
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural features of FAP174, a MYCBP-1 orthologue from Chlamydomonas reinhardtii, revealed by computational and experimental analyses†
M. Yogeshaa,
Venkatramanan G. Raob,
Elvis A. F. Martis c,
Evans C. Coutinhoc,
Holger Gohlked,
Santhosh Chidangila,
Prabhakar M. Dongree and
Jacinta S. D'Souza*ab
c,
Evans C. Coutinhoc,
Holger Gohlked,
Santhosh Chidangila,
Prabhakar M. Dongree and
Jacinta S. D'Souza*ab
aDepartment of Atomic & Molecular Physics, Manipal University, Manipal 576104, India
bUM-DAE Centre for Excellence in Basic Sciences, Kalina Campus, Santacruz (E), Mumbai-400098, India. E-mail: jacinta@cbs.ac.in; Fax: +91-22-26524983; Tel: +91-22-26524978
cMolecular Simulations Group, Department of Pharmaceutical Chemistry, Bombay College of Pharmacy, Kalina, Santacruz (E), Mumbai 400098, India
dMathematisch-Naturwissenschaftliche Fakultät, Institut für Pharmazeutische und Medizinische Chemie, Heinrich-Heine-Universität Düsseldorf, 40225 Düsseldorf, Germany
eDepartment of Biophysics, University of Mumbai, Kalina Campus, Santacruz (E), Mumbai-400098, India
First published on 7th November 2017
Abstract
The dimers of regulatory sub-units of protein kinase A (RIα, RIIα, RIβ, and RIIβ isoforms) and DPY-30 interact with their respective A-Kinase Anchoring Proteins (AKAPs) via their Dimerization and Docking domains (D/D). The mammalian MYCBP-1 protein, which stimulates the E-box-dependent transcriptional activity of the c-Myc protein, also interacts with AKAPs present in various sub-cellular locales. We have earlier shown that the Flagellar Associated Protein 174 (FAP174) from Chlamydomonas reinhardtii is an orthologue of human MYCBP-1 and binds to AKAP240. Here, we set out to identify the D/D domain of FAP174 and the amino acid residues involved in oligomerization. By means of electrophoresis of recombinant proteins with an increasing concentration of a reducing agent (DTT), MALDI-TOF, FT-IR, and micro-Raman spectroscopy, we show that the D/D domain resides in and around the C46 residue. Using in silico methods, we infer that the neighboring amino acids of C46, viz. A39, L40, Y42, and L47, might also be involved in dimerization. The secondary structure of FAP174 was investigated using computer-assisted prediction, CD, micro-Raman spectroscopy, and FT-IR. Accordingly, FAP174 is made of four α-helices. Helix B contains two VLV stretches at positions 21–23 and 25–27. Mutating these VLV stretches to alanines followed by native gel electrophoresis of the respective recombinant proteins indicated their role in oligomerization. The results imply that FAP174, unlike most of the other AKAP-interacting proteins, harbors two different domains, one for dimerization and the other for oligomerization.
1. Introduction
cAMP is a second messenger that mediates most of its effects through cAMP-dependent protein kinase (PKA) and controls a broad spectrum of cellular events1 through crucial intracellular organization involving the A-Kinase Anchoring Proteins (AKAPs).2 AKAPs can bind to multiple signaling proteins forming modules or units that provide the ‘anchor’ or ‘scaffold’ for the integration of different signaling pathways. To understand this complex signaling assemblies, it is crucial to identify and isolate these units, the interacting partners, and their interacting domains.A less known protein in one such AKAP complex is MYCBP-1 or Associate of Myc-1 (AMY-1). Originally identified as a c-Myc-binding protein, it stimulates the E-box-dependent transcriptional activity of the c-Myc protein, which plays key roles in several cellular processes. After expression, MYCBP-1 translocates from the cytoplasm to the nucleus,3 acting there as a co-transcription factor.4 For regulation, MYCBP-1 forms a ternary complex with S-AKAP84/AKAP149 and the regulatory subunit (RII) of PKA5–7 in the mitochondria. In addition, MYCBP-1 independently binds to the RII-Binding Domain (RII-BD) of AKAP149 and AKAP95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6,7 and interacts with various other AKAPs.5–8 All known AKAPs harbor a RII-BD that comprises an amphipathic helix.9 However, the mechanistic details of these interactions have remained elusive. Meanwhile, there has been an immense progress on the structure elucidation of PKA and AKAPs; and, with no understanding of the biochemical features (including dimerization and/or oligomerization) and the structure of MYCBP-1 and its orthologues such as FAP174, the biological function of this signaling unit has remained unanswered.
6,7 and interacts with various other AKAPs.5–8 All known AKAPs harbor a RII-BD that comprises an amphipathic helix.9 However, the mechanistic details of these interactions have remained elusive. Meanwhile, there has been an immense progress on the structure elucidation of PKA and AKAPs; and, with no understanding of the biochemical features (including dimerization and/or oligomerization) and the structure of MYCBP-1 and its orthologues such as FAP174, the biological function of this signaling unit has remained unanswered.
Interestingly, MYCBP-1 was also found in the human ciliary proteome (NCBI accession no. ACR55627).10 Subsequently, Flagellar Associated Protein 174 (FAP174), a component of the C. reinhardtii ciliary proteome, was identified as a MYCBP-1 homologue.11 Our experiments revealed that FAP174 binds to AKAP240; both these proteins being localized to the central pair of the axoneme. Additionally, FAP174 was found to be localized to the nucleus and basal body.12 We then set out to search for a D/D domain on FAP174; this domain allows AKAP-binding proteins to interact with the amphipathic helix of all AKAPs as dimers.13 We hypothesized that the only cysteine at position 46 of the FAP174 sequence could contribute to forming a dimer via a disulfide bridge. Furthermore, a multiple sequence alignment of FAP174 with other AKAP-interacting proteins such as RII and DPY-30 revealed two conserved VLV stretches at positions 21–23 and 25–27.12
Building upon these previous results, here, we investigate the role of C46 and the two conserved VLV stretches on the secondary structure and oligomerization properties of FAP174. We did so by applying conventional gel electrophoresis after subjecting wild-type (WT) FAP174 and FAP174 variant proteins (FAP174VLV21-23AAA, FAP174VLV25-27AAA and FAP174C46A) with an increasing concentration of a reducing agent such as DTT, biophysical techniques such as MALDI-TOF, CD, FT-IR, and micro-Raman spectroscopy, as well as ab initio protein structure prediction and molecular dynamics (MD) simulations. Our results imply that FAP174, unlike most of the other AKAP-interacting proteins, harbors two different domains, one for dimerization and the other for oligomerization.
