
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Theoretical investigation on proton transfer mechanism of extradiol dioxygenase†
Yang Liu ab,
Ningyu Tub,
Wenyu Xieb and
Youming Li*a
ab,
Ningyu Tub,
Wenyu Xieb and
Youming Li*a
aState Key Laboratory of Pulp and Paper Engineering, South China University of Technology, Guangzhou 510640, P. R. China. E-mail: ymli3@scut.edu.cn
bFaculty of Environmental & Biological Engineering, Guangdong University of Petrochemical Technology, Maoming 525000, P. R. China
First published on 6th September 2017
Abstract
A QM/MM method ONIOM (B3LYP: Amber) was employed to discuss the catalytic mechanism of non-heme iron extradiol dioxygenases (HPCD). Previous research suggested that protonation of alkylperoxo intermediate was achieved by transferring the proton from monoanionic catechol substrate to superoxide anion via the histidine residue near the active site. Herein, our results demonstrated that the proton was transferred from the monoanionic catechol substrate to the superoxide anion directly. The catalytic mechanism could be performed via two parallel pathways, named pathway A and B. Both of them consisted of a proton-transfer process and distal oxygen attack procedure but occurred in a different sequence. Our key mechanistic discovery for catalytic reactions revealed a two-state reactivity (TSR) scenario, in which quintet state crossed over the septet state. Pathway A was more kinetically and thermodynamically favorable.
1 Introduction
The mechanism for catechol ring-cleavage catalyzed by non-heme iron (or manganese) extradiol dioxygenases has attracted much attention for many years due to its significance for degradation of aromatic compounds.1–5 Extradiol dioxygenases include 2,3-dihydroxybiphenyl 1,2-dioxygenase (DHBD), catechol 2,3-dioxygenase (2,3-CTD), homoprotocatechuate 2,3-dioxygenase (HPCD), protocatechuate 4,5-dioxygenase (4,5-PCD), and homogentisate 1,2-dioxygenase (HGO), which all contain a 2-His-1-carboxylate facial triad.6,7 Prior studies have mostly focused on two extradiol dioxygenases: HPCD8–12 and DHBD.13–15 Remarkably, three key intermediates (superoxo, alkylperoxo and bound product) of HPCD with a slow substrate 4-nitrocatechol (4NC) was captured by experimental methods,16 the whole catalytic mechanism became more and more clear.13,17,18 Several consensus reaction steps are shown in Scheme 1. Firstly, Fe–O2 adduct generates a key intermediate (alkylperoxo species) after a series of catalytic reactions. An O–O bond cleavage and a Criegee rearrangement process follow subsequently to yield an oxepine intermediate. Finally, attack on the substrate by an OH group leads to C–O bond breaking and forms the final product.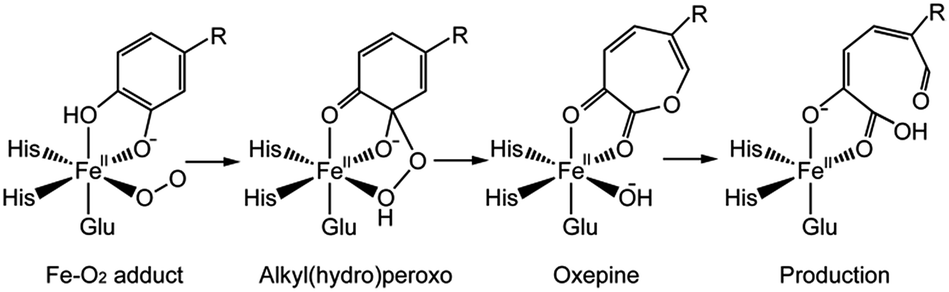 | ||
| Scheme 1 Proposed catalytic mechanism of extradiol dioxygenase. | ||
On the one hand, experimental studies have proved that the protonation of alkylperoxo is very important for O–O bond cleavage.19,20 It is generally accepted that the proton caused the protonation of alkylperoxo comes from the monoanionic catechol substrate, which has been confirmed by the available UV resonance Raman spectroscopic and electronic absorption spectroscopic data13,21–23 and the asymmetric binding X-ray crystal structure of the enzyme–substrate complex.24,25 However, the protonation mechanism of alkylperoxo is still under debate, because no intermediate is captured by experimental methods before the formation of Fe(II)–alkyl(hydro)peroxo species in wild-type (WT) enzyme reaction. On the other hand, research results have showed that the histidine residue (His200 of HPCD) near the active site has a marked influence on the activity of catalytic reaction.25–27 Considering the above two aspects, several researchers suggested that His200 could participate in the proton-transfer process and speculated that the monoanionic substrate gave a proton to the His200, which was then passed to superoxide anion.17,19 In order to explain the above mechanism in more detail, attempts based on DFT calculation have been carried out.28–31 Christian and co-workers28 suggested that the proton-transfer and oxygen addition process consisted of several steps (Scheme 2): (a) His200 abstracted a proton from the hydroxyl group of the catecholic substrate. (b) Formed H-bonding between distal oxygen atom (Od) of superoxide anion and the proton of His200 by rotational transition state of protonated His200. (c) Formed H-bonding between proximal oxygen atom (Op) of dioxygen and His200 by reorientation of His200. (d) Od added on aromatic ring, meanwhile, and the proton of His200 transferred to Op via the same transition state.
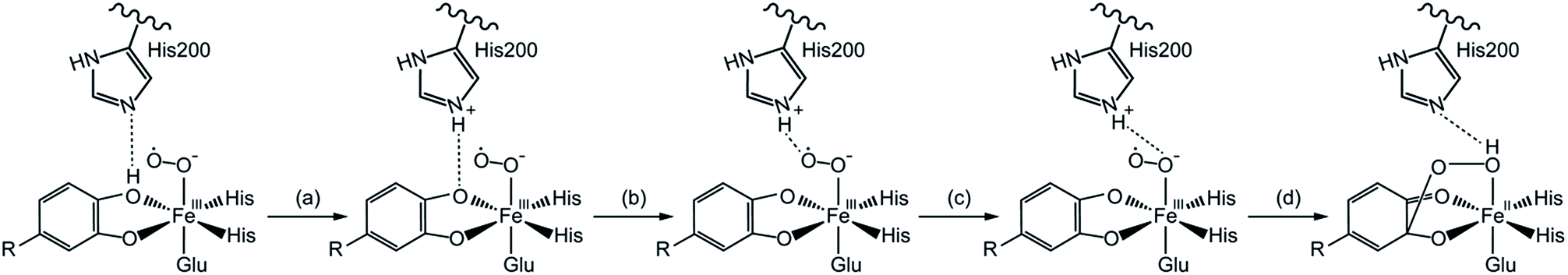 | ||
| Scheme 2 The proton transfers from the substrate to hydrogen-bonding for the Fe–O2 adduct.28 | ||
In the light of the results for recent mutant experiment of HPCD,32 the researchers speculated two different catalytic mechanisms of proton-transfer process for WT enzyme and mutant ones respectively. For WT enzyme, the proton passed from monoanionic substrate to superoxo anion via His200 as in the reaction pathway showed in Scheme 2. For mutant enzyme, as His200 was replaced by Asn200 or Gln200 which had no ability of accepting or providing proton, the proton-transfer process would perform without the help of His200 variants. However, another possibility, that the His200 or His200 variants didn't play a role in carrying proton, has been ignored. The proton-transfer process was carried out between the monoanionic substrates and superoxide anion directly, and the weak interaction between residue200 and the proton still has an effect on the whole system. It seemed a good interpretation on why the proton-transfer procedure still occurred when the His200 was replaced by Asn200 or Gln200.
