
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photo-induced tandem cyclization of 3-iodoflavones with electron rich five-membered heteroarenes†
Qian Yangab,
Rui Wanga,
Jie Hana,
Chenchen Lia,
Tao Wang a,
Yong Liangc and
Zunting Zhang*a
a,
Yong Liangc and
Zunting Zhang*a
aKey Laboratory of the Ministry of Education for Medicinal Resources and Natural Pharmaceutical Chemistry, National Engineering Laboratory for Resource Development of Endangered Crude Drugs in Northwest of China, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an 710062, People's Republic of China. E-mail: zhangzunting@sina.com; Fax: +86-29-81530727; Tel: +86-29-85303940
bSchool of Sciences, Xi'an University of Technology, Xi'an 710054, China
cDepartment of Molecular Medicine, Beckman Research Institute, Hope Duarte, CA 91010, USA
First published on 5th September 2017
Abstract
Vinyl radicals were generated from 3-iodoflavones under a mercury lamp and tandem cyclization reactions occurred with five-membered heteroarenes entailing two consecutive C–C bond formations to synthesize benzo[e]chromeno[2,3-g]indol-13(1H)-one derivatives. The tandem cyclization reactions worked in acetonitrile without any additives such as transition metals, ligands and oxidants, giving rise to a broad variety of novel polycyclic xanthone frameworks in good yield under mild and environmentally friendly reaction conditions.
Introduction
Flavones are a class of natural products and can be found in numerous plants. They display various biological activities, including antioxidant, anti-tumor, anti-proliferative and anti-inflammatory activities, and have been used in the treatment of cancer, cardiovascular diseases, neurodegenerative disorders, etc. Flavones are also an important scaffold for medicinal chemistry and versatile building blocks for the construction of various heteroaromatics.1–3Tandem cyclizations, as a powerful method for the synthesis of polycyclic compounds, have been successfully applied to the Diels–Alder reactions,4–7 Michael addition,8 rearrangements9–11 and radical cyclizations.12–14 During the last decade, extensive investigations have been made on the photo-induced tandem vinyl radical cyclization, which could build multiple C–C bonds in a single step under mild conditions.15–17 In 2014, O. Reiser reported a radical tandem addition–cyclization of α-bromochalcones or α-bromocinnamates with heteroarenes using an iridium complex as the catalyst and irradiating with visible light (Scheme 1, top).15 Similarly, O. Reiser reported a visible light-mediated coupling of α-bromochalcones with alkenes under the visible-light photo-redox conditions in the presence of fac-Ir(ppy)3 at room temperature (Scheme 1, middle).16 In 2017, another transition-metal catalyzed tandem cyclization of vinyl radicals used for the synthesis of substituted indolines was developed via a photoredox-mediated reductive quenching cycle (Scheme 1, bottom).17
 | ||
| Scheme 1 The reported photo-induced tandem radical cyclization. | ||
Following our interest in the photo-reactions of chromone analogues18–21 as well as nucleoside substrates,22 we herein report a new photo-induced tandem cyclization of 3-iodoflavones with π-excessive five-membered heteroaromatics at ambient temperature without the requirement of any transition-metal-catalyst and/or oxidant additives.
Results and discussion
Initially, a stirred solution of 3-iodoflavone 1a (0.2 mmol) and N-methylpyrrole 2a (6 mmol) in DMF (40 mL) was irradiated using a high-pressure mercury lamp (500 W) at room temperature under an argon atmosphere for 5 h. The cyclization product 3a was isolated (13%) and characterized by NMR and HRMS (Table 1, entry 1). Inspired by this promising result, various solvents, such as THF, t-BuOH, Me2CO and MeCN, were screened. However, only a trace amount of the cyclization product 3a was observed in THF, while the yields of 3a were slightly increased in other solvents (entries 2–5). In order to avoid the coupling of 1a with benzene and toluene, such aromatic solvents were not used. As expected, the cyclization yields increased upon increasing the loading of 2a (entries 6–8). In addition, it was noteworthy that the yield of 3a dropped to 35% when 0.35 M of 2a was employed (entry 9), which clearly indicated that solvent free conditions will not work very well. Finally, the irradiation time was optimized and the yield of 3a was increased to 47% when the reaction time was extended to 7 h (entries 10–14).