2. Materials and methods
The biochemical reagents and media components were obtained from Amresco (USA), SRL (India), Merck Millipore (India) and Qiagen (Germany).2.1 Mass determination of proteins
The FAP174 WT and mutant genes were cloned in pET28a vector and over-expressed in E. coli BL21(DE3) cells. The 6XHis-tagged proteins were individually purified using a NiNTA-agarose column.12 The purity of the proteins was determined by electrophoresis of the purified fractions on a 15% SDS-PAGE gel followed by the standard silver staining method. The masses of the samples were determined using the Mass Spectrometry facility at the Tata Institute of Fundamental Research (Mumbai, India). Briefly, ∼3000 ng μl−1 of the purified proteins were mixed individually with the matrix sinapinic acid and spotted onto the MALDI-TOF plate. The samples were allowed to dry at room temperature. The plate was then loaded on the machine and spectra developed in the linear mode.2.2 Effect of DTT on dimerization of FAP174 WT and variant proteins
For this assay, 10 μg of each protein sample was treated with an increasing concentration (0–250 mM) of DTT followed by incubation at room temperature for 5 minutes. The samples were then treated with Laemmli buffer (without any reducing agent) followed by electrophoresis on a 15% SDS-PAGE gel. The polypeptides were evident after Coomassie Blue R-250 staining.2.3 Biophysical characterization of FAP174 and its variants
The secondary structure of FAP174 WT and three variants were determined using a JASCO CD Polarimeter J-810. The spectra were measured using a cuvette with 1 mm path length. The CD spectra of these proteins were measured from 190–260 nm with a band width of 1 nm and a speed of 50 nm s−1. The samples, at a concentration of 0.1 mg ml−1, were individually suspended in 10 mM phosphate buffer, pH 7.0 and the chamber was continuously flushed with N2 gas. The resultant spectral values are expressed in units of delta epsilon (Δε). The secondary structure of FAP174 WT and variants were analyzed by deconvolution of the CD spectrum using the online BeStSel analysis program14 applied at the wavelength range of 190–250 nm.FT-IR spectra of the lyophilized proteins were recorded at room temperature by mixing with KBr and pressing to form discs. The FTIR measurement was carried out using a JASCO 6300 FT-IR spectrometer equipped with a DLATGS detector and a standard source. Two hundred scans with 2 cm−1 spectral resolution were obtained. Approximately, 2 mg of pure lyophilized protein powder was used to obtain the Raman spectra using a Horiba Jobin-Yvon Spectrometer with an inverted microscope (Nikon Eclipse Ti-U, Japan). A diode laser (Starbright Diode Laser, Torsana Laser Tech, Denmark) with wavelength of 785 nm and 47 mW power was used to illuminate the protein samples sandwiched between the quartz coverslip and the borosilicate glass slide. Symphony CCD detector cooled under liquid nitrogen was utilized to collect the Raman scattered radiation from the sample.
2.4 Secondary structure prediction
The predictions of the secondary structure and the helical content of FAP174 WT and three variants were studied using five well-known methods15–22 available as online webservers. Table 1 enlists the webservers and their methodologies used to predict the secondary structures of proteins. Since predictions are highly sensitive to the method used, results from all five servers were used to draw a consensus picture of the secondary structure.| Webserver | Method |
|---|---|
| LOMETS implemented in I-TASSER21 | Local meta-threading approach |
| PSIPRED17, 18 | Two-stage neural network based on protein specific scoring matrices generated by PSI-BLAST |
| QUARK22 | Neural networks |
| GOR V20 | Combination of information theory, Bayesian statistics and evolutionary information |
| JPRED15, 16 | Jnet algorithm |
2.5 Tertiary structure prediction
The tertiary structure of FAP174 WT and its three variants were predicted by the ab initio method implemented in the QUARK program available as an online server22 and by the threading method implemented in the I-TASSER webserver.19 QUARK requires no templates for structure prediction, as it assembles the protein structure from models of small fragments (1–20 residues long) by replica-exchange Monte Carlo simulation guided by an atomic-level knowledge-based force field. I-TASSER retrieves templates from the Protein Data Bank and threads the query sequence over the folds on the template proteins. For the unaligned regions in the query sequence, I-TASSER employs the QUARK methodology to predict the folds.Our previous studies showed that FAP174 WT binds to AKAPs12 akin to DPY-30 (PDB id 3G3623) and RIIα D/D (PDB id 2IZX24) proteins. To determine the relationship between the structures of FAP174 WT, DPY-30 and RIIα D/D, all predicted 3D-models of FAP174 WT were superimposed onto the crystal structures of DPY-30 and RIIαD/D proteins. The superimposition (protein structure alignment) was performed with Maestro – the graphical user interface in Schrödinger Suite 2015-1. The QUARK model 3, which showed the lowest all atom RMSD with respect to DPY-30 and RIIα D/D (see results and discussion) was selected, and from it the 3D models for the variants (FAP174VLV21-23AAA, FAP174VLV25-27AAA and FAP174C46A) were generated by replacing the sidechain atoms of valine/leucine/cysteine by CH3 of alanine.
2.6 Refinement of the 3D models by molecular dynamics simulations
The AMBER14 suite25 was used to perform molecular dynamics (MD) simulations. The Amber force field 14 (ff14SB)26,27 was employed to represent the proteins. The TIP3P water model28 was used to solvate the proteins in a periodic truncated octahedral box. The dimensions of the solvent box were extended 12.0 Å beyond the solute atoms. The system was relaxed using three cycles of minimization of 50000 steps each, wherein the first 10000 steps adopted the steepest descent minimizer and the remaining 40000 steps used the conjugate gradient minimizer, with the cut-off for non-bonded interactions set to 8.0 Å. The first cycle of minimization was performed with a restraining harmonic potential and a force constant of 25 kcal (mol−1 Å−2) on the solute atoms, the force constant was lowered to 5 kcal (mol−1 Å−2) in the next cycle, and the last cycle was performed without any restraining forces. The system was heated, under the NVT ensemble, from 0 K to 300 K over a period of 1 ns with a restraining harmonic potential and a force constant of 5 kcal (mol−1 Å−2) on the solute atoms by Langevin dynamics and a collisional frequency of 2.0 ps−1. After heating, the system was equilibrated, under the NVT ensemble, for six cycles of 1 ns each, where the force constants of the restraining potentials on the solute atoms were gradually reduced from 5.0 to 0.0 kcal (mol−1 Å−2). The next step employed the NPT ensemble for equilibration for another six cycles (1 ns each), wherein the force constants of the restraining potentials on the solute atoms was gradually lowered from 5.0 to 0.0 kcal (mol−1 Å−2). In both equilibration phases (NVT and NPT), the cut-off for the non-bonded interactions was set to 9.0 Å.