To confirm our ideas on the catalytic mechanism of HPCD, quantum mechanics/molecular mechanics (QM/MM) calculations were implemented. We focused on the mechanism of proton transfer between the monoanionic substrate and dioxygen, and revealed the most favorable pathway for the formation of Fe–alkylperoxo species in WT HPCD by computational methods.
2 Computational methods and models
2.1 System build and classical MM calculations
The initial structure of HPCD (EC 1.13.11.15, and PDB code 4GHG) was obtained from the Protein Data Bank. The whole protein was used to study the oxygenation reaction. The water molecule above the Fe atom in the active site was replaced by one dioxygen molecule. All classical molecular mechanics (MM) calculations have been performed with AMBER ff14SB force field and TIP3P water model.33 The protonated states of the protein residues were predicted using the H++ website.34,35 The prepared structure was surrounded by a cubic box of TIP3P water molecules having a minimum distance of 8 Å between the protein and the end of the box, and neutralized by adding Na+ counter ions with the AMBER Leap module.36,37 Ligand was treated by gaff AMBER force field, charges and atom types for all amino acid residues and Fe atom were obtained from the AMBER library.Three steps of system minimizations were performed before molecular dynamics simulation. Firstly, position constraints were enforced for the whole protein and ligand with the 500 kcal mol−1 Å−2 harmonic potentials, the water molecules and counterions were first optimized. Secondly, a further minimization using a force constant of 10 kcal mol−1 Å−2 on the atoms in His200, His155, His214, Glu267, Arg243, Tyr257, Trp192, native substrate (homoprotocatechuate, HPCA), Fe atom, and dioxygen. Thirdly, the entire system was minimized without restraint. A cutoff radius of 10 Å was used for nonbonding interactions, and long-range electrostatic interactions were treated by the Particle Mesh Ewald (PME) method.38 A 50 ps simulation of the heating from 0 K to 300 K with force constants was taken place, followed by 200 ps simulation with constant pressure. Finally, a 14 ns molecular dynamics simulation in the NVT (300 K, 1 atm) ensemble was carried out. In order to obtain the initial structure for QM/MM work, a 50 ps cooling simulation was applied from 300 K to 0 K. Counterions and water molecules 5 Å away from the protein were removed. Thus, the final system containing 7775 atoms was the initial model for the next ONIOM study. An additional 6 ns MD simulation was performed to test the stability of the whole system. The 3D structure of HPCD reached a stable state after 14 ns where the RMSD of the protein backbone atoms converged to 2.2 Å (Fig. S1†).
2.2 QM/MM models and methods
In order to present the QM/MM calculations, ONIOM method implemented in the Gaussian 09 D.01 was used.39 The ONIOM method40 works by approximating the energy of the real system as a combination of the energies computed by less computationally expensive means. Specifically, the energy was computed as the energy of the model with corrections for the size difference between model and real and for the method accuracy difference between the molecular mechanics and the quantum mechanics method used for model (denoted MM and QM in the following equation):| EQM,real ≈ EMM,model + (EMM,real − EMM,model) + (EQM,model − EMM,model) |
| EONIOM,QM:MM = EMM,real + EQM,model − EMM,model |
A two-layer ONIOM method was used for the QM/MM study of the catalytic mechanism of HPCD. The geometry structures of QM parts were optimized by the B3LYP density functional theory method and MM parts were treated by AMBER ff14SB force field at the mechanical embedding level. Fe atom, imidazole rings of the His155, His214 and His200, CH2CO2− part of the Glu267, guanidine of the Arg243, HPCA and dioxygen were all selected as QM region. A valence double-zeta polarized basis set 6-31G(d) was used. The charge value of QM part was zero, and the total net charge of the whole system was −16.
The atomic charges of the QM region were treated by the RESP41 (restrained electrostatic potential) program at the B3LYP/6-31G(d) level of theory. The change of the QM region charge distribution during the reaction could reduce the accuracy of the ONIOM energies. Therefore, geometry optimizations of all stationary points were carried out by several steps. First of all, intermediates and transition structures were optimized by ONIOM calculations at mechanical embedding level with an initial set of charges. Secondly, an improved set of atomic charges of QM region were obtained by using the RESP procedure, after that, a further optimization was carried out with the improved charges. Finally, the above step could be repeated several times until the ONIOM energy difference between the last two rounds was very slight. The ONIOM energy of the optimized structure was computed at the mechanical embedding (ME) levels with a larger triple-zeta basis sets 6-311+G(d,p). Meanwhile, the ONIOM energy was combined with a thermal correction to the Gibbs free energy computed at the ONIOM-ME (B3LYP/6-31G(d):Amber) level.
