In comparison with the predicted values, the experimental values in the [N1888][POAA] extraction system were closer than that of the HPOAA extraction system as the reaction proceeds. The relative errors of REEs are less than 43% in the HPOAA extraction system and 25% in the [N1888][POAA] extraction system after 80 minutes, respectively. Further studies on plots of −ln(1 − [RE](o)/[RE](o)e)V/A/(1 + kd) vs. time (Fig. 4) are found to lie on straight lines which indicated that the mass transfer process could be treated as a pseudo-first-order reversible reaction.
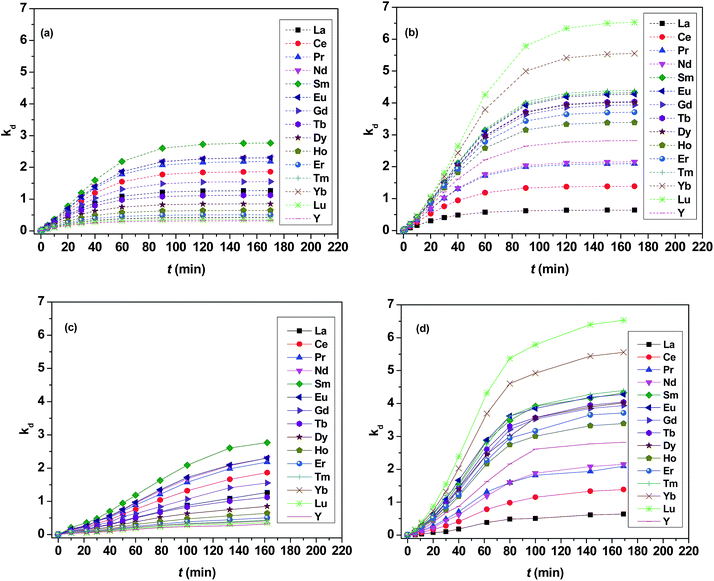 |
| | Fig. 3 Plots of distribution ratio versus time for the extraction of REEs. (a) and (b) are the predicted kd value by theoretical calculations. (a) V = 90 cm3, A = 12 cm2, Cini = 0.002 mol L−1, δ = 0.00179 cm, 300 rpm, T = 298 K. Set kd values of La–Lu plus Y after extraction equilibrium equal to 1.27, 1.86, 2.18, 2.30, 2.77, 2.30, 1.55, 1.12, 0.849, 0.641, 0.505, 0.430, 0.415, 0.358, 0.315, respectively. (b) V = 90 cm3, A = 12 cm2, Cini = 0.002 mol L−1, δ = 0.00179 cm, 300 rpm, T = 298 K. Set kd values of La–Lu plus Y after extraction equilibrium equal to 0.640, 1.39, 2.10, 2.15, 4.33, 4.28, 3.94, 4.04, 4.02, 3.39, 3.71, 4.40, 5.56, 6.53, 2.82, respectively. Organic phase: (c) 0.20 mol L−1 HPOAA in n-heptane with saponification degree of 30%. (d) 0.20 mol L−1 [N1888][POAA] in n-heptane without saponification. Aqueous phase: ∑RECl3 = 0.030 mol L−1 (0.0020 mol L−1 each. RE = La–Lu plus Y except Pm), pH = 4.5. O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :A = 90 mL:90 mL, A = 12 cm2, 300 rpm, T = 298 K. :A = 90 mL:90 mL, A = 12 cm2, 300 rpm, T = 298 K. | |
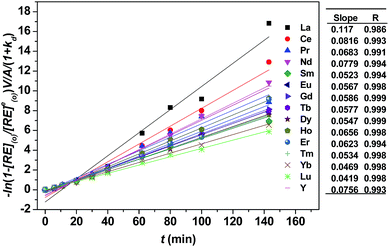 |
| | Fig. 4 Plots of −ln(1 − [RE](o)/[RE](o)e)V/A/(1 + kd) vs. time in [N1888][POAA] system. Organic phase: 0.20 mol L−1 [N1888][POAA] in n-heptane. Aqueous phase: ∑RECl3 = 0.030 mol L−1 (0.0020 mol L−1 each), pH = 4.5, O:A = 90 mL:90 mL, A = 12 cm2, 300 rpm, T = 298 K. | |
Next, the extraction rate in the HPOAA and [N1888][POAA] system have been deduced and the results are presented in Table 2. The literature value of the extraction rates with constant interfacial area and rapid mixing using HDEHP as extractant are also listed in Table 2. Although [N1888][POAA] synthesized from HPOAA and [N1888]Cl, there were large difference between [N1888][POAA] extraction system and HPOAA system. The position of yttrium lies without the lanthanide series in the HPOAA system and the extraction rate order of HPOAA followed the sequence, Y < Lu < Yb < Tm < Er < Ho < Dy < Tb < La < Gd < Ce < Pr < Eu < Nd < Sm. When it was developed to the [N1888][POAA], the extraction rate order follows, La < Ce < Pr < Nd < Y < Ho < Er < Gd < Dy < Tb < Eu < Tm < Sm < Yb < Lu.