|
||||
|---|---|---|---|---|
| Entry | Solvent | Concentration of 2a [mol L−1] | Time [h] | Yieldb [%] |
| a A mixture of 1a (0.2 mmol, 0.005 M) and 2a (2–14 mmol, 0.05–0.35 M) in anhydrous MeCN (40 mL) was irradiated using a high-pressure mercury lamp (500 W) at ambient temperature under an Ar atmosphere.b Isolated yield. | ||||
| 1 | DMF | 0.15 | 5 | 13 |
| 2 | THF | 0.15 | 5 | Trace |
| 3 | t-BuOH | 0.15 | 5 | 16 |
| 4 | Me2CO | 0.15 | 5 | 20 |
| 5 | MeCN | 0.15 | 5 | 24 |
| 6 | MeCN | 0.05 | 5 | 15 |
| 7 | MeCN | 0.2 | 5 | 31 |
| 8 | MeCN | 0.25 | 5 | 40 |
| 9 | MeCN | 0.35 | 5 | 35 |
| 10 | MeCN | 0.25 | 1 | 18 |
| 11 | MeCN | 0.25 | 3 | 30 |
| 12 | MeCN | 0.25 | 6 | 43 |
| 13 | MeCN | 0.25 | 7 | 47 |
| 14 | MeCN | 0.25 | 10 | 43 |

Cyclization of 3-iodoflavones with various five membered heteroarenes
In order to screen the scope of the substrates and compatibility of functional groups, various 3-iodoflavones 1 and π-excessive five-membered heteroarenes 2 were screened, and the cyclization yields are listed in Table 2. Thus, irradiation of 1a and N-methylpyrrole 2a in MeCN using a high-pressure mercury lamp (500 W) at room temperature for 7 h gave 3a in 47% yield (entry 1). Analogously, treatment of 1b, bearing an additional methyl group at the R2 position, gave the cyclization product 3b in a lower yield (entry 2). However, treatment of 1c, bearing an additional fluoro group at the R2 position, gave the cyclization product 3c in a higher yield (entry 3). Various combinations of hydrogen, electron-withdrawing and electron-donating groups were explored for the 3-iodoflavone 1 substrates (entries 4–16). As shown in the table, the yields of 3 were significantly affected by the functional groups attached. The presence of electron-donating groups on both R1 and R2 gave the cyclization products 3 in much lower yields (entries 2, 4, 6 and 7) when compared to the substrate bearing only one electron-donating group at either R1 or R2 (entries 1, 5 and 10–11). For example, the yield of 3d was decreased to 38% when –OCH3 was present at both R1 and R2 (entry 4). On the other hand, the yield of 3 could be easily increased to over 68% with the introduction of an electron-withdrawing group to either R1 or R2 (entries 3, 8 and 13–15).
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | 1 | 2 | 3 | Yieldsb [%] |
| a A mixture of 1 (0.2 mmol, 0.005 M) and 2 (10 mmol, 0.25 M) in anhydrous MeCN (40 mL) was irradiated using a high-pressure mercury lamp (500 W) at ambient temperature for 7 h under an Ar atmosphere.b Isolated yield.c Irradiation for 10 h. | ||||||
| 1 | OCH3 | H | 1a | 2a | 3a | 47 |
| 2 | OCH3 | 6-CH3 | 1b | 2a | 3b | 40 |
| 3 | OCH3 | 6-F | 1c | 2a | 3c | 49 |
| 4 | OCH3 | 7-OCH3 | 1d | 2a | 3d | 38 |
| 5 | CH3 | H | 1e | 2a | 3e | 50 |
| 6 | CH3 | 6-CH3 | 1f | 2a | 3f | 46 |
| 7 | CH3 | OCH3 | 1g | 2a | 3g | 42 |
| 8 | CH3 | 6-F | 1h | 2a | 3h | 56 |
| 9 | H | H | 1i | 2a | 3i | 61 |
| 10 | H | 6-CH3 | 1j | 2a | 3j | 51 |
| 11 | H | 7-OCH3 | 1k | 2a | 3k | 45 |
| 12 | H | 6-F | 1l | 2a | 3l | 66 |
| 13 | F | H | 1m | 2a | 3m | 68 |
| 14 | F | 6-CH3 | 1n | 2a | 3n | 54 |
| 15 | F | 7-OCH3 | 1o | 2a | 3o | 49 |
| 16 | COOC2H5 | H | 1p | 2a | 3p | 64 |
| 17 | H | H | 1i | 2b | 3q | 40c |
| 18 | OCH3 | H | 1a | 2c | 3r | 25 |
| 19 | OCH3 | H | 1a | 2d | 3s | 18 |

Various π-excessive five-membered heteroarenes 2 were also explored to examine their compatibility and reactivity (entries 17–19). Thus, irradiation of 1i with pyrrole 2b in MeCN at room temperature for 10 h gave the cyclization product 3q (entry 17). Due to the ring closure for the formation of an aromatic system (vide infra), the stability issue22 was not observed with the current protocol. The cyclization of 3-iodoflavone 1a also proceeded smoothly with furan 2c and thiophene 2d to give the corresponding products in fair yields (entries 18–19). It is noteworthy that no cyclization products were observed for benzene or other six-membered aromatics under the optimal conditions. Generally, the cyclization efficiency follows the electrophilic reactivity N-methylpyrrole > pyrrole > furan > thiophene, which can easily explain why no cyclization product was observed for the six-membered aromatics.