3 Computational results and discussion
Previous evidences42,43 proved that the Fe ion was formed as quintet Fe(II) in the crystal, since the combination of quintet Fe(II) with triplet O2 led to three possibilities of spin combinations, the high-spin (septet), intermediate spin (quintet), and low spin (triplet). The previous DFT model studies28–30 suggested that the quintet spin state or septet state might be the ground state. Meanwhile, the experimental result indicated a quintet ground state by characterizing a high-spin S1 = 5/2 Fe(III) antiferromagnetically (AF) coupled to S2 = 1/2 superoxo radical in H200N mutant experiment of HPCD with 4NC substrate.27 However, for one thing, the electronic structure of Fe–superoxo species for mutant enzyme may be different from the WT one. For another, the catalytic mechanism suggested in this study is different from the former ones. Hence, it is necessary to discuss all possibilities for triplet state, quintet state and septet state.3.1 The character of reactive species (Fe–O2 adduct)
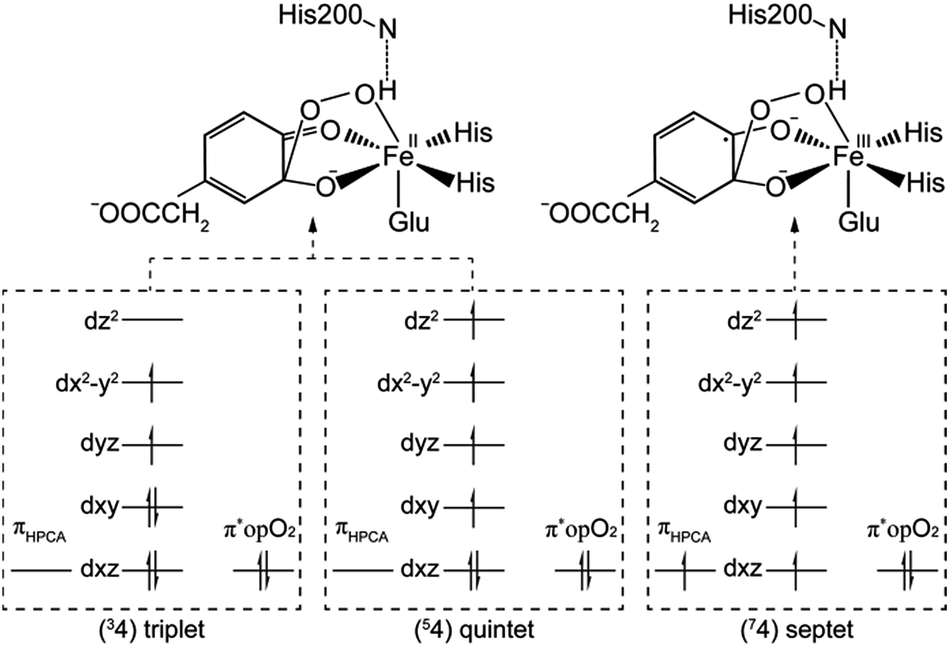
After further analysis, different orientation of Od form different reactive species. There are two possibilities orientations, the Op–Od bond axes near the plane of Fe–Op–O2 for 1, and the Op–Od bond axes near the plane of Fe–Op–O1 for 1′, as shown as in Scheme 4. A DFT calculation is carried out to obtain optimized structures (shown in Fig. S2†) and relative energies for 1 and 1′ in quintet state. The result shows that the difference of relative energy between 51 and 51′ is very slight (less than 0.5 kcal mol−1). Because Od is nearly located between the proton and Op in 51′, the proton is very hard to move to Op directly around Od. Hence, it seems that 51′ is not the reactive species of the catalytic mechanism we suggest. Differently, the orientation of Od is vertical in plane of H1–O1–Op in 51, the movement of proton in plane of H1–O1–Op is unblocked. Therefore, we put our attention on the 1 as reactive species in this study. The valence electron configurations of reactive species 1 for different spin states are shown in Fig. 1, which are unambiguously supported by the analysis of spin population distribution (Table S1†). For triplet state, three unpaired α-spins are located on the iron, and one β-spin is situated on the dioxygen group (the unpaired electron occupies the out of Fe–Op–Od plane (op) π*-orbital). For quintet state, five unpaired α-spins are located on the iron, and one