Table 2 The difference of the extraction rate between HPOAA system and [N1888][POAA] system
| RE3+ |
kao (mm s−1) |
HDEHP ref. 25 |
Δkao = b/a |
| (a) HPOAA |
(b) [N1888][POAA] |
| La |
1.18 × 10−2 |
1.25 × 10−2 |
2.1 × 10−1 |
1.05 |
| Ce |
1.70 × 10−2 |
1.89 × 10−2 |
1.6 × 10−1 |
1.11 |
| Pr |
1.95 × 10−2 |
2.39 × 10−2 |
9.0 × 10−2 |
1.22 |
| Nd |
1.98 × 10−2 |
2.80 × 10−2 |
3.7 × 10−2 |
1.42 |
| Sm |
2.53 × 10−2 |
3.77 × 10−2 |
1.5 × 10−2 |
1.49 |
| Eu |
2.06 × 10−2 |
4.04 × 10−2 |
1.2 × 10−2 |
1.96 |
| Gd |
1.58 × 10−2 |
4.13 × 10−2 |
1.2 × 10−2 |
2.61 |
| Tb |
1.29 × 10−2 |
3.89 × 10−2 |
7.8 × 10−3 |
3.02 |
| Dy |
1.02 × 10−2 |
3.82 × 10−2 |
7.9 × 10−3 |
3.74 |
| Ho |
8.12 × 10−3 |
3.71 × 10−2 |
1.0 × 10−2 |
4.57 |
| Er |
6.80 × 10−3 |
3.86 × 10−2 |
1.6 × 10−2 |
5.67 |
| Tm |
6.38 × 10−3 |
4.64 × 10−2 |
3.4 × 10−2 |
7.27 |
| Yb |
5.90 × 10−3 |
4.34 × 10−2 |
7.2 × 10−2 |
7.37 |
| Lu |
5.56 × 10−3 |
4.57 × 10−2 |
9.9 × 10−2 |
8.21 |
| Y |
4.21 × 10−3 |
3.55 × 10−2 |
1.0 × 10−1 |
8.44 |
Comparing with HPOAA extraction system, the forward extraction rates (kao, mm s−1) of heavy REEs in [N1888][POAA] extraction system increased significantly. It is interesting to note that the extraction rate of yttrium raised 8.44 times to the original extraction system. In other word, the extraction of heavy REEs were quite effective using the current in [N1888][POAA] extraction system. It is reported that when the acidic extractants were prepared as BILs, their extractabilities could be increased.39 Thus, the kinetic results were consistent with the thermodynamic reaction data.