Electrophilic aromatic substitution (EAS)
On the basis of the experimental results and literature,19,20,24,25 a two-step mechanism for the cyclization of 3-iodoflavones 1 with π-excessive five-membered heteroarenes 2 has been proposed. Initially, the photo-induced hemolytic cleavage of the C–I bond of 1 under UV light irradiation gave the radical intermediate A. Coupling of A with heteroarenes 2 afforded the reactive intermediate B, followed by the abstraction of hydrogen with an iodine radical to give the coupling product C with the release of HI (Scheme 2).19,20 | ||
| Scheme 2 Proposed mechanism for the preparation of coupling product C. | ||
The cyclization step (second step, Scheme 3) started with the UV irradiation of C via the formation of the ring closed intermediate D,21,23 followed by a thermal suprafacial [1,5]-H shift to form intermediate E. Furthermore, keto–enol tautomerization of E led to the formation of the enol intermediate F, which generated a more stable syn-isomer G via another keto–enol tautomerization.21–24 Finally, the cyclization product 3 was formed via the syn-elimination of a hydrogen molecule from intermediate G along with the restoration of the aromaticity of the conjugated system.
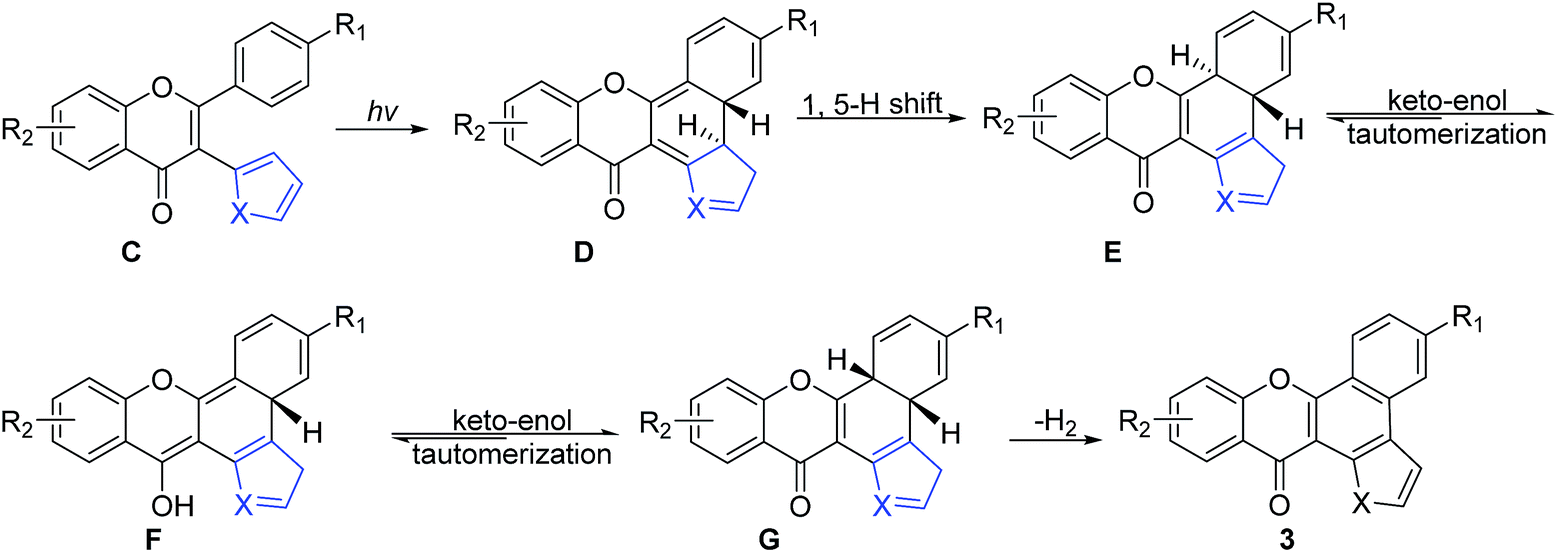 | ||
| Scheme 3 Proposed mechanism for the preparation of the photocyclization product. | ||
In order to study the importance of keto–enol tautomerization and figure out the rate limiting step, coupling product 1aa was isolated and subjected to the cyclization conditions separately (Scheme 4). Thus, irradiation of 1a and 2a under a high-pressure mercury lamp at ambient temperature for 1 h yielded the coupling product 1aa with 25% along with a trace amount of the cyclization product 3a. A solution of 1aa (5 mM) in MeCN was irradiated under a high-pressure mercury lamp at ambient temperature in parallel with a solution of 1a (5 mM) and 2a (0.25 M) in MeCN. After 7 h, substrate 1aa gave the cyclization product 3a in 95% yield, while only 47% of 3a was obtained for 3-iodoflavone 1a. Thus, we could easily conclude that coupling of 3-iodoflavone with heteroarenes was the rate-limiting step.