β-spin is situated on the dioxygen group. For septet state, five unpaired α-spins are located on the iron, and one α-spin is situated on the dioxygen group. Obviously, the Fe ions are high spins in both quintet state and septet state. And the 51 is presented as the character of an high spin S1 = 5/2 Fe(III) AF coupled to S2 = 1/2 superoxo radical which coincide with the experimental result of trapping and characterizing Fe(III)–superoxo intermediate of HPCD.27 However, the septet Fe(III)–superoxide intermediate (71) involving a ferromagnetic coupling of Fe(III) with superoxo radical is the most stable form, lower in energy than that in the quintet state (51) and the triplet state (31) by 10.5 and 19.2 kcal mol−1, respectively (Fig. 3).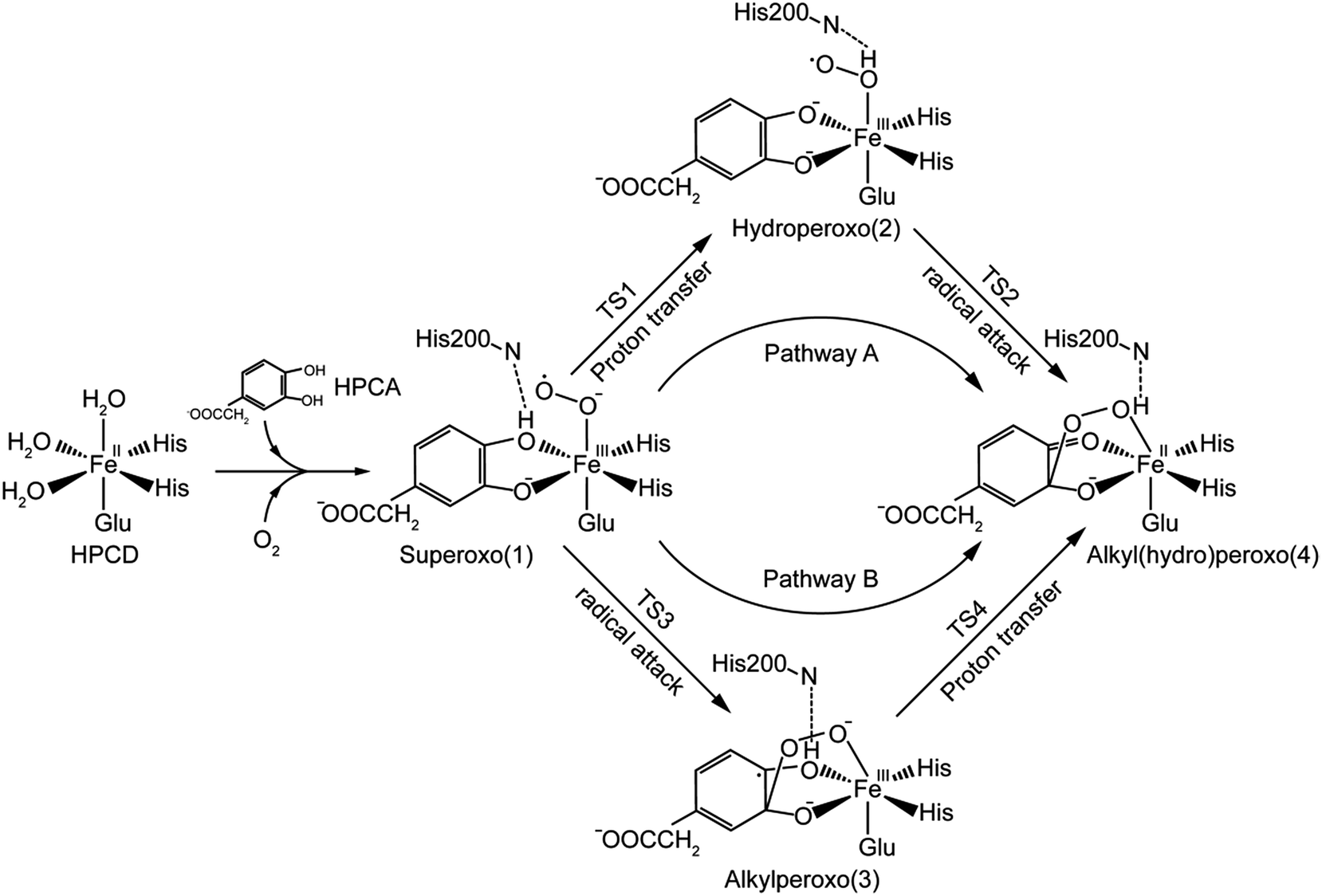 | ||
| Scheme 3 The catalytic mechanism for the formation of alkyl(hydro)peroxo species. | ||
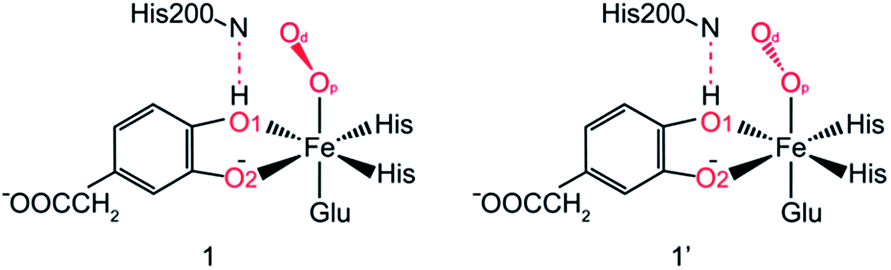 | ||
| Scheme 4 Different Od orientation of Fe–O2 adducts for WT HPCD. | ||
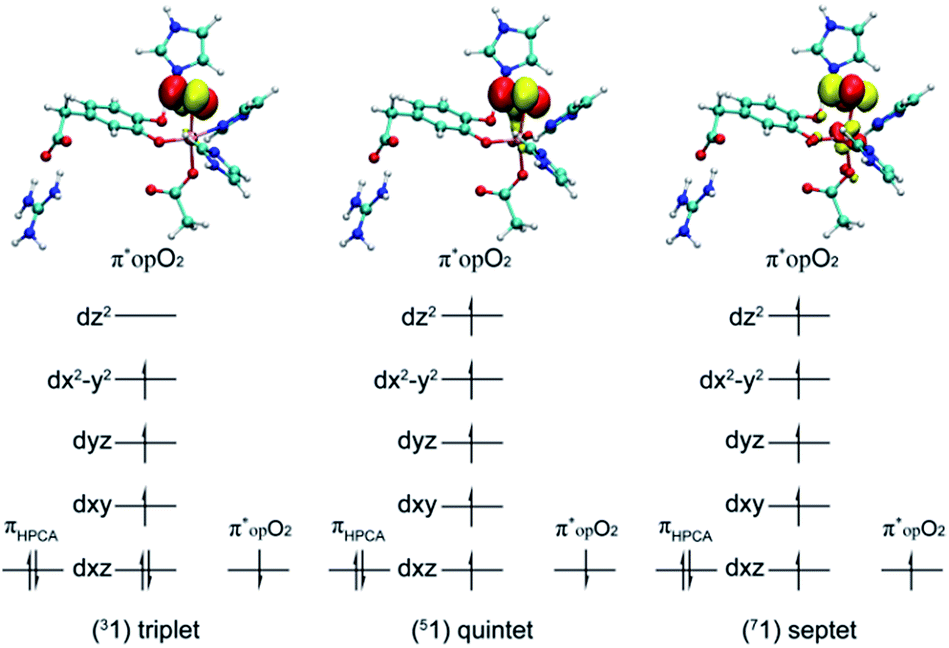 | ||
| Fig. 1 The valence electron configurations of Fe–O2 adducts for triplet state, quintet state and septet state. | ||
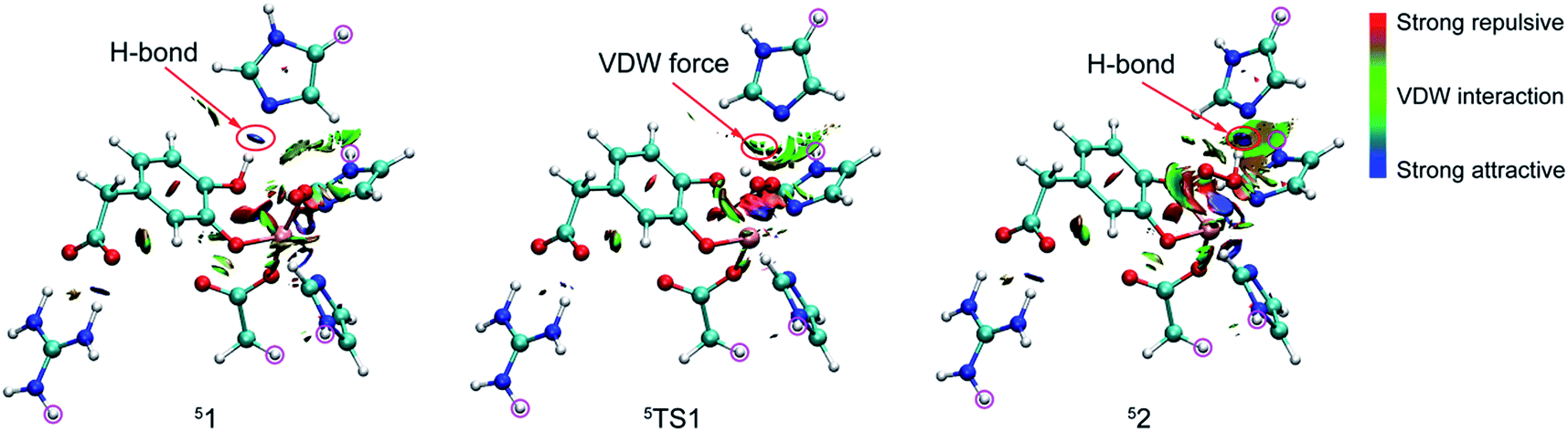 | ||
| Fig. 2 RDG isosurface map for weak interaction of 51, 5TS1, and 52. | ||
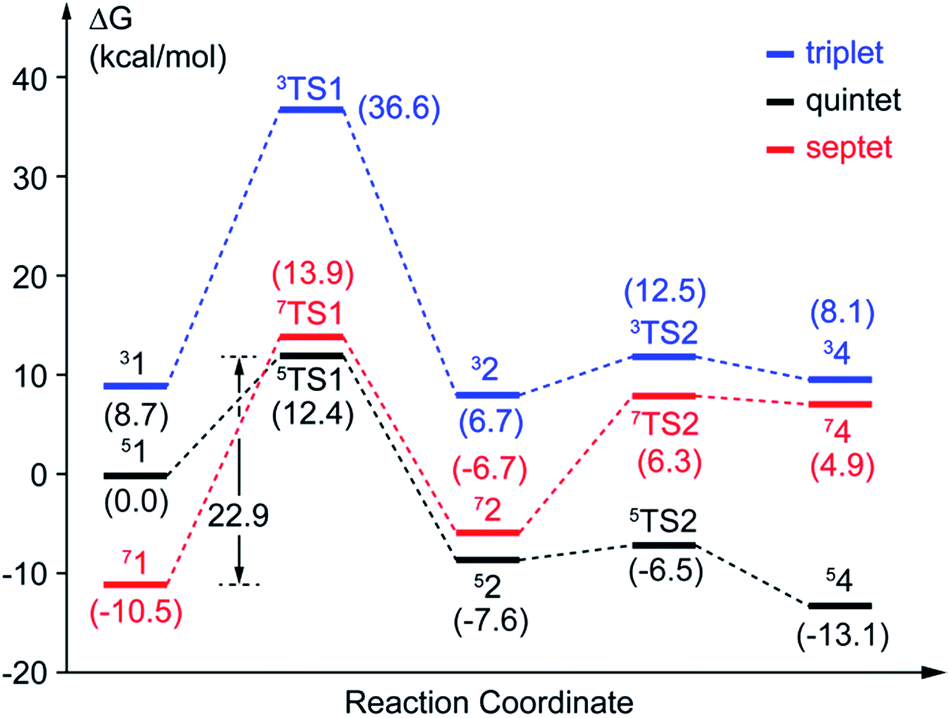 | ||
| Fig. 3 Potential energy profiles of pathway A. The values with parentheses are relative free energies. | ||
3.2 Reaction mechanism
In this study, two parallel pathways contain proton-transfer and oxo attack process are carried out to form alkyl(hydro)peroxo species. It is described as pathway A and B in Scheme 3. In pathway A, proton-transfer reaction takes place followed by Od attacks on the aromatic ring. In pathway B, Od attacks and then proton transfers.(a) Proton transfer. The above research illustrates that 31, 51, and 71 are all presented as superoxo radicals. By attacking of the superoxo radical anion, the proton transfers from O1 of HPCA to Op of dioxygen. As shown in Fig. 3, the DFT-calculated barrier (ΔG‡Q) on quintet state potential energy surface (PES) from 51 to 5TS1 is 12.4 kcal mol−1, which can be compared with that (ΔG‡T) of 27.9 kcal mol−1 on triplet state PES from 31 to 3TS1. This transition state stabilization of about 15.5 kcal mol−1 makes 5TS1 the energetically lowest transition state among all three proton-transfer transition states. This reaction picture is a typical TSR scenario. Starting from the septet ground state 