3.2 Dependence of the extraction rate on stirring speed
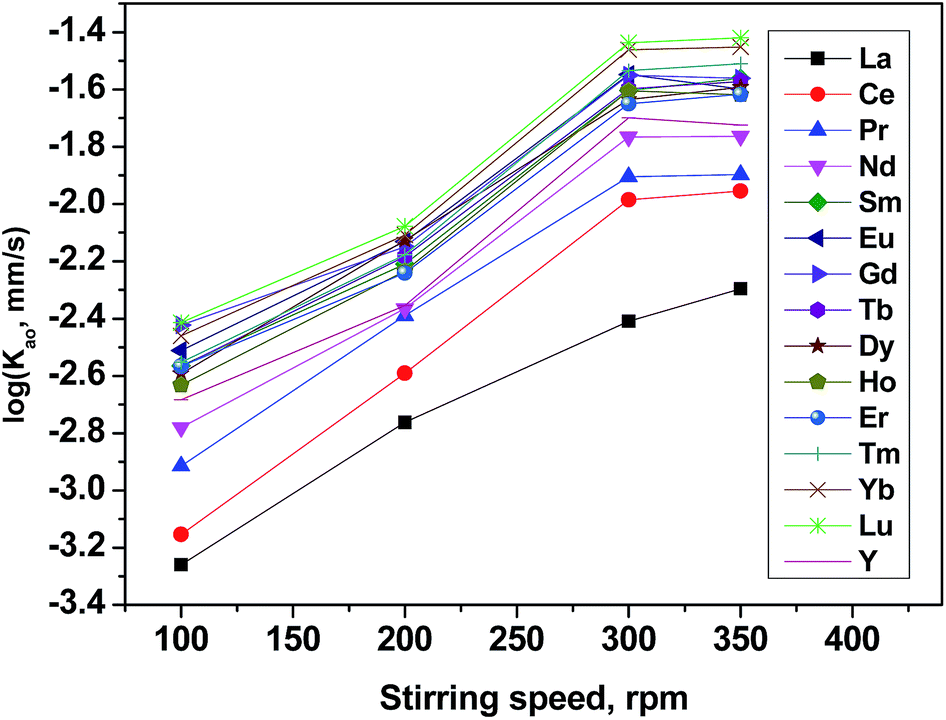
It is known that, the mass transfer rate of metal cations in liquid–liquid solvent extraction systems was a function of both the diffusion of the involved species and the kinetics of the chemical reaction in the two liquid phases. Therefore, diffusion-controlled and chemical reaction-controlled regimes were two representative types of extraction regimes.
The effect of the extraction rate on the stirring rate was adopted to identify the reaction regime of the current extraction system and the results are shown in Fig. 5. It is found that as the mixing speed increases, the extraction rates reached asymptotes from 100 to 300 rpm but was nearly constant in the range of mixing speed exceeds 300 rpm. When the stirring speed exceeded 350 rpm, it was hard to maintain the quiescent interface. The independence of extraction rate from the stirring speed suggests that with increased stirring rate and average velocity (U), the diffusion film (δ) became thinner and the diffusion resistance became smaller. Detailed discussion on the effect of stirring speed on δ and U are in ESI (Fig. S1†). Beyond a distinct stirring rate, the diffusion resistance may be ignored and only the chemical reaction needs to be considered. Therefore, other kinetic experiments were measured at 300 rpm to maintain the same hydrodynamic conditions in this work.
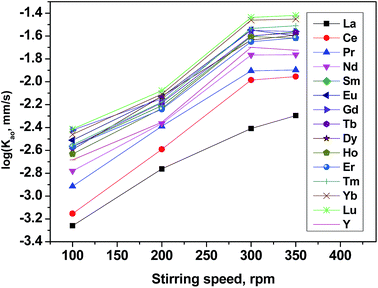 |
| | Fig. 5 Effect of stirring speed on extraction rate of mixed REEs. Organic phase: 0.20 mol L−1 [N1888][POAA] in n-heptane. Aqueous phase: ∑RECl3 = 0.030 mol L−1 (0.0020 mol L−1 each), pH = 4.5, O:A = 90 mL:90 mL, A = 12 cm2, T = 298 K. | |
Follow-up studies have shown that, the stirring speed did not affect the separation factors of REEs. The extraction order followed the same sequence in the [N1888][POAA] system at different stirring speed.