The production MD runs, in triplicate, for all the systems were performed at 300 K and 1 bar for a timescale of 1 μs (total of 3 μs for each system), the integration time step being 2 fs. Long-range electrostatic interactions were evaluated with the particle mesh Ewald method,29 and the cut-off for non-bonded interactions during the production phase was fixed at 10.0 Å. All bonds involving hydrogen atoms were restrained using the SHAKE algorithm.30 The coordinates were sampled every 50 ps and then analyzed with respect to mass-weighted backbone-RMSD and all-atom RMSF (root mean square fluctuation) along with the radius of gyration, using the CPPTRAJv15.0031 tool available in AmberTools15.
2.7 Validation of 3D models
The models predicted by QUARK were validated using the program ANOLEA.32 It performs an assessment of protein structures by several methods. The quality of the models before and after MD refinement were assessed using the QMEAN6 score.33 This is a consensus score built from a linear combination of six terms (Cβ interaction energy, all-atom pairwise energy, solvation energy, torsion angle energy, secondary structure agreement and solvent accessibility agreement). The Z-scores were calculated from high-resolution experimental structures of similar size, which have been resolved by X-ray crystallography. A higher QMEAN6 score, with a positive Z-score, indicates a superior quality of the model. The Cβ interaction energy evaluates long-range interactions at the residue level for the β carbon atoms of the proteins, whereas the all-atom pairwise energy evaluates the long-range interactions for all the atoms. The local backbone geometry was evaluated by the torsion angle potential, which is computed over three consecutive amino acids. The burial of residues was analyzed by the solvation energy. The secondary structure agreement term relates the predicted secondary structure of the target sequence (calculated by PSIPRED)17 with the DSSP predicted secondary structure of the model. Finally, the solvent accessibility agreement term connects the predicted relative solvent accessibility (buried/exposed) from ACCpro34 with the relative solvent accessibility derived from DSSP.353. Results and discussion
3.1 Identification of the oligomerization control motifs and dimerization via the disulfide bond
FAP174 WT harbors only one cysteine at position 46 that could be involved in dimerization through formation of a disulfide bond. When recombinant FAP174 WT protein was purified using NiNTA-agarose with >95% purity and freshly electrophoresed on a denaturing SDS-PAGE gel, two species were observed; a monomer and a dimer at ∼12.5 and ∼25 kDa, respectively (Fig. S1,† lane 1). The same species were observed at 12.59 kDa and 25.12 kDa when analyzed using MALDI-TOF (Fig. S2†). We assume that this results from the presence of the reducing agent 2-mercaptoethanol that does not completely reduce the protein to its monomeric state. In fact, under non-denaturing and non-reducing conditions, the protein forms higher order oligomers that are poorly resolved by electrophoresis on a 10% native gel (Fig. S3,† lane 1). To understand how certain residues in the protein affect the dimerization/oligomerization status, three FAP174 variants were generated, including a C46A mutation as well as alanine mutations of the conserved VLV stretches at position 21–23 and 25–27 (FAP174VLV21-23AAA, FAP174VLV25-27AAA and FAP174C46A). The mutant genes were synthesized, sub-cloned in a pET28a expression vector and over-expressed in E. coli. Individual recombinant variant proteins were then purified using NiNTA-agarose affinity chromatography (Fig. S1†). These samples were prepared in Laemmli buffer containing 50 mM of the reducing agent 2-mercaptoethanol that does not completely reduce the protein. Therefore, both monomers and dimers were evident in all the samples, except in the case of FAP174C46A. Again, these samples, after dialysis, were analyzed for their masses using MALDI-TOF. Their molecular weights were 12.36 kDa and 24.66 kDa for both the VLV variants (FAP174VLV21-23AAA and FAP174VLV25-27AAA). However, for FAP174C46A, only one species at 12.35 kDa was obtained (Fig. S2†). Therefore, under the partly reducing conditions, there exists an equilibrium between the populations of monomers and dimers of FAP174 WT, FAP174VLV21-23AAA, and FAP174VLV25-27AAA, but not FAP174C46A. Hence, these results demonstrate that C46 is the primary cause for dimerization of FAP174. Retaining C46 but removing the VLVs individually leads exclusively to the formation of dimers under non-reducing conditions (Fig. S2†), contributing further evidence to the fact that C46 is the only cause for dimerization.In addition, the cysteine and VLV stretches were observed to have a function in promoting the protein to oligomerize as seen in the WT protein on the native gel (Fig. S3,† lane 1). Further, these oligomers exist as monomers and dimers in the presence of a partly reducing agent and denaturing conditions, where C46 sometimes retains a disulfide bridge (Fig. S1†). When the cysteine is substituted with an alanine, the VLVs alone are unable to promote oligomerization (Fig. S3†), indicating that oligomers form from dimers, rather than monomers. In turn, the presence of cysteine and the absence of either one of the VLVs forces the protein to form dimers (Fig. S3†). This is the first evidence that shows the effect of the VLV residues on the formation of oligomers. We, therefore, suggest that the VLV stretches are the ‘oligomerization control motifs’ of FAP174. As of now, oligomerization and/or dimerization properties of MYCBP-1 and its orthologue FAP174 proteins had not been studied.
To confirm the role of C46 in dimer formation, dithiothreitol (DTT) was used as a reducing agent to cleave the disulfide bridge in the dimer, thus producing a monomer. Upon increasing the molarity of DTT, the concentration of a given protein dimer decreased for FAP147 WT, FAP174VLV21-23AAA and FAP174VLV25-27AAA (Fig. 1A, C and D), requiring ∼50 mM for the complete dissociation of the dimer. However, the FAP174C46A variant did not show any tendency to dimerize even at zero DTT (Fig. 1B), confirming the direct involvement of C46 in the dimerization. Similar experiments using the reducing agent DTT were conducted for RIa, wherein 100 mM reduced the dimer to the monomer form.36
 | ||
| Fig. 1 Effect of increasing concentration of dithiothreitol (DTT) on the dimerization of (A) FAP174 WT, (B) FAP174C46A, (C) FAP174VLV21-23AAA and (D) FAP174VLV25AAA. The numbers above each lane are concentrations of DTT in mM. | ||
Raman, CD, and FTIR spectroscopy aided us to understand the structural changes and dimerization in FAP174 WT and its variants (Fig. 2, 4 and 5). The Raman signatures (Fig. 2B) of the dimeric states in the region 500–550 cm−1 are the peaks at 523 and 546 cm−1 that correspond to the S–S stretch vibrations of the intermolecular disulfide bond, suggesting a trans–gauche-trans geometry37,38 (Fig. 2B). These two peaks are restricted to FAP174 WT, FAP174VLV21-23AAA and FAP174VLV25-27AAA, in accordance with the formation of dimers.