1, a spin state transition to the quintet surface has to occur during this energetically most favorable proton-transfer step. The effective activation barrier ΔG‡eff from 71 to 5TS1 is 22.9 kcal mol−1. Additionally, triplet state is not likely to be involved during the proton-transfer process since it remains above the quintet and septet PESs. The key parameter of the intermediates and transition states are shown in Fig. S3.† According to the analysis of spin population distribution (Table S1†), the spin population distribution has little change in 1, TS1 and 2. It is indicated that there is little electron migration for this elementary reaction process. All optimized structures and key structural parameters of quintet state are shown in Fig. 4.
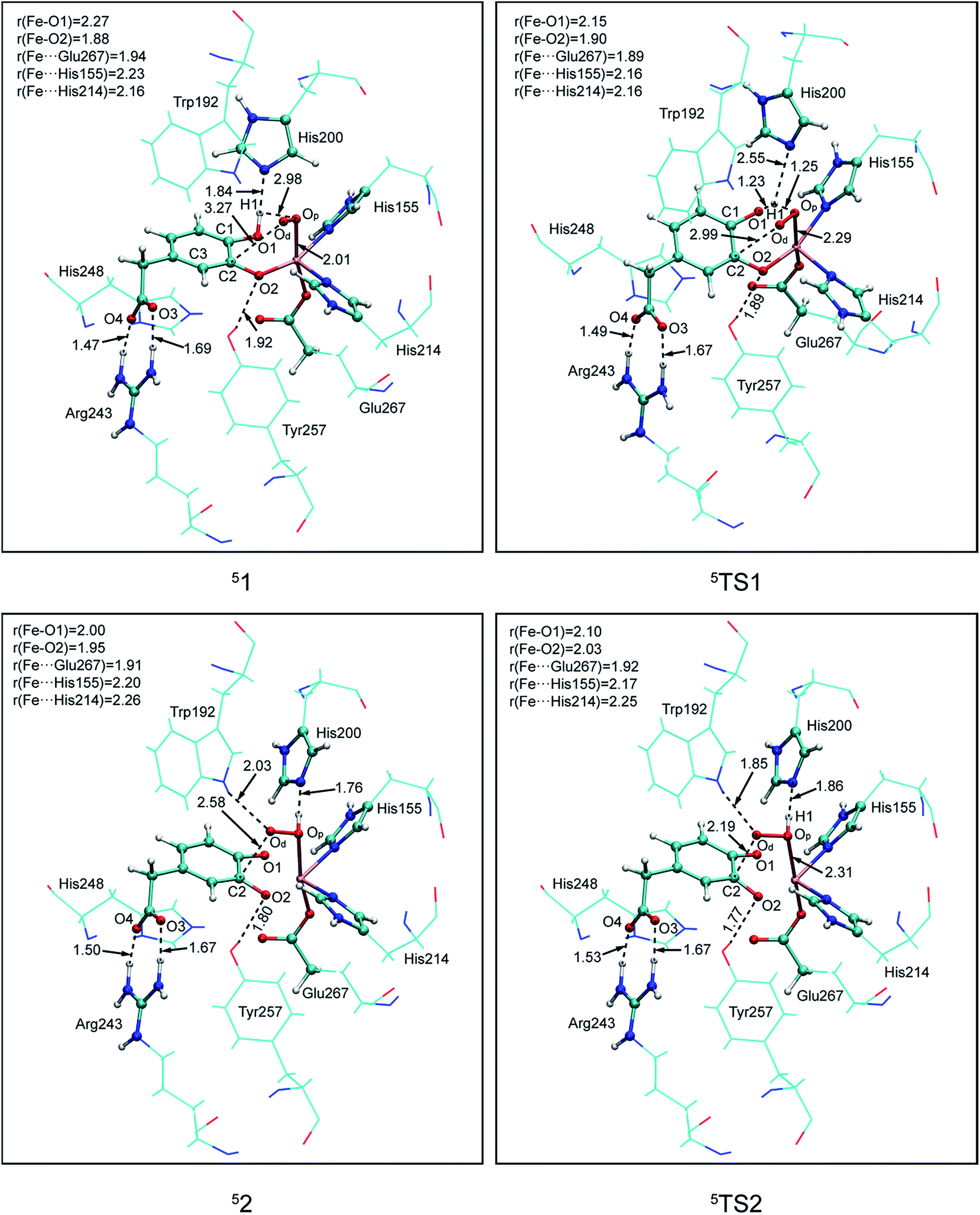 | ||
| Fig. 4 Calculated key structural parameters for intermediates and transition states of pathway A in quintet. Bond lengths (Å) are shown. | ||
Reduced density gradient (RDG) and sign(λ2)ρ function is employed to investigate the weak interaction between His200 and the proton. All calculations are carried out at B3LYP/6-31G(d) level for the QM region and the link atoms are signed by purple circle. As shown in Fig. 2, the blue, green, and red colors of RDG surfaces indicate the strong attractive, van der Waals interaction, and strong repulsive respectively. There is a strong H-bond between His200 and the proton with 1.84 Å in 51. And then length of the H-bond increases to 2.55 Å in 5TS1. The color of the RDG surface is situated between blue and green, which indicates that the weak interaction is correspondingly weakened accompanied by an increase of system energy. Finally, the H-bond interaction enhances gradually in pace with the free energy of the system cut down, and the product (52) is formed. Obviously, the result indicates that the weak interaction makes contribution to the free energy barrier. All analysis are performed and visualized by means of Multiwfn software.44
(b) Hydroperoxo attack. In this step, the distal oxygen of hydroperoxide radical attacks the C2 of aromatic ring to give the intermediate 4. The theoretical calculation shows that different electron-transfer mechanisms lead to different electron structures of alkyl(hydro)peroxo (shown in Fig. 7). For triplet state, one electron transfers from aromatic ring to superoxo accompanied with the gradually reducing distance of C2 and Od (2.84 Å in 32, 2.01 Å in 3TS2, and 1.54 Å in 34, in