3.3 Dependence of the extraction rate on temperature
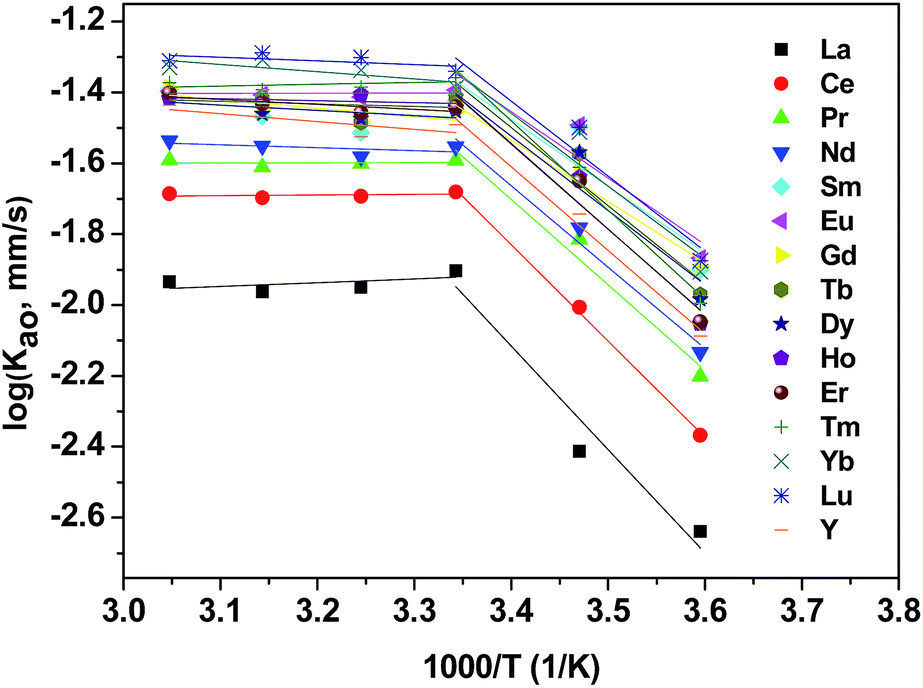
In the most general case, the extraction rate increases with the increasing temperature. Effect of temperature on extraction rate of mixed rare earths have been studied and results are shown in Fig. 6. Present study demonstrates increased sharply in the extraction of REEs with an increase at temperatures lower than 298 K and slowly at temperatures higher than 298 K.
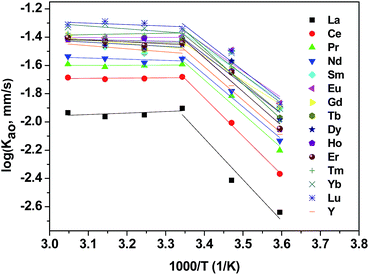 |
| | Fig. 6 Effect of temperature on extraction rate of mixed REEs. Organic phase: 0.20 mol L−1 [N1888][POAA] in n-heptane. Aqueous phase: ∑RECl3 = 0.030 mol L−1 (0.0020 mol L−1 each). pH = 4.5. O:A = 90 mL:90 mL, A = 12 cm2, 300 rpm. | |
The activation energy, Ea, was calculated according to the Arrhenius equation and the results are listed in Table 3.
| |
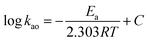 | (8) |
where
C is known as the pre-exponential factor.
Table 3 The activation energy calculated in the [N1888][POAA] system
| RE3+ |
Ea, kJ mol−1 |
| <298 K |
>298 K |
| La |
58.1 |
1.57 |
| Ce |
54.5 |
0.78 |
| Pr |
48.5 |
0.95 |
| Nd |
46.2 |
4.38 |
| Sm |
38.1 |
11.9 |
| Eu |
37.8 |
1.52 |
| Gd |
34.7 |
9.31 |
| Tb |
44.5 |
7.11 |
| Dy |
42.3 |
6.33 |
| Ho |
48.6 |
−1.07 |
| Er |
48.3 |
5.32 |
| Tm |
50.6 |
1.15 |
| Yb |
41.4 |
0.76 |
| Lu |
42.6 |
−0.88 |
| Y |
47.4 |
8.59 |
It is reported that a process was characterized as diffusion controlled when Ea was <20 kJ mol−1 or as chemical reaction controlled when Ea was >42 kJ mol−1.40 Correspondingly, it suggests that a chemical reaction controlled regime when temperatures were lower than 298 K and a diffusion controlled regime when temperatures exceeded 298 K.
The temperature does affects the separation factors of REEs. As shown in Table 4, the separation factor of heavy REEs of Tm, Yb and Lu in the [N1888][POAA] system increased 16.2%, 29.5% and 34.6% when the temperature increased from 278 K to 328 K, respectively. When it comes to light REEs, there are different rate decrease of the separation factor of La, Ce, Pr, Nd, Sm, Eu, Gd and Tb. Similar results were reported that when the temperature increased from 278 K to 333 K, the separation factor of Tm/Yb and Yb/Lu in the P507 and C272 extraction system increased form 1.13 and 1.18 to 2.31 and 1.68, respectively.41 In combination with Fig. 6, it might be explained that although the absolute extraction rates of all REEs increase with the increasing temperature, the absolute extraction rates of heavy REEs increases higher than that of light REEs which result in the growing gaps of separation factors between heavy REEs and light REEs.