 | ||
| Fig. 2 Raman Spectroscopy of FAP174 and variant proteins. (A) Raman spectra of (a) FAP174 WT, (b) FAP174VLV21-23AAA, (c) FAP174VLV25-27AAA and (d) FAP1s74C46A recorded in the spectral region of 500–1700 cm−1. (B) Micro-Raman spectra of (a) FAP174 WT, (b) FAP174VLV21-23AAA, (c) FAP174VLV25-27AAA and (d) FAP174C46A in the spectral region of 880–1000 cm−1. (C) Micro-Raman spectra of (a) FAP174 WT, (b) FAP174VLV21-23AAA, (c) FAP174VLV25-27AAA and (d) FAP174C46A in the spectral region of 500–1000 cm−1. | ||
In all, FAP174 clearly exhibits a tendency to dimerize. Hence, we propose that it belongs to the families of proteins that share certain PPI motifs that mediate dimerization. The dimerization status of FAP174 WT, and the role of the VLV stretches in promoting oligomerization, need to be probed further to understand the implications in cellular function. As of now, no oligomerization motif has been assigned to MYCBP-1 proteins from any organism; the evidence from this study leads us to conclude that FAP174, a ciliary MYCBP-1-like protein, possesses an oligomerization control motif in terms of the VLV stretches.
3.2 FAP174 and the variants are helical proteins
Molecular modeling was carried out primarily to understand the tertiary structure of FAP174 and its three variants. Our predictions show that the replacement of the VLV regions with alanine (two triple variants), or C46 with alanine, have no significant effect on the secondary structure of the three variants vis-à-vis the WT protein (Fig. 3). Among the five methods used to predict the secondary structure, we observed a high consensus (Fig. S4–S7†), strengthening our confidence in the predictions. We observed a low/medium agreement in the predictions for regions of random coil, more specifically at the interface of the helix and the random coil. These ambiguities are common, as these uncertainties are also observed in experimentally solved structures; consequently, these uncertainties are also reflected in the computational predictions that are based on those experimental data. The predicted helical content for all structures is in the range of 68–70%. | ||
| Fig. 3 Consensus secondary structure of WT FAP 174 and its three variants. Prediction of the secondary structure of FAP174 WT and its variant proteins using multiple prediction tools. | ||
The helical content was validated experimentally using three techniques (CD spectroscopy, FT-IR, and micro-Raman spectroscopy). FT-IR spectroscopy confirmed the predictions (Fig. 4). The FT-IR spectra of FAP174 WT and the variant proteins in the spectral region 1000–4000 cm−1 indicate the presence of the α-helix amide I band at ∼1658 cm−139 and the amide II band near 1540 cm−1, which refers to bending of the N–H groups.40 The band due to Tyr (νC–C mode) is seen at ∼1520 cm−1 and overlaps with the amide II band.41 The micro-Raman spectra of all proteins were recorded in the 500–1800 cm−1 range (Fig. 2A); detailed assignments are listed in the ESI Table S1.† In all spectra, a broad band around 1600–1700 cm−1 is very prominent. The peak near 1650–1660 cm−1 is the amide I band, a signature for the α-helix as previously reported by Spiro and Gaber.37,42 This observation is in accordance with the computational work that shows all FAP174 proteins (WT and the variants) are largely made of α-helices with no signs of β-sheets. The band in the region 1200–1300 cm−1 corresponds to amide III.37,43 A low frequency amide III band (∼1250 cm−1) indicates a lower α-helical content, whereas the high frequency (∼1300 cm−1) band is an indicator of higher α-helical content. A band at 1280 cm−1 for the FAP174 WT and the variant proteins indicate an intermediate α-helical content, in tune with the computational predictions (vide supra). Inspecting the amide I band for all proteins shows that the FAP174VLV21-23AAA and FAP174VLV25-27AAA variants have pronounced peaks at 1655 and 1657 cm−1, in addition to the peak at 1651 cm−1. These additional frequencies at 1655 and 1657 cm−1 arise due to the contribution of ordered α-helix. Thus, compared to FAP174 WT and the FAP174C46A variant, the FAP174VLV21-23AAA and FAP174VLV25-27AAA variants have more ordered α-helical structure. These observations are comparable with BSA containing high amount of alpha helix as secondary structure.37 In contrast, the FAP174 WT and the FAP174C46A variant proteins show a single peak at 1651 cm−1. Besides differences in the preference for oligomerization, this is the second prominent difference between FAP174 WT and the FAP174C46A variant on the one hand and the two VLV variants on the other. Another important band which appears around 930–945 cm−1 in the micro-Raman spectra is due to C–C and C–N stretching and is also ascribed to the α-helical structure of proteins;42,44 all proteins exhibit a band at 934 cm−1.