Fig. S3†), resulting in the formation of C2–Od bond. But at the same time, another electron transfers from the electron-rich aromatic ring to the iron along with the increasing Fe–O1 bond length (1.87 Å in 32, 2.17 Å in 3TS2, and 2.31 Å in 34, in Fig. S3†). For quintet state, though the electron transfers in an analogous mechanism as in the triplet state, the bond lengths are different. When one electron transfers from aromatic ring to superoxo, the distance of C2 and Od reduces gradually from 2.58 Å in 52, 2.19 Å in 5TS2, to 1.53 Å in 54, as shown in Fig. 4, accompanied with the increasing Fe–O1 bond length (2.00 Å in 52, 2.10 Å in 5TS2, and 2.24 Å in 54). For the septet state, one electron transfers from aromatic ring to superoxo accompanied with the gradually reducing distance of C2 and Od (2.86 Å in 72, 1.84 Å in 7TS2, and 1.52 Å in 74, in Fig. S4†). The mechanism for the septet state differs from the former two. Since there is little electron transfer from the aromatic ring to the iron, the bond length of Fe–O1 changes very slightly (1.99 Å in 72, 2.00 Å in 7TS2, and 2.00 Å in 74) and Fe(III)–alkyl(hydro)peroxo is formed. This hydroperoxo attack process is thermodynamically endothermic in triplet and septet state but exothermic in quintet state. The quintet state is ground state for lowest energy profiles (shown in Fig. 3), with a very tiny barrier of about 1.1 kcal mol−1. It seems that this step happens very easily.
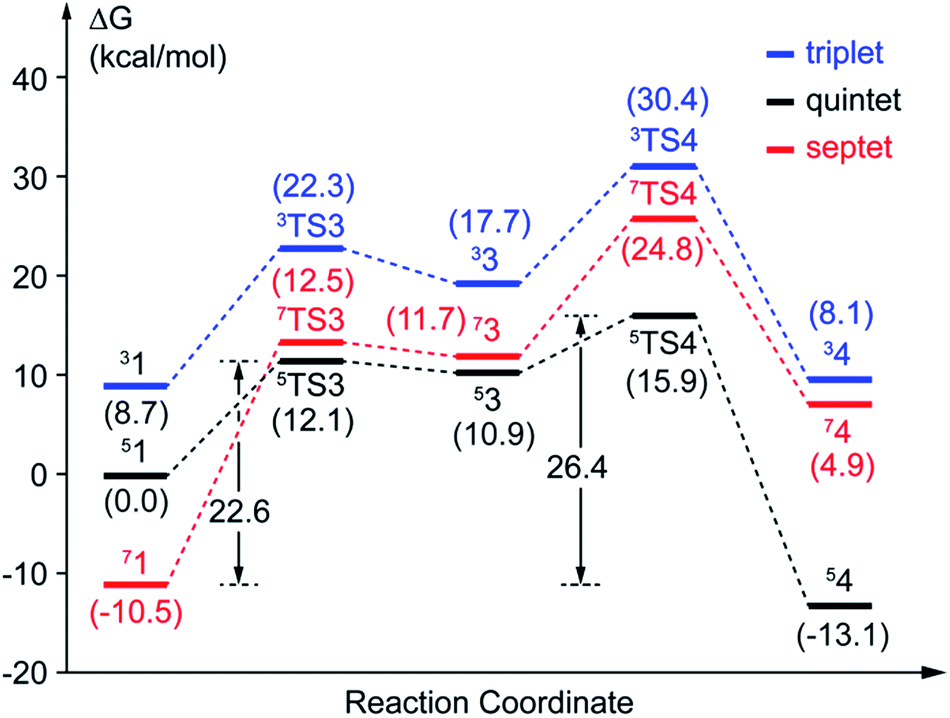
(a) Superoxo attack. This step involves a radical attack of superoxo radical at C2 position of the aromatic ring, and it begins from intermediate 1, ends in intermediate 3 via TS3. The result of attack is the bonding of C2–Od, and the alkylperoxo species (intermediate 3) is formed as follow. The distance of C2 and Op is decreased (from 3.16 Å to 1.45 Å via 1.81 Å in triplet state, from 3.27 Å to 1.49 Å via 1.76 Å in quintet state, as shown in Fig. 5, and from 3.27 Å to 1.48 Å via 1.77 Å in septet state) along with one electron transfer from substrate to superoxo. Seeing from the potential energy profiles in Fig. 6, 5TS3 is the lowest-lying transition state for superoxo attack among all three spin states. But the septet state is the ground state of reactive species. This process is also a TSR scenario, in which the initially excited quintet states, crossover through the high-spin ground septet states to promote superoxo attack. The effective activation barrier is 22.6 kcal mol−1 (shown in Fig. 6).