Table 4 The separation factor calculated at 278 K, 288 K, 298 K, 308 K, 318 K and 328 K in the [N1888][POAA] system
| βLn/Y |
278 K |
288 K |
298 K |
308 K |
318 K |
328 K |
| La |
0.29 |
0.28 |
0.23 |
0.23 |
0.21 |
0.21 |
| Ce |
0.53 |
0.61 |
0.49 |
0.54 |
0.48 |
0.47 |
| Pr |
0.78 |
0.90 |
0.74 |
0.71 |
0.77 |
0.74 |
| Nd |
0.90 |
0.94 |
0.76 |
0.83 |
0.83 |
0.78 |
| Sm |
1.82 |
1.87 |
1.53 |
1.61 |
1.54 |
1.39 |
| Eu |
1.95 |
1.99 |
1.52 |
1.67 |
1.56 |
1.37 |
| Gd |
1.49 |
1.64 |
1.40 |
1.32 |
1.23 |
1.14 |
| Tb |
1.66 |
1.71 |
1.43 |
1.48 |
1.53 |
1.43 |
| Dy |
1.64 |
1.61 |
1.43 |
1.65 |
1.58 |
1.61 |
| Ho |
1.37 |
1.41 |
1.20 |
1.38 |
1.49 |
1.39 |
| Er |
1.45 |
1.41 |
1.32 |
1.49 |
1.55 |
1.47 |
| Tm |
1.66 |
1.59 |
1.55 |
1.82 |
1.96 |
1.93 |
| Yb |
2.12 |
2.12 |
2.09 |
2.43 |
2.68 |
2.74 |
| Lu |
2.28 |
2.33 |
2.32 |
2.67 |
2.87 |
3.07 |
3.4 Dependence of the extraction rate on specific interfacial area
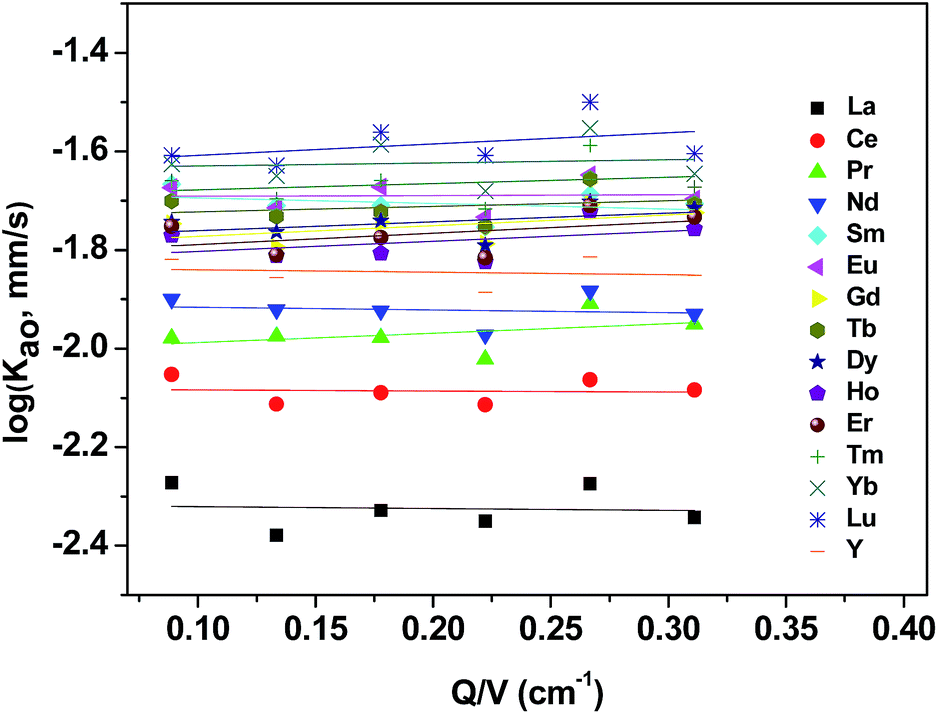
To examine the dependence of the extraction rate, mass transfer rates were obtained as function of the specific interfacial area (A/V) at a constant stirring speed (300 rpm). As shown in Fig. 7, the extraction rate was independent of the specific interfacial area, which indicated that reactions occurring in the bulk of the phases rather than rate determining.