 | ||
| Scheme 4 The stepwise reaction of the tandem cyclization reaction. | ||
To obtain further insight into reaction mechanism pathway and validate the rationality of the proposed mechanism, a substrate without the presence of C4![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O was synthesized and subjected to the cyclization conditions. Initially, 1aa was reduced to 1ab using LiAlH4–AlCl3 according to the procedure reported in literature26,27 (Scheme 5). Then, the 4,4-dihydro substrate 1ab was subjected to the optimal conditions. As expected, no cyclization product 1ac was detected, which could be explained by the lack of keto–enol tautomerization.
O was synthesized and subjected to the cyclization conditions. Initially, 1aa was reduced to 1ab using LiAlH4–AlCl3 according to the procedure reported in literature26,27 (Scheme 5). Then, the 4,4-dihydro substrate 1ab was subjected to the optimal conditions. As expected, no cyclization product 1ac was detected, which could be explained by the lack of keto–enol tautomerization.
 | ||
| Scheme 5 Reductive carbonyl experiments. | ||
In order to expand the photo-induced tandem cyclization of 3-iodoflavones with π-excessive five-membered heteroarenes methodology to other systems bearing a similar functional moiety, we subjected the (Z)-α-bromo-β-phenylvinylketone analogues 4 to our cyclization protocol (Scheme 6). Thus, irradiation of α-bromochalcones 4a and the pyrrole analogues 2 under the optimal conditions for 10 h yielded 5a and 5b in 57% and 42% yield, respectively. Similarly, irradiation of 4b with pyrrole analogues 2 gave 5c and 5d in 47% and 38% yield, respectively. Due to the stabilization of the additional phenyl ring through the larger conjugation system, α-bromochalcone 4a gave the cyclization product in an improved yield compared to the methyl substrate 4b. Even though a similar cyclization has been reported,15 it is important to mention that the current protocol avoids the use of transition-metal complexes and high boiling solvent, which could be problematic during the purification stage.