 | ||
| Fig. 5 Calculated key structural parameters for intermediates and transition states of pathway B in quintet. Bond lengths (Å) are shown. | ||
 | ||
| Fig. 6 Potential energy profiles of pathway B. The values with parentheses are relative free energies. | ||
(b) Proton transfer. The proton transfer occurs between O1 and Op. There are two different type of reaction mechanism: one is a proton-coupled electron transfer (PCET) process with one net electron flows toward Fe in triplet and quintet states; and the other is a proton transfer procedure without electron migration in septet state. This reaction step also results in producing three kinds of electron characteristic of alkyl(hydro)peroxo species (Fig. 7). The relative free energy barrier is 12.7 kcal mol−1 in triplet state, 5.0 kcal mol−1 in quintet state, and 13.1 kcal mol−1 in septet state, respectively (shown in Fig. 6). However, the relative energy of TS4 is much higher than TS3. The reaction rate of the pathway B is affected by both superoxo attack and proton transfer.
 | ||
| Fig. 7 The electron configurations of alkyl(hydro)peroxo for triplet state, quintet state and septet state. | ||
Additionally, oxo attack can also probably occur between distal oxygen and C1 of HPCA. But this possibility has been ruled out on the basis of a recent research.31
3.3 The favorable reaction pathway
In comparison with the total effective activation barrier of the two pathways, we find that the pathway A (ΔG‡teff is 22.9 kcal mol−1) is more favorable than pathway B (ΔG‡teff is 26.2 kcal mol−1). Additionally, we also do some research on the pathway of proton transfer via His200, which is mentioned in previous studies. It is regrettable that we have not located a transition state for this proton-transfer process, and no stable intermediate structure for protonation of His200 is found. Instead, a PES scan of proton transfer from substrate to His200 is carried out by changing the distance of O1 and H1 in 0.1 Å increments. Meantime, the intrinsic reaction coordinate (IRC) connecting 1, TS1, and 2 is generated by the same abscissa as the distance of O1 and H1. The results are shown in Fig. 8, the relative energy of proton transfer from substrate to His200 is monotone increasing along with the increase of the r(O1–H1), and the estimated value of relative energy barrier is no less than 30 kcal mol−1. On the contrary, the relative energy barrier of 1–TS1–2 is no more than 25 kcal mol−1. Thus, proton transfer from substrate to proximal oxygen is more favorable than to His200. In summary, pathway A is both thermodynamically and kinetically more favorable than the others.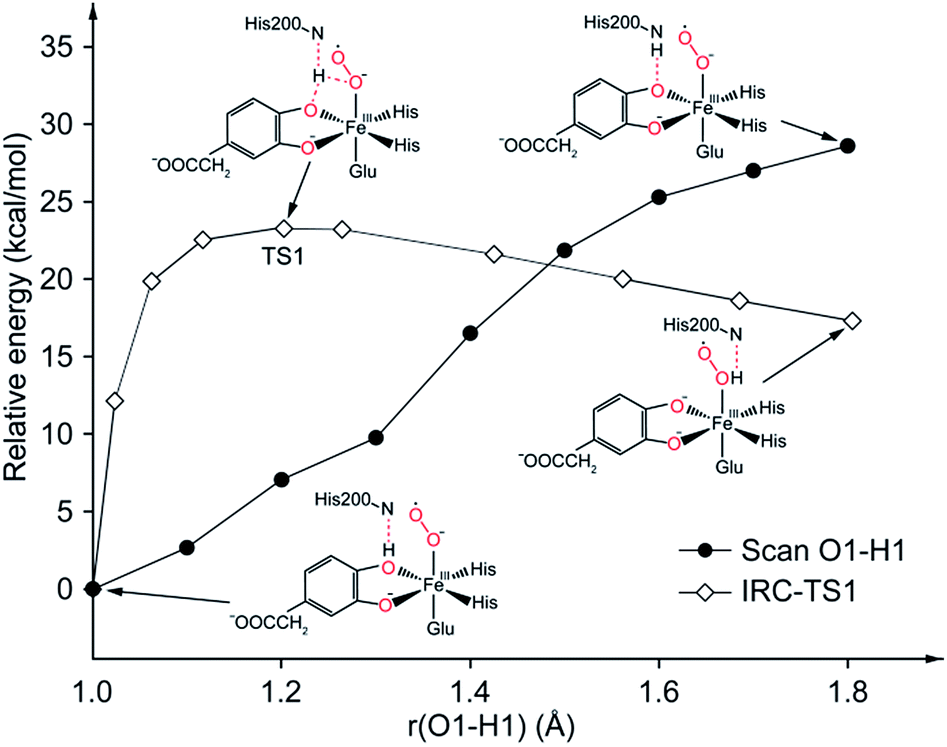 | ||
| Fig. 8 PES scan of proton transfer from substrate to His200 and IRC connecting 1, TS1, and 2. All calculations are performed at ONIOM (B3LYP/6-31G(d):Amber) level in quintet state. | ||
4 Conclusions
The two parallel pathways to form consensus alkyl(hydro)peroxo species are shown in this study. Pathway A can be described as proton transfer from monoanionic catechol substrate to proximal oxygen of dioxygen with subsequently distal oxygen of hydroperoxo attacks on the aromatic ring. Pathway B is performed by the attack of distal oxygen and followed with a PCET procedure. From the different potential energy profiles of three spin states, we can draw the conclusion that the triplet state is no catalytically relevance for the much higher energy profile than the others. Meanwhile, the quintet is the ground state for the whole process except intermediate 1. The first step of the two pathways is a TSR scenario.His200 is found not to work as a proton carrier, but to provide a weak interaction with the proton to stabilize the active center. The proton transfer takes place between the catecholic substrates and superoxide anion directly. By comparing the pathway A, pathway B, and proton-transfer process via His200 from the viewpoint of chemical thermodynamics and kinetics, the pathway A is the most favorable one of the three.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 21476091), Natural Science Foundation of Guangdong province, China (No. 2015-A030313801), Natural Science Foundation for Young Talent Project of Guangdong, China. We thank Richard Chang and Ziang Li for technical assistance.Notes and references
- G. Lin, G. Reid and T. D. H. Bugg, Chem. Commun., 2000, 1119–1120 RSC.
- F. H. Vaillancourt, J. T. Bolin and L. D. Eltis, Crit. Rev. Biochem. Mol. Biol., 2006, 41, 241–267 CrossRef CAS PubMed.
- E. G. Kovaleva and J. D. Lipscomb, Nat. Chem. Biol., 2008, 4, 186–193 CrossRef CAS PubMed.
- T. D. H. Bugg, in Iron-Containing Enzymes: Versatile Catalysts of Hydroxylation Reactions in Nature, The Royal Society of Chemistry, 2011, pp. 42–66 Search PubMed.
- M. D. White and E. Flashman, Curr. Opin. Chem. Biol., 2016, 31, 126–135 CrossRef CAS PubMed.
- K. D. Koehntop, J. P. Emerson and L. Que, JBIC, J. Biol. Inorg. Chem., 2005, 10, 87–93 CrossRef CAS PubMed.