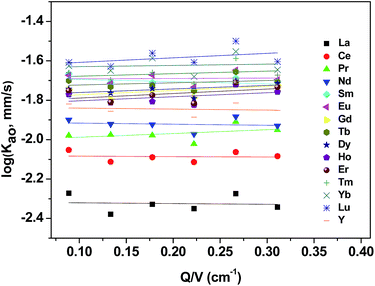 |
| | Fig. 7 Effect of specific interfacial area on extraction rate of mixed REEs. Organic phase: 0.20 mol L−1 [N1888][POAA] in n-heptane. Aqueous phase: ∑RECl3 = 0.030 mol L−1 (0.0020 mol L−1 each), pH = 4.5, O:A = 90 mL:90 mL, A = 8–28 cm2, 300 rpm, T = 298 K. | |
3.5 Extraction rate equation of REEs
Based on the mechanism of neutral extraction combined with ion association in the bifunctional ionic liquid system,17 the extraction equation of REEs with [N1888][POAA] can be presented as follows:| | |
RECl3 + [N1888][POAA](o) ⇄ [N1888]Cl·REPOAACl2
| (9) |
Accordingly, the extraction rate equation of REEs can be proposed as:
| |
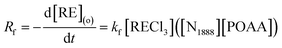 | (10) |
where,
kf is the forward extraction constant.
To simplify the calculation model, the reverse extraction rate can be ignored when the concentration of extractant in the organic phase is much higher than the concentration of metal ions in the aqueous phase. Combining eqn (2) and (10), one can write the following equation:
| |
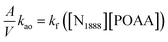 | (11) |
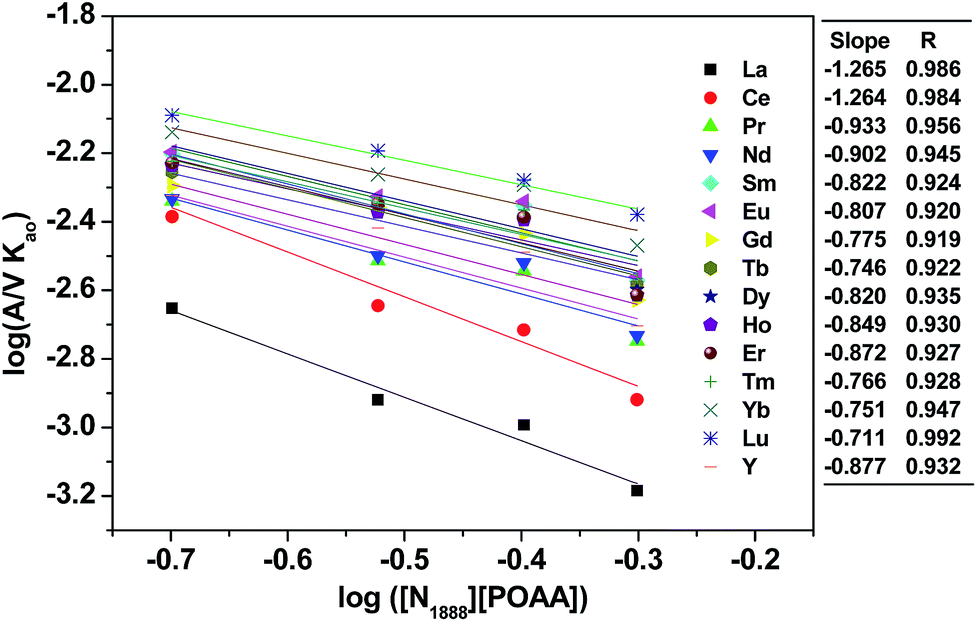
By investigating the effect of [N1888][POAA] concentration on extraction rate of mixed REEs (Fig. 8), kf of REEs were obtained and the results are shown in Table 5. Therefore, the average extraction rate equation of REEs can be expressed as: −d[RE](o)/dt = 0.141[RECl3]([N1888][POAA]).