 | ||
| Scheme 6 Cyclization of α-bromo-β-phenylvinylketone analogues 4 with pyrrole 2. | ||
Conclusion
In conclusion, we have demonstrated a new tandem cyclization protocol for the synthesis of fused heterocyclic aromatic systems via the coupling of both 3-iodoflavone and α-bromochalcone derivatives with various π-excessive five-membered heteroarenes. The described methodology provides rapid access to polycyclic xanthones with excellent regioselectivity under mild and environment friendly conditions without the requirement of any catalyst or additives.Experimental section
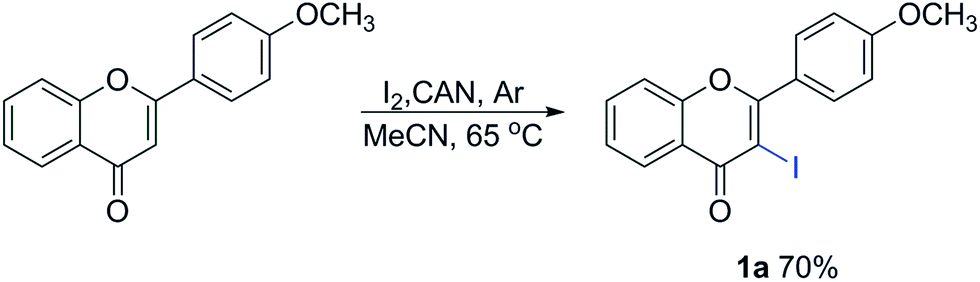
Unless otherwise noted, all reagents were purchased from commercial suppliers and used without purification. MeCN was distilled from CaH2 just prior to use. Thin-layer chromatography (TLC) was performed using silica gel 60 GF254 plates. The silica gel (size 200–300 mesh) used for the column chromatography was purchased from Qingdao Haiyang Chemistry Plant (China). 1H NMR and 13C NMR spectra were recorded on 400 (1H) and 100 (13C) MHz spectrometers. The spectra were referenced internally to CDCl3 or DMSO-d6. High-resolution mass spectrometry (HRMS) was recorded using the electron-spray ionization (ESI) technique. The melting points were measured using X-5 micro-melting point apparatus and were uncorrected. IR spectra were recorded on a Nicolet 170SX FT-IR spectrophotometer using KBr pellets. All the irradiation experiments were performed on a BL-GHX-V photochemical reactor equipped with a 500 W high-pressure Hg lamp.General procedure for the preparation of 3-iodoflavones 128
A solution of 2-(4-methoxyphenyl)-4H-chromen-4-one (1.0 mmol), I2 (1.2 mmol) and CAN (1.1 mmol) in anhydrous MeCN (100 mL) was stirred at 65 °C (oil/water bath) under an Ar atmosphere until the disappearance of the substrate. After being cooled to ambient temperature, the reaction mixture was poured into a cold solution of Na2S2O3 (50 mL, 0.1 M) and extracted with CH2Cl2 (20 mL, ×3). The organic layers were combined, washed (H2O), dried (MgSO4) and concentrated. The residue was purified by column chromatography (hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc = 9:1) to afford the desired product 1a (70%) as a white solid.
:EtOAc = 9:1) to afford the desired product 1a (70%) as a white solid.
Analogously, compounds 1b–1p were prepared using the same method as described for 1a.
General procedure for the synthesis of α-bromochalcones 416
Bromine (Br2, 4.0 mmol) was added to a stirred solution of chalcone (2.0 mmol) in CH2Cl2 (40.0 mL) over 10 min at ambient temperature. Triethylamine (10.0 mmol) was slowly added to the resulting mixture when chalcone was completely converted to dibromide (judged by TLC). After stirring for another 12 h, the reaction mixture was extracted with CH2Cl2 (20 mL, ×3). The combined organic layers were washed with brine, dried over anhydrous Na2SO4 and the volatiles were removed under reduced pressure. The oily residue was purified using column chromatography to give α-bromochalcone 4a (82%) as a white solid.Analogously, 4b was obtained in 92% yield using the same method described for 4a.
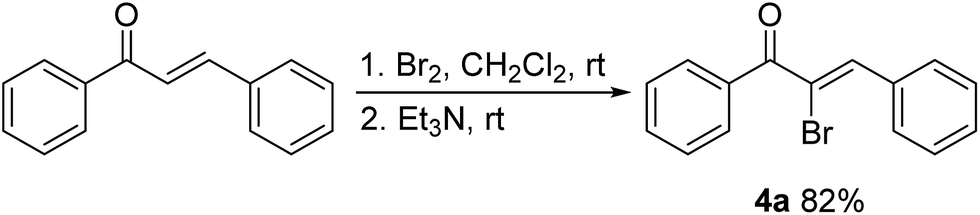
General procedure for the cyclization of 3-iodoflavones or α-bromochalcones with heteroarenes
A solution of 3-iodo-2-(4-methoxyphenyl)-4H-chromen-4-one 1 (0.2 mmol) and N-methylpyrrole 2a (10 mmol) in anhydrous MeCN (40 mL) in a 50 mL quartz tube was degassed (bubbling Ar for 30 min) and irradiated for 7 h using a high-pressure mercury lamp (500 W) at ambient temperature. The volatiles were removed under reduced pressure and the residue was purified using column chromatography (hexane:EtOAc = 20:1) to give the corresponding product 3a (47%) as a white solid.
Analogously, 3b–3s, 5a–5d were prepared using the same method described for 3a.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (No. 21672132 and 21502110).Notes and references
- S. Kumar and A. K. Pandey, Sci. World J., 2013, 16 Search PubMed.