- P. C. A. Bruijnincx, G. van Koten and R. J. M. Klein Gebbink, Chem. Soc. Rev., 2008, 37, 2716–2744 RSC.
- E. G. Kovaleva, M. B. Neibergall, S. Chakrabarty and J. D. Lipscomb, Acc. Chem. Res., 2007, 40, 475–483 CrossRef CAS PubMed.
- M. M. Mbughuni, M. Chakrabarti, J. A. Hayden, K. K. Meier, J. J. Dalluge, M. P. Hendrich, E. Münck and J. D. Lipscomb, Biochemistry, 2011, 50, 10262–10274 CrossRef CAS PubMed.
- K. L. Henderson, V. H. Le, E. A. Lewis and J. P. Emerson, JBIC, J. Biol. Inorg. Chem., 2012, 17, 991–994 CrossRef CAS PubMed.
- S. L. Groce and J. D. Lipscomb, J. Am. Chem. Soc., 2003, 125, 11780–11781 CrossRef CAS PubMed.
- M. W. Vetting, L. P. Wackett, L. Que, J. D. Lipscomb and D. H. Ohlendorf, J. Bacteriol., 2004, 186, 1945–1958 CrossRef CAS PubMed.
- F. H. Vaillancourt, G. Labbé, N. M. Drouin, P. D. Fortin and L. D. Eltis, J. Biol. Chem., 2002, 277, 2019–2027 CrossRef CAS PubMed.
- M. Sylvestre, T. Macek and M. Mackova, Curr. Opin. Biotechnol., 2009, 20, 242–247 CrossRef CAS PubMed.
- S. Dai, F. H. Vaillancourt, H. Maaroufi, N. M. Drouin, D. B. Neau, V. Snieckus, J. T. Bolin and L. D. Eltis, Nat. Struct. Biol., 2002, 9, 934–939 CrossRef CAS PubMed.
- E. G. Kovaleva and J. D. Lipscomb, Science, 2007, 316, 453–457 CrossRef CAS PubMed.
- J. D. Lipscomb, Curr. Opin. Struct. Biol., 2008, 18, 644–649 CrossRef CAS PubMed.
- E. G. Kovaleva and J. D. Lipscomb, Biochemistry, 2008, 47, 11168–11170 CrossRef CAS PubMed.
- H. J. Cho, K. Kim, S. Y. Sohn, H. Y. Cho, K. J. Kim, M. H. Kim, D. Kim, E. Kim and B. S. Kang, J. Biol. Chem., 2010, 285, 34643–34652 CrossRef CAS PubMed.
- S. Chatterjee, D. Sheet and T. K. Paine, Chem. Commun., 2013, 49, 10251–10253 RSC.
- F. H. Vaillancourt, S. Han, P. D. Fortin, J. T. Bolin and L. D. Eltis, J. Biol. Chem., 1998, 273, 34887–34895 CrossRef CAS PubMed.
- F. H. Vaillancourt, C. J. Barbosa, T. G. Spiro, J. T. Bolin, M. W. Blades, R. F. B. Turner and L. D. Eltis, J. Am. Chem. Soc., 2002, 124, 2485–2496 CrossRef CAS PubMed.
- D. M. Arciero and J. D. Lipscomb, J. Biol. Chem., 1986, 261, 2170–2178 CAS.
- E. G. Kovaleva and J. D. Lipscomb, Biochemistry, 2012, 51, 8755–8763 CrossRef CAS PubMed.
- S. L. Groce and J. D. Lipscomb, Biochemistry, 2005, 44, 7175–7188 CrossRef CAS PubMed.
- J. P. Emerson, E. G. Kovaleva, E. R. Farquhar, J. D. Lipscomb and L. Que, Proc. Natl. Acad. Sci., 2008, 105, 7347–7352 CrossRef CAS PubMed.
- M. M. Mbughuni, M. Chakrabarti, J. A. Hayden, E. L. Bominaar, M. P. Hendrich, E. Münck and J. D. Lipscomb, Proc. Natl. Acad. Sci., 2010, 107, 16788–16793 CrossRef CAS PubMed.
- G. J. Christian, S. Ye and F. Neese, Chem. Sci., 2011, 3, 1600–1611 RSC.
- G. Dong, S. Shaik and W. Lai, Chem. Sci., 2013, 4, 3624–3635 RSC.
- G. Dong and W. Lai, J. Phys. Chem. B, 2014, 118, 1791–1798 CrossRef CAS PubMed.
- G. J. Christian, F. Neese and S. Ye, Inorg. Chem., 2016, 55, 3853–3864 CrossRef CAS PubMed.
- E. G. Kovaleva, M. S. Rogers and J. D. Lipscomb, Biochemistry, 2015, 54, 5329–5339 CrossRef CAS PubMed.
- D. A. Case, J. T. Berryman, R. M. Betz, D. S. Cerutti, T. E. Cheatham III, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, T. Luchko, R. Luo, B. Madej, K. M. Merz, G. Monard, P. Needham, H. Nguyen, H. T. Nguyen, I. Omelyan, A. Onufriev, D. R. Roe, A. Roitberg, R. Salomon-Ferrer, C. L. Simmerling, W. Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, D. M. York and P. A. Kollman, AMBER 2015, University of California, San Francisco, 2015 Search PubMed.
- J. C. Gordon, J. B. Myers, T. Folta, V. Shoja, L. S. Heath and A. Onufriev, Nucleic Acids Res., 2005, 33, W368–W371 CrossRef CAS PubMed.
- R. Anandakrishnan, B. Aguilar and A. V. Onufriev, Nucleic Acids Res., 2012, 40, W537–W541 CrossRef CAS PubMed.
- P. Campomanes, U. Rothlisberger, M. Alfonso-Prieto and C. Rovira, J. Am. Chem. Soc., 2015, 137, 11170–11178 CrossRef CAS PubMed.
- K. G. Sprenger, V. W. Jaeger and J. Pfaendtner, J. Phys. Chem. B, 2015, 119, 5882–5895 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas,J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford, CT, 2009.
- T. Vreven, K. S. Byun, I. Komáromi, S. Dapprich, J. A. Montgomery, K. Morokuma and M. J. Frisch, J. Chem. Theory Comput., 2006, 2, 815–826 CrossRef CAS PubMed.
- J. Wang, P. Cieplak and P. A. Kollman, J. Comput. Chem., 2000, 21, 1049–1074 CrossRef CAS.
- A. Marquez and J. Fernandez Sanz, J. Am. Chem. Soc., 1992, 114, 10019–10024 CrossRef CAS.
- S. Ye and F. Neese, Proc. Natl. Acad. Sci., 2011, 108, 1228–1233 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spin population distribution, optimized structures in triplet and septet states, and Cartesian coordinates of optimized structures. See DOI: 10.1039/c7ra08080h |
| This journal is © The Royal Society of Chemistry 2017 |