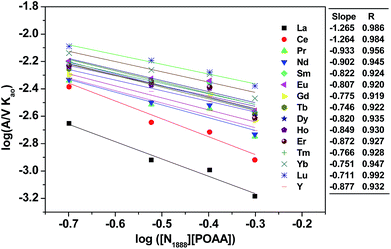 |
| | Fig. 8 Effect of [N1888][POAA] concentration on extraction rate of mixed REEs. Organic phase: 0.20–0.50 mol L−1 [N1888][POAA] in n-heptane. Aqueous phase: ∑RECl3 = 0.030 mol L−1 (0.0020 mol L−1 each), pH = 4.5, O:A = 90 mL:90 mL, A = 12 cm2, 300 rpm, T = 298 K. | |
Table 5 The forward extraction constant of REEs
| |
La |
Ce |
Pr |
Nd |
Sm |
Eu |
Gd |
Tb |
Dy |
Ho |
Er |
Tm |
Yb |
Lu |
Y |
Average |
| kf |
0.054 |
0.054 |
0.117 |
0.125 |
0.151 |
0.156 |
0.168 |
0.179 |
0.151 |
0.142 |
0.134 |
0.171 |
0.178 |
0.194 |
0.133 |
0.141 |
4 Conclusions
The extraction kinetics of mixed REEs with bifunctional ionic liquid [N1888][POAA] using a constant interfacial area cell were reported and characterized. The experimental results show that practical kinetic values obtained from kinetic experiments fit the data predicted by theoretical calculations very well. The relative errors of experimental values and theoretical values are less than 43% and 25% in the HPOAA and [N1888][POAA] extraction system after 80 minutes, respectively. Effect of stirring speed, temperature specific interfacial area and extractant concentration on the extraction rate of REEs were studied. 300 rpm was suggested to ensure the extraction regimes were chemical reaction-controlled rather than diffusion-controlled when the temperatures were lower than 298 K. The extraction rate was independent of the specific interfacial area, which indicated that reactions occurring in the bulk of the phases. The average extraction rate equation of REEs have also been obtained using a constant interfacial area cell as: −d[RE](o)/dt = 0.141 [RECl3]([N1888][POAA]). Kinetic data obtained above will be used to recognize the thermodynamic mechanism and optimize extraction processes.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by National Natural Science Foundation of China (21571179), Science and Technology Major Projects of Fujian Province (2015HZ0001-3), Natural Science Foundation of Fujian Province (2016J0102) and Science and Technology Service Network Initiative.
Notes and references
- A. Rout, S. Wellens and K. Binnemans, RSC Adv., 2014, 4, 5753–5758 RSC.
- Q. Su, Presented in part at the Proceedings of the 9th China rare earth Entrepreneur Association Conference, 2008 Search PubMed.
- D. Q. Li, Prog. Chem., 1995, 7, 209–213 CAS.
- C. H. Yan, J. T. Jia, C. S. Liao, S. Wu and G. X. Xu, Tsinghua Sci. Technol., 2006, 11, 241–247 CrossRef CAS.
- J. C. Xiao, Presented in part at the Proceedings of the Ninth National Symposium on Inorganic Chemistry of China Chemical Society, Nanchang, China, 2015-07-25, 2015 Search PubMed.
- X. Q. Sun, B. Peng, J. Chen, D. Q. Li and F. Luo, Talanta, 2008, 74, 1071–1074 CrossRef CAS PubMed.
- J. G. Huddleston, A. E. Visser, W. M. Reichert, H. D. Willauer, G. A. Broker and R. D. Rogers, Green Chem., 2001, 3, 156–164 RSC.
- X. Q. Sun and K. E. Waters, ACS Sustainable Chem. Eng., 2014, 2, 2758–2764 CrossRef CAS.
- A. Rout, K. A. Venkatesan, T. G. Srinivasan and P. V. Rao, Sep. Purif. Technol., 2012, 95, 26–31 CrossRef CAS.
- X. Q. Sun, Y. Ji, L. Zhang, J. Chen and D. Q. Li, J. Hazard. Mater., 2010, 182, 447–452 CrossRef CAS PubMed.
- Y. H. Chen, H. Y. Wang, Y. C. Pei, J. Ren and J. J. Wang, ACS Sustainable Chem. Eng., 2015, 3, 3167–3174 CrossRef CAS.
- W. Wang, H. L. Yang, H. M. Cui, D. L. Zhang, Y. Liu and J. Chen, Ind. Eng. Chem. Res., 2011, 50, 7534–7541 CrossRef CAS.
- H. L. Yang, J. Chen, W. Wang, H. M. Cui, W. G. Liu and Y. Liu, Sci. China: Chem., 2016, 59, 532–537 CrossRef CAS.
- W. Z. Ye, in Advances in Rare Earth New Materials and New Processes, Science Press, Beijing, 1998, pp. 3–10 Search PubMed.