 | ||
| Fig. 4 Secondary structure of FAP174 and variants. The secondary structures of (A) FAP174 WT, (B) FAP174VLV21-23AAA, (C) FAP174VLV25-27AAA and (D) FAP174C46A were studied using FTIR spectroscopy – expressed in the units of absorption (Abs) in the spectral range of 1000–4000 cm−1. | ||
The Raman spectra of proteins usually display prominent modes corresponding to the aromatic amino acids. The ratio of the intensity of the tyrosine bands at 850 cm−1 and 828 cm−1 (I850/I828), which usually varies between 0.3 and 2.5, can be used as a marker to investigate the H-bonding state of the tyrosine hydroxyl group. In all four proteins, two bands due to Tyr are observed near 848 and 826 cm−1, possibly due to the Fermi resonance splitting. The I850/I828 ratio for all proteins is ∼1.1, indicating that Tyr is exposed and acts both as a donor and acceptor. Of the 93 a.a. in the FAP174 WT, 12 are alanine, FAP174C46A possesses 13, and FAP174VLV21-23AAA and FAP174VLV25-27AAA contain 15 alanines each. Typically, alanine exhibits a N–Cα–Cβ bond stretch at 908 cm−1, Cα–Cβ–H valence angle bending at 1105 cm−1 and CH3 asymmetric bending at ∼1457 cm−1 as characteristic frequencies. Peaks at ∼1105 and ∼1457 cm−1 are observed in all proteins, indicating Cα–Cβ–H valence angle bending and CH3 asymmetric bending of the methyl group of alanine. Although replacing the VLV with alanine did not lead to major secondary structural changes, a strong peak at 902 cm−1 (Fig. 2C), which as stated previously is the stretching of the N–Cα–Cβ bond of alanine, is observed only in the FAP174VLV21-23AAA and FAP174VLV25-27AAA variants;45 this is the third prominent difference between the VLV variants vis-à-vis FAP174 WT and the FAP174C46A variant. It may be emphasized that all proteins of the RII superfamily, including RIIa of PKA and DPY-30, contain two helices in their N-terminus that form the D/D fold.46,47
 | ||
| Fig. 5 Secondary structure of FAP174 and variants. The secondary structures of FAP174 WT, FAP174VLV21-23AAA, FAP174VLV25-27AAA and FAP174C46A were studied using CD spectroscopy – expressed in the units of delta epsilon (Δε) in the spectral range of 190–250 nm. (A) Far UV CD spectra of FAP174 WT and mutants in 10 mM sodium phosphate buffer, pH 7.0. Experimental and fitted CD spectra of (B) FAP174 WT, (C) FAP174C46A, (D) FAP174VLV21-23AAA, and (E) FAP174VLV25-27AAA. Note the goodness of fit. | ||
Further quantification of the secondary structure of recombinant FAP174 WT and its variants was carried out by deconvoluting the spectra using the BeStSel webserver. This webserver provides a reference set that contains CD spectra of 73 proteins.14 The quantification (Fig. 5B–E) revealed, in agreement with the qualitative analysis, a relatively high amount of α-helix, i.e. up to 65%. Of these, the regular helix (H1, Table 2) was about 48%, while the shorter helix (H2, Table 2) was about 18%. In contrast, only ∼4% of anti-parallel β-sheets, no parallel β-sheets, ∼8% of β-turns, and ∼27% of unordered structures were present in the protein (Table 2). The goodness of fit obtained for the given experimental data is shown in Fig. 5B–E and corroborated by a normalized root mean square deviation (NRMSD) as low as 0.010.50,51
| Protein | Fraction of each secondary structure in percentages | RMSD | NRMSD | |||||
|---|---|---|---|---|---|---|---|---|
| H1 | H2 | APB | PB | Trn | Unrd | |||
| FAP WT | 47 | 18 | 2.0 | 0.0 | 6.0 | 27 | 0.312 | 0.010 |
| % total SS | 65 | 2.0 | ||||||
| FAP174C46A | 46 | 16 | 4.0 | 0.0 | 7.0 | 27 | 0.295 | 0.010 |
| % total SS | 62 | 4.0 | ||||||
| FAP174 VLV21-23AAA | 48 | 17 | 2.0 | 0.0 | 6.0 | 27 | 0.291 | 0.009 |
| % total SS | 65 | 2.0 | ||||||
| FAP174 VLV25-27AAA | 45 | 18 | 2.0 | 0.0 | 8.0 | 27 | 0.199 | 0.007 |
| % total SS | 63 | 2.0 | ||||||
In all, the qualitative and quantitative results obtained from the CD spectra of FAP WT and variants are in good agreement with MD simulations, Raman and FT-IR spectroscopy studies.
3.3 Tertiary structure prediction and refinement by MD simulations
As FAP174 is an orthologue of MYCBP-1, similarities in structure and function between these two proteins are expected. In vitro experiments have shown that recombinant FAP174 interacts with the ciliary AKAP240.12 Of specific interest is the fact that, just like DPY-30 and RIIα Dimerization and Docking domains (D/D), MYCBP-1 also binds to AKAPs.13,52 Thus, it was of interest to unravel the relationship between FAP174 and DPY-30 as well as RIIα D/D. The structures of DPY-30 and RIIα D/D have been resolved by X-ray crystallography, and a comparison of the 3D structures of FAP174 WT with DPY-30 and RIIa D/D facilitated identification of the best model for FAP174 WT. Moreover, the FAP174 protein has a topology like DPY-30 and RIIα D/D (Fig. 6A–F), at least in the regions we hypothesize to be the oligomerization control motifs (cyan regions in Fig. 6A–F) and the regions in the vicinity of C46 that are involved in dimerization (magenta regions in Fig. 6A–F). The similarity between FAP174 and DPY-30 and RIIα D/D is seen for both the QUARK (model ranked 3) and I-TASSER (model ranked 4) models (all atom RMSD ∼3.9 Å). Furthermore, the helices B and C superimpose well over the DPY-30 and RIIα D/D domains. This suggests that the predicted FAP174 tertiary structures possess the necessary structural arrangement prerequisite for binding to AKAPs and dimerization. The QUARK model, which bears a closer resemblance than the I-TASSER model to DPY-30 (all atom RMSD ∼3.0 Å) and RIIα D/D (all atom RMSD ∼2.7 Å), was selected for refinement by MD simulations and further analysis. An overall topology of the structure of FAP174 WT is presented in Table 3. | ||
| Fig. 6 Models of FAP174 (red ribbons) superimposed on DPY-30 (green ribbons) and RIIα Dimerization and Docking domain (blue ribbons). (A and B) FAP174 WT, (C and D) FAP174VLV21-23AAA, and (E and F) FAP174VLV25-27AAA. The cyan region indicates the oligomerization control motifs (VLV regions), and the magenta region depicts residues C46, Y42, and I43. | ||
| Helix | Sequence | Loop/turn | Sequence |
|---|---|---|---|
| A | E3 – A16 | N-terminal coil | M1 – S2 |
| B | A18 – E31 | Turn ab | G17 |
| C | T38 – C46 | Loop bc | E32 – K37 |
| D | P52 – R62 | Loop cd | L47 – T51 |
| E | Q66 – A89 | Loop de | D63 – L65 |
| — | — | C-terminal coil | A90 – A93 |
To refine our models and understand their dynamical behavior, three independent MD simulations of 1 μs length each in explicit TIP3P water were carried out. The changes and the time evolution of the conformations were studied by the backbone RMSD considering three regions – the complete protein (Fig. 7A), non-terminal amino acids (residues 5–87, Fig. 7B), and residues spanning the core (residues 15–65, Fig. 7C). A large backbone-RMSD (Fig. 7A), computed for all 93 amino acid residues, is observed for the FAP174C46A and FAP174VLV21-23AAA variants. This is attributed to large fluctuations in the terminal regions because ignoring the terminal residues from the RMSD analysis resulted in lower values (Fig. 7B and C). Moreover, it is seen that after ∼200 ns, the core region of all FAP174 variants are structurally stable. However, the core region in the FAP174WT showed the greatest deviation from the initial conformation exemplified by a large average backbone-RMSD of 5.08 Å (±0.78), but reaches a plateau beyond ∼250 ns.