- R. S. Keri, S. Budagumpi, R. K. Pai and R. G. Balakrishna, Eur. J. Med. Chem., 2014, 78, 340–374 CrossRef CAS PubMed.
- M. Singh, M. Kaur and O. Silakari, Eur. J. Med. Chem., 2014, 84, 206–239 CrossRef CAS PubMed.
- H. B. Kagan and O. Riant, Chem. Rev., 1992, 92, 1007–1019 CrossRef CAS.
- J. D. Winkler, Chem. Rev., 1996, 96, 167–176 CrossRef CAS PubMed.
- K.-i. Takao, R. Munakata and K.-i. Tadano, Chem. Rev., 2005, 105, 4779–4807 CrossRef CAS PubMed.
- P. Wessig and G. Müller, Chem. Rev., 2008, 108, 2051–2063 CrossRef CAS PubMed.
- A. Stelmakh, T. Stellfeld and M. Kalesse, Org. Lett., 2006, 8, 3485–3488 CrossRef CAS PubMed.
- T. Nakamura, T. Matsui, K. Tanino and I. Kuwajima, J. Org. Chem., 1997, 62, 3032–3033 CrossRef CAS PubMed.
- J. Stephen Clark and P. B. Hodgson, Tetrahedron Lett., 1995, 36, 2519–2522 CrossRef.
- M. Alajarín, M.-M. Ortín, P. Sánchez-Andrada and Á. Vidal, J. Org. Chem., 2006, 71, 8126–8139 CrossRef PubMed.
- S. Handa and G. Pattenden, Chem. Commun., 1998, 311–312 RSC.
- R. Leardini, D. Nanni, A. Tundo and G. Zanardi, Tetrahedron Lett., 1998, 39, 2441–2442 CrossRef CAS.
- D. Yang, S. Gu, Y.-L. Yan, H.-W. Zhao and N.-Y. Zhu, Angew. Chem., Int. Ed., 2002, 41, 3014–3017 CrossRef CAS PubMed.
- S. Paria and O. Reiser, Adv. Synth. Catal., 2014, 356, 557–562 CrossRef CAS.
- S. Paria, V. Kais and O. Reiser, Adv. Synth. Catal., 2014, 356, 2853–2858 CrossRef CAS.
- S. K. Pagire and O. Reiser, Green Chem., 2017, 19, 1721–1725 RSC.
- Q. Wang, Z. Zhang, Z. Du, H. Hua and S. Chen, Green Chem., 2013, 15, 1048–1054 RSC.
- Q. Yang, Y. He, T. Wang, L. Zeng and Z. Zhang, Mol. Diversity, 2016, 20, 9–16 CrossRef CAS PubMed.
- L. Liu, Q. Wang, Z. Zhang, Q. Zhang, Z. Du, D. Xue and T. Wang, Mol. Diversity, 2014, 18, 777–785 CrossRef CAS PubMed.
- J. Han, T. Wang, Y. Liang, Y. Li, C. C. Li, R. Wang, S. Q. Feng and Z. T. Zhang, Org. Lett., 2017, 19, 3552–3555 CrossRef CAS PubMed.
- Q. Yang, T. Wei, Y. He, Y. Liang and Z.-T. Zhang, Helv. Chim. Acta, 2015, 98, 953–960 CrossRef CAS.
- Y. Liang, J. Gloudeman and S. F. Wnuk, J. Org. Chem., 2014, 79, 4094–4103 CrossRef CAS PubMed.
- A. G. Lvov, V. Z. Shirinian, V. V. Kachala, A. M. Kavun, I. V. Zavarzin and M. M. Krayushkin, Org. Lett., 2014, 16, 4532–4535 CrossRef CAS PubMed.
- A. G. Lvov, V. Z. Shirinian, A. V. Zakharov, M. M. Krayushkin, V. V. Kachala and I. V. Zavarzin, J. Org. Chem., 2015, 80, 11491–11500 CrossRef CAS PubMed.
- C. Valla, A. Baeza, F. Menges and A. Pfaltz, Synlett, 2008, 3167–3171 CAS.
- T. G. C. Bird, B. R. Brown, I. A. Stuart and A. W. R. Tyrrell, J. Chem. Soc., Perkin Trans. 1, 1983, 1831–1846 RSC.
- F. J. Zhang and Y. L. Li, Synthesis, 1993, 565–567 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07793a |
| This journal is © The Royal Society of Chemistry 2017 |