- Y. G. Wang, S. T. Yue, D. Q. Li, M. J. Jin and C. Z. Li, Solvent Extr. Ion Exch., 2002, 20, 701–716 CrossRef CAS.
- Y. L. Wang, H. Y. Zhou, Y. B. Wang, F. J. Li and X. Q. Sun, Sep. Purif. Technol., 2017, 184, 280–287 CrossRef CAS.
- Y. L. Wang, C. Huang, F. J. Li, Y. M. Dong, Z. Y. Zhao and X. Q. Sun, Sep. Purif. Technol., 2016, 162, 106–113 CrossRef CAS.
- M. A. Jeannot and F. F. Cantwell, Anal. Chem., 1997, 69, 235–239 CrossRef CAS.
- N. S. Awwad, Chem. Eng. Process., 2004, 43, 1503–1509 CrossRef CAS.
- F. Kubota, M. Goto and F. Nakashio, Sep. Sci. Technol., 1995, 30, 777–792 CrossRef CAS.
- S. H. Lin and R. S. Juang, Ind. Eng. Chem. Res., 2002, 41, 853–861 CrossRef CAS.
- C. Ungureanu, L. Marchal, A. A. Chirvase and A. Foucault, Bioresour. Technol., 2013, 132, 406–409 CrossRef CAS PubMed.
- J. J. Liu, Y. L. Wang and D. Q. Li, J. Chem. Technol. Biotechnol., 2007, 82, 949–955 CrossRef CAS.
- D. Datta and S. Kumar, Ind. Eng. Chem. Res., 2013, 52, 14680–14686 CrossRef CAS.
- K. P. Nichols, R. R. Pompano, L. Li, A. V. Gelis and R. F. Ismagilov, J. Am. Chem. Soc., 2011, 133, 15721–15729 CrossRef CAS PubMed.
- S. H. Yin, S. W. Li, J. H. Peng and L. B. Zhang, RSC Adv., 2015, 5, 48659–48664 RSC.
- C. Q. Chen and T. Zhu, Solvent Extr. Ion Exch., 1994, 12, 1013–1032 CrossRef CAS.
- J. Lu, G. X. Ma, D. Q. Li and M. S. Ni, Chin. J. Appl. Chem., 1998, 15, 43–46 CAS.
- Y. L. Wang, Y. L. Li, D. Q. Li and W. P. Liao, Hydrometallurgy, 2013, 140, 66–70 CrossRef CAS.
- S. T. Yue, W. P. Liao, D. Q. Li and Q. Su, Chin. J. Chem., 2002, 20, 545–549 CrossRef CAS.
- Y. G. Wang, S. T. Yue and D. Q. Li, Solvent Extr. Ion Exch., 2002, 20, 345–358 CrossRef CAS.
- W. P. Liao, G. H. Yu, S. T. Yue and D. Q. Li, Talanta, 2002, 56, 613–618 CrossRef CAS PubMed.
- B. Wei, Master of Science, University of Jinan, 2005.
- H. L. Yang, J. Chen, W. Wang, H. M. Cui, D. L. Zhang and Y. Liu, Chin. J. Chem. Eng., 2014, 22, 1174–1177 CrossRef CAS.
- L. Chen, Doctor of Natural Science, The University of Chinese Academy of Sciences, 2016.
- F. KneißI, A. Aeist and W. Nitsch, Solvent Extr. Ion Exch., 1999, 17, 475–493 CrossRef.
- P. R. Danesi, R. Chiarizia and C. F. Coleman, CRC Crit. Rev. Anal. Chem., 1980, 10, 1–126 CrossRef.
- Z. Zheng, J. Lu, D. Q. Li and G. X. Ma, Chem. Eng. Sci., 1998, 53, 2327–2333 CrossRef CAS.
- H. L. Yang, W. Wang, H. M. Cui, D. L. Zhang, Y. Liu and J. Chen, J. Chem. Technol. Biotechnol., 2012, 87, 198–205 CrossRef CAS.
- J. F. Yu and C. Ji, Chem. Res. Chin. Univ., 1992, 13, 224–226 CAS.
- W. H. Yang, S. H. Zhang, S. Wu, H. P. Li and Q. Z. Fan, China Pat., CN201110408624.3, 09-12-2011.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07851j |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab
*ab