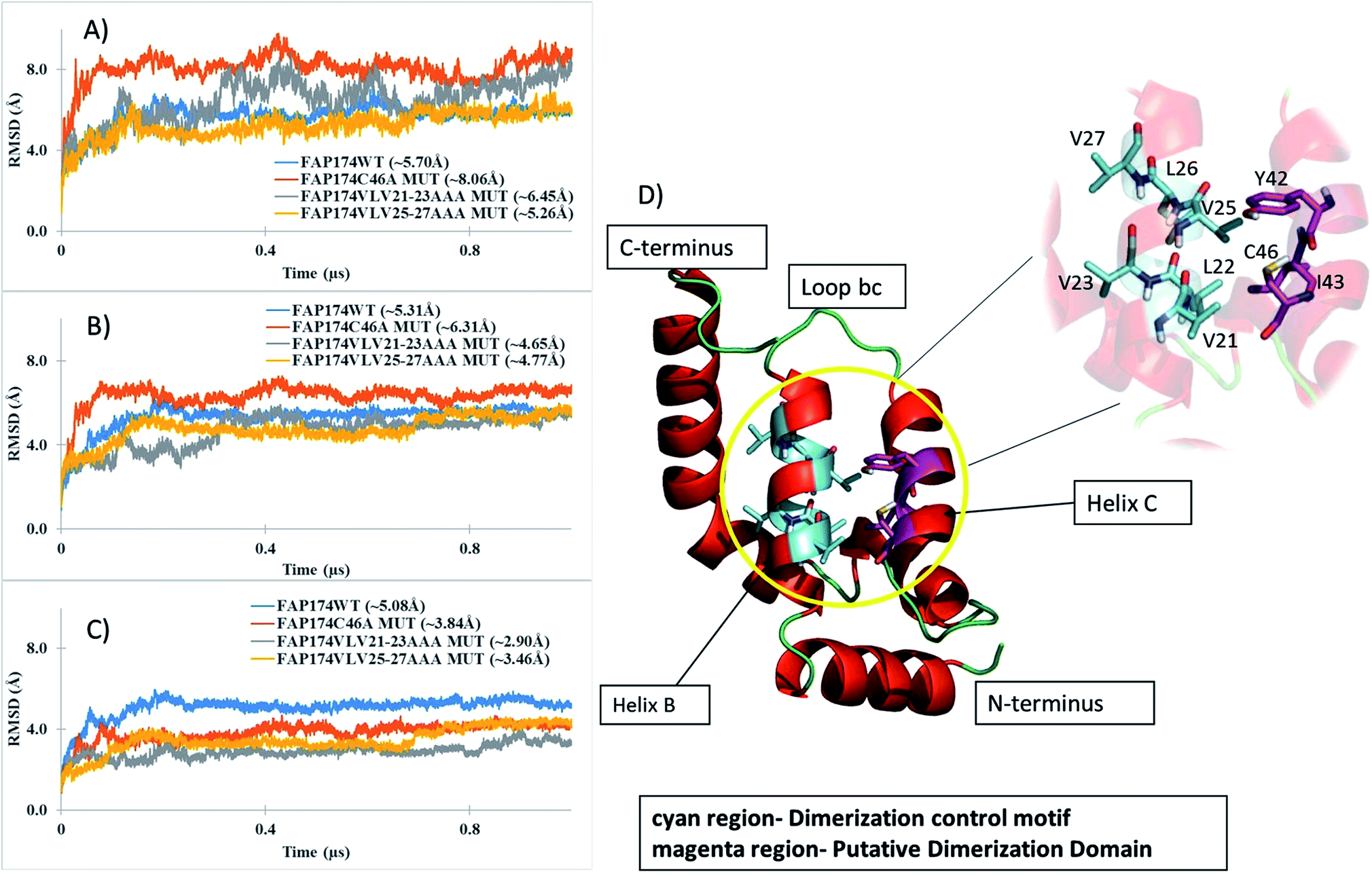 | ||
| Fig. 7 Backbone-RMSD plots from 1 μs MD simulations for (A) all 93 residues of WT FAP174 and the three variants, (B) non-terminal amino acids (residues 5–87), (C) core amino acids (residues 15–65). Blue trace backbone atoms from the FAP174 WT; orange trace backbone atoms from FAP174C46A; grey trace backbone atoms from FAP174VLV21-23AAA and yellow trace backbone atoms from FAP174VLV25-27AAA. (D) Ribbon representation of the FAP174 WT highlighting the oligomerization control motif (cyan region) and residues C46, Y42 and I43 (magenta region). Values in parenthesis in panels (A–C) indicate the mean RMSD over three independent MD trajectories, respectively. | ||
The radius of gyration (RoG) is a good indicator to understand structural changes at a global level of a protein; a decreasing trend indicates compacting tendencies whereas an increasing trend indicates loosening of the structure. The RoG versus normalized distribution plot was used to understand the packing tendency of the FAP174 proteins. All FAP174 variants achieved a similar packing distribution that shows sharp maxima at ∼11 Å for the core regions (Fig. S8A†) and relatively broad maxima that ranged from 12–13 Å for non-terminal amino acids (Fig. S8B†). However, the FAP174VLV21-23AAA mutant shows a slightly skewed distribution for RoG for non-terminal residues with a relatively sharp maximum at ∼14 Å (Fig. S6B†). The core region compacts well for the VLV mutants compared to the WT and the C46 variant. The primary reason may be the decrease in the bulky groups by substituting VLV with AAA. This pattern is consistent with both VLV mutants whereas the WT and C46 variant (not involving the VLV regions) show a skewed distribution indicating that the core region is not as compact as the VLV variants.
3.4 Validation of models using QMEAN server
The QMEAN6 scores with the accompanying Z-scores were employed to assess the improvement in the quality of the tertiary models after refinement with MD simulations. QMEAN6 scores range from 0 to 1, where a score close to 1 indicates good quality models. The Z-scores describe the comparison of the QMEAN6 score for the query model with those of high-resolution X-ray structures of similar size; herein a positive value indicates a better quality model. Table 4 lists the QMEAN6 scores for all models before and after refinement using MD simulations. The QMEAN6 scores indicate that the MD has either improved the model quality, or the model quality is as good as the model prior to MD simulations. However, the Z-scores indicate that our models do not achieve the high-resolution quality that X-ray structures of similar size are able to achieve. This is to be expected for small proteins, like in the current case, that share low sequence homology with any other experimentally solved structure, such that only ab initio structure prediction could be applied.| Model | QMEAN6 score (Z-score) | |
|---|---|---|
| Before MD refinement | After MD refinement | |
| FAP174 WT | 0.68 (−0.53) | 0.79 (−0.03) |
| FAP174VLV21-23AAA | 0.72 (−0.26) | 0.72 (−1.10) |
| FAP174VLV25-27AAA | 0.70 (−0.37) | 0.69 (−1.5) |
| FAP174C46A | 0.71 (−0.30) | 0.71 (−1.4) |
3.5 Tertiary structure of FAP174
FAP174 WT comprises five helices (helices A–E) and four connecting loops (bc, cd, de and ef). The oligomerization control motif (Fig. 6D magenta colored region) and the cysteine residue (Fig. 6D cyan colored region), by which dimerization occurs, lie in helix B (A18-E31) and helix C (T38-C46), respectively. Helix B forms the central core of the FAP174 WT structure, being partially surrounded by all other helices.Like DPY-30 and RIIα D/D, our models for FAP174 (including the variants) also harbor two α-helices (helix B and C) connected by a loop bc. These arrangements in DPY-30 and RIIα D/D facilitate dimerization.22,23 All atom RMSDs of ∼1.4 Å and ∼1.8 Å are obtained when helices B, C and loop bc of FAP174 are superimposed with those of DPY-30 and RIIα D/D, respectively, indicating very similar folds in this region. Based on these results, we propose that the predicted FAP174 structure possesses the DPY-30 and RIIα D/D like motif, which can explain the ability of FAP14 to dimerize and then to bind to AKAPs. It has been reported that the hydrophobic groove in the D/D domain forming the dimer interface is made up of L12, L13, Y16, T17, V20, L21, L28, V29, F31, A32, V33, Y35, F36, T37, and L39.46 Upon comparison of this groove with FAP174, we found 6 out of 15 amino acid residues being similar; these are V25, L26, A39, L40, Y42, and L47. We thus infer that residues A39, L40, Y42, and L47 in the vicinity of C46 might be involved in dimerization of FAP174.
4. Conclusion
The evidences obtained from computer simulations and biophysical experiments indicate that the ciliary MYCBP-1-like protein FAP174 is largely made up of helices and under partly reducing and denaturing conditions exists as an equilibrium mixture of dimer and monomer. Otherwise, it exists as an oligomer that is probably made of dimers. Experiments indicate that C46 is responsible for the dimerization of FAP174 by forming a disulfide bridge linking the two monomers. We have also identified an oligomerization control motif (stretches of VLVs) that plays a crucial role for the oligomerization ability of FAP174 that has not been determined in the mammalian MYCBP-1 (Table 5). This oligomerization tendency of FAP174 appears to have hampered our previous attempts on determining the solution structure using NMR. The FAP174 structure predicted by the ab initio method QUARK is suggested to possess a DPY-30- and RIIα-like D/D motif, which can explain the ability of FAP14 to dimerize and then to bind to AKAPs (Table 5).| Features | Mammalian MYCBP-1 | C. reinhardtii FAP174 |
|---|---|---|
| Molecular weight (kDa) | 12 | 10 |
| Secondary structure | α-helices, only C-terminus | α-helices (current study) |
| Presence of cysteine | None | One |
| Dimerization and oligomerization status | Not determined | This study |
| Transcript expression in tissues/cells | Heart, placenta, pancreas, kidney, skeletal muscle | C. reinhardtii cells |
| Sub-cellular locations & AKAPs that bind | Nucleus (AKAP95), sperm mitochondria (S AKAP84/149), trans-golgi network (BIG1 and BIG2), motile cilia | Flagellar central Pair (AKAP240), basal body (AKAP unidentified) and nucleus (AKAP unidentified) |
| References | 3–8 and 11 | 12 |
Conflicts of interest
The authors have no conflict of interest to declare.Abbreviations
| FAP174 | Flagellar associated protein 174 |
| MYCBP-1 | MYC-Binding protein-1 |
| AKAP | A-Kinase anchoring protein |
| PPI | Protein–protein interactions |
| MD | Molecular dynamics |
| RoG | Radius of gyration |
| Cα-RMSD | Root mean squared deviation of alpha carbons |
| RMSF | Root mean squared fluctuation |
Acknowledgements
This work was supported by a grant (Ref No: BT/R3159/RB/10/960/2011) awarded by the Department of Biotechnology, India to SC and JSD. YM acknowledges the stipend received through this grant. The idea of carrying out a study on micro-Raman spectroscopy of a ciliary protein was conceived by Prof. Deepak Mathur from TIFR, India; SC and JD thank him for providing encouragement. JD wishes to thank Mrs Gitanjali Dhotre of the Department of Chemical Sciences, TIFR, Mumbai for help provided in recording the MALDI-TOF spectra of all proteins used in this study. The authors also thank Dr Sameer Maji, Prof. Swati Patankar, and Mr Pradeep Kumar Singh from Indian IIT-Bombay for help in using the CD instrument. EAFM and ECC are indebted to the Department of Science and Technology (DST), Department of Biotechnology (DBT), and Council of Scientific and Industrial Research (CSIR) for financial assistance for building the computing facilities at the Department of Pharmaceutical Chemistry, Bombay College of Pharmacy. EAFM thanks whole-heartedly the MCBR4 consortium [Heidelberg 2015, Prof. Dr Peter Comba (Heidelberg), Prof. Dr Hendrik Zipse (Munich) and Prof. Dr G. Narahari Sastry (Hyderabad)] for the financial assistance to carry out research at Heinrich Heine University Düsseldorf, Germany.References
- D. Cooper, Biochem. Soc. Trans., 2005, 33, 1319–1322 CrossRef CAS PubMed.
- A. Perino, A. Ghigo, J. D. Scott and E. Hirsch, Circ. Res., 2012, 111, 482–492 CrossRef CAS PubMed.
- T. Taira, J. Maëda, T. Onishi, H. Kitaura, S. Yoshida, H. Kato, M. Ikeda, K. Tamai, M. Iguchi-Ariga and M. Sanae, Genes Cells, 1998, 3, 549–565 CrossRef CAS PubMed.
- M. Furusawa, T. Onishi, T. Taira, S. Iguchi-Ariga and H. Ariga, Int. J. Oncol., 2000, 16, 339–384 CAS.
- M. Furusawa, T. Ohnishi, T. Taira, S. M. Iguchi-Ariga and H. Ariga, J. Biol. Chem., 2001, 276, 36647–36651 CrossRef CAS PubMed.
- M. Furusawa, T. Taira, S. M. Iguchi-Ariga and H. Ariga, J. Biol. Chem., 2002, 277, 50885–50892 CrossRef CAS PubMed.
- H. Yukitake, M. Furusawa, T. Taira, S. M. Iguchi-Ariga and H. Ariga, J. Biol. Chem., 2002, 277, 45480–45492 CrossRef CAS PubMed.
- R. Ishizaki, H.-W. Shin, S. M. Iguchi-Ariga, H. Ariga and K. Nakayama, Genes Cells, 2006, 11, 949–959 CrossRef CAS PubMed.
- M. S. Kapiloff, M. Rigatti and K. L. Dodge-Kafka, J. Gen. Physiol., 2014, 143, 9–15 CrossRef CAS PubMed.
- Y. Xie, H. Wang, Ihsanawati, S. Kishishita, C. Takemoto, K. Murayama, M. Shirozu and S. Yokoyama, 2011, 10.2210/pdb2yy0/pdb.
- G. J. Pazour, N. Agrin, J. Leszyk and G. B. Witman, J. Cell Biol., 2005, 170, 103–113 CrossRef CAS PubMed.
- V. G. Rao, R. B. Sarafdar, T. S. Chowdhury, P. Sivadas, P. Yang, P. M. Dongre and J. S. D'Souza, BMC Cell Biol., 2016, 17, 24 CrossRef PubMed.
- Y. Chen, B. Wan, K. C. Wang, F. Cao, Y. Yang, A. Protacio, Y. Dou, H. Y. Chang and M. Lei, EMBO Rep., 2011, 12, 797–803 CrossRef CAS PubMed.
- A. Micsonai, F. Wien, L. Kernya, Y.-H. Lee, Y. Goto, M. Réfrégiers and J. Kardos, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3095–E3103 CrossRef CAS PubMed.
- C. Cole, J. D. Barber and G. J. Barton, Nucleic Acids Res., 2008, 36, W197–W201 CrossRef CAS PubMed.
- J. A. Cuff, M. E. Clamp, A. S. Siddiqui, M. Finlay and G. J. Barton, Bioinformatics, 1998, 14, 892–893 CrossRef CAS PubMed.
- D. T. Jones, J. Mol. Biol., 1999, 292, 195–202 CrossRef CAS PubMed.
- L. J. McGuffin, K. Bryson and D. T. Jones, Bioinformatics, 2000, 16, 404–405 CrossRef CAS PubMed.
- A. Roy, A. Kucukural and Y. Zhang, Nat. Protoc., 2010, 5, 725–738 CrossRef CAS PubMed.
- T. Z. Sen, R. L. Jernigan, J. Garnier and A. Kloczkowski, Bioinformatics, 2005, 21, 2787–2788 CrossRef CAS PubMed.
- S. Wu and Y. Zhang, Nucleic Acids Res., 2007, 35, 3375–3382 CrossRef CAS PubMed.
- D. Xu and Y. Zhang, Proteins, 2012, 80, 1715–1735 CrossRef CAS PubMed.
- X. Wang, Z. Lou, X. Dong, W. Yang, Y. Peng, B. Yin, Y. Gong, J. Yuan, W. Zhou and M. Bartlam, J. Mol. Biol., 2009, 390, 530–537 CrossRef CAS PubMed.
- M. G. Gold, B. Lygren, P. Dokurno, N. Hoshi, G. McConnachie, K. Taskén, C. R. Carlson, J. D. Scott and D. Barford, Mol. Cell, 2006, 24, 383–395 CrossRef CAS PubMed.
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS PubMed.
- D. Case, Q. Cai, D. Cerutti, T. Cheatham III, T. Darden, R. Duke, H. Gohlke, A. Goetz and S. Gusarov, Journal, 2014, 29–31 Search PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015 Search PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- J.-P. Ryckaert, G. Ciccotti and H. J. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- D. R. Roe and T. E. Cheatham III, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
- F. Melo and E. Feytmans, J. Mol. Biol., 1998, 277, 1141–1152 CrossRef CAS PubMed.
- P. Benkert, M. Biasini and T. Schwede, Bioinformatics, 2011, 27, 343–350 CrossRef CAS PubMed.
- J. Cheng, A. Z. Randall, M. J. Sweredoski and P. Baldi, Nucleic Acids Res., 2005, 33, W72–W76 CrossRef CAS PubMed.
- W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577–2637 CrossRef CAS PubMed.
- P. Banky, L. J.-S. Huang and S. S. Taylor, J. Biol. Chem., 1998, 273, 35048–35055 CrossRef CAS PubMed.
- A. Rygula, K. Majzner, K. M. Marzec, A. Kaczor, M. Pilarczyk and M. Baranska, J. Raman Spectrosc., 2013, 44, 1061–1076 CrossRef CAS.
- R. W. Williams, Methods Enzymol., 1986, 130, 311–331 CAS.
- C. L. Wilder, A. D. Friedrich, R. O. Potts, G. O. Daumy and M. L. Francoeur, Biochemistry, 1992, 31, 27–31 CrossRef CAS PubMed.
- J. Grdadolnik and Y. Maréchal, Biopolymers, 2001, 62, 40–53 CrossRef CAS PubMed.
- A. Barth, Prog. Biophys. Mol. Biol., 2000, 74, 141–173 CrossRef CAS PubMed.
- T. G. Spiro and B. P. Gaber, Annu. Rev. Biochem., 1977, 46, 553–570 CrossRef CAS PubMed.
- K. C. Ravindra, B. R. Selvi, M. Arif, B. A. Reddy, G. R. Thanuja, S. Agrawal, S. K. Pradhan, N. Nagashayana, D. Dasgupta and T. K. Kundu, J. Biol. Chem., 2009, 284, 24453–24464 CrossRef CAS PubMed.
- V. Lin and J. Koenig, Biopolymers, 1976, 15, 203–218 CrossRef CAS PubMed.
- W. Qian, J. Bandekar and S. Krimm, Biopolymers, 1991, 31, 193–210 CrossRef CAS PubMed.
- F. S. Kinderman, C. Kim, S. von Daake, Y. Ma, B. Q. Pham, G. Spraggon, N. H. Xuong, P. A. Jennings and S. S. Taylor, Mol. Cell, 2006, 24, 397–408 CrossRef CAS PubMed.
- M. G. Newlon, M. Roy, D. Morikis, D. W. Carr, R. Westphal, J. D. Scott and P. A. Jennings, EMBO J., 2001, 20, 1651–1662 CrossRef CAS PubMed.
- K. Bakshi, M. R. Liyanage, D. B. Volkin and C. R. Middaugh, Methods and Protocols, Therapeutic Peptides, 2014, pp. 247–253 Search PubMed.
- G. Holzwarth and P. Doty, J. Am. Chem. Soc., 1965, 87, 218–228 CrossRef CAS PubMed.
- L. Whitmore and B. A. Wallace, Biopolymers, 2008, 89, 392–400 CrossRef CAS PubMed.
- D. Mao, E. Wachter and B. Wallace, Biochemistry, 1982, 21, 4960–4968 CrossRef CAS PubMed.
- W. E. Theurkauf and R. B. Vallee, J. Biol. Chem., 1982, 257, 3284–3290 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07836f |
| This journal is © The Royal Society of Chemistry 2017 |