
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Temperature, ultrasound and redox triple-responsive poly(N-isopropylacrylamide) block copolymer: synthesis, characterization and controlled release
Yin-Ku Lina,
Yung-Ching Yub,
Shiu-Wei Wangb and
Ren-Shen Lee *b
*b
aDepartment of Traditional Chinese Medicine, Chang Gung Memorial Hospital at Keelung, Keelung, Taiwan
bDivision of Natural Science, Center of General Education, Chang Gung University, 259 Wen-Hwa 1st Road, Guishan Dist., Tao-Yuan 33302, Taiwan. E-mail: shen21@mail.cgu.edu.tw; Fax: +886-3-2118700
First published on 6th September 2017
Abstract
Triple stimuli-responsive polymers PNiPAAm-S-S-PXCL containing a disulfide (–S–S–) bond as a junction point between hydrophilic and hydrophobic chains were synthesized through ring-opening polymerization (ROP) and nucleophilic substitution. When the polymer solutions were treated with reducing agents and exposed to ultrasound, significant hydrolysis of the amide bonds was observed. The morphology of the particles and the changes in the fluorescence intensity of Nile red revealed that the copolymers underwent micellization or dissociation transitions in water in response to temperature variation, reducing agent treatment, and ultrasound. The release rate could be greatly increased and adjusted remotely under high intensity focused ultrasound in the presence of dithiothreitol. The nanoparticles exhibited slight toxicity against HeLa cells in the concentration range 10–1000 μg mL−1. The DOX-loaded PNiPAAm21-S-S-PMCL25 micelles effectively inhibited the proliferation of HeLa cells with a half-maximal inhibitory concentration of 2.33 μg mL−1. The multiple stimuli-responsive polymeric nanoparticles show promising potential as new nanocarriers for controlled release.
Introduction
For optimal drug release, stimuli-responsive amphiphilic copolymers are of great interest and have been extensively investigated as potential nanocarrier systems.1 The stimuli that trigger drug release from nanocarriers can be broadly classified as internal (e.g., pH, glutathione, and enzymes) and external (e.g., physical stimuli, heat, light, magnetic fields, electrical fields and ultrasound).2 Recently, multi-responsive polymers, which combine two or more responsive functions, have attracted academic attention. Various stimuli-responsive systems capable of responding to two or more environmental changes have attracted particular attention for two reasons: (i) the multi-responsive feature can be elicited concurrently or sequentially using more than one stimulus, providing more effective spatiotemporal control; (ii) because behavioral changes in biological systems are often a result of a combination of environmental changes rather than a single factor, multi-responsive materials offer an ideal artificial platform for mimicking natural biological processes.3–16 Because of the implications, the development of simple synthetic methods that can obtain multi-stimuli-responsive materials with precise control over their architecture and functionality is imperative.Poly(N-isopropylacrylamide) (PNiPAAm) is water soluble at room temperature but undergoes transition from a hydrated coil to a dehydrated globule at its lower critical solution temperature (LCST) approximately 32 °C, close to body temperature. Because of their temperature-sensitivity and favorable biocompatibility, PNiPAAm-containing materials have been widely utilized in drug delivery systems and temperature-targeted therapy materials.17–19
Redox is a type of internal stimulus for copolymer micelles. The redox responsive copolymer micelle containing a central disulfide (–S–S–) bond has attracted increasing attention because the –S–S– bond can be cleaved specifically in the reducing environment inside the cell by using redox reagents such as glutathione (GSH), whereas it is stable in the oxidizing environment outside the cell. Moreover, cancer cells often exhibit elevated levels of glutathione, which may enhance the therapeutic efficacy and reduce the probability of drug resistance in cancer cells.20–22 However, physical stimuli, including light irradiation, magnetic field, and ultrasonic treatment, have attracted much attention lately, because they possess certain advantages related to their remote and spatiotemporally controlled application.23
Ultrasound is one of the most promising external triggers for controlled drug release from polymeric matrixes because of its deep penetration, non-invasiveness and controllable properties.24 High-intensity focused ultrasound (HIFU) was originally developed as an extracorporeal tool for the treatment of tumors because of its thermal effects.25–29 The ultrasonic wave can be focused in a small area. This means that the intensity is quite high only in the focal spot, while in other areas the intensity can be sufficiently low and accepted by the human body. Previously, Xia et al. synthesized the block copolymer PEG-S-S-PLA containing the central labile disulfide linkage between poly(ethylene glycol) (PEG) and poly(L-lactic acid) (PLA) segments and proved that copolymer degradation occurs after HIFU irradiation or GSH treatment.30
In our previous research, we have found that PNiPAAm block copolymer showed intriguing changes in structures and properties in response to temperature and pH/or photo-dual-changes.31,32 While, the synthesis of multi-functional nanocarriers for drug delivery and the design of new multi-stimuli responsive means with precise control are equally vital and must be realized. In this paper, we report the synthesis of multi-responsive amphiphilic Poly(N-isopropyl acrylamide) block poly(4-substituted-ε-caprolactone) (PNiPAAm-S-S-PXCL) copolymer containing disulfide (–S–S–) bonds as junction points between hydrophilic and hydrophobic chains. Here, the disulfide moieties, carboxylate groups and PPNiPAAm block endow the block copolymer with redox-, ultrasound, and thermal responsive properties, respectively. We investigated the physicochemical and stimuli–degradable properties of these micelles in an aqueous phase through fluorescence spectroscopy, dynamic light scattering (DLS), and transmission electron microscopy (TEM). Because of the combined treatment of HIFU irradiation and DTT, the release rate of entrapped payloads could be greatly enhanced and remotely controlled. Finally, we examined the effectiveness of PNiPAAm-S-S-PXCL nanoparticles as drug carriers using hydrophobic indomethacin as a model drug and studied the cytotoxicity of these nanoparticles when internalized into human cervical cancer HeLa cells.
Experimental section
Materials
N-Isopropyl acrylamide (NiPAAm), 2-mercaptoethanol, benzoyl peroxide, 4-methylcyclohexanone, 4-phenylcyclohexanone, 3-mercapto propionic acid (MPA), hafnium(IV) chloride tetrahydrofuran complex (HfCl4·2THF), pyrene, indomethacin (IMC), and Nile red (NR) were purchased from Aldrich Chemical Co. (Milwaukee, WI). In addition, m-chloroperoxybenzoic acid was purchased from Fluka Chemical Co. (Buchs SG1, Switzerland), and stannous octoate (SnOct2) was purchased from Strem Chemical Inc. (Newburyport, MA). NiPAAm (97%) was recrystallized twice from toluene and dried under vacuum prior to use. PNiPAAm-OH, 4-methyl-ε-caprolactone (MCL), and 4-phenyl-ε-caprolactone (BCL) were prepared according to previously described methods,33,34 and 2-(2-pyridyldithio)-ethanol and PNiPAAm-SH were prepared according to reported procedures.35 Doxorubicin hydrochloride (99%) (Aldrich, Saint Louis, MO) was deprotonated to obtain hydrophobic DOX, as previously described.36 N,N-Dimethyl formamide (DMF), and toluene were dried and distilled under calcium hydride. Other high-pressure liquid chromatography (HPLC) grade solvents, such as tetrahydrofuran (THF), dimethylsulfoxide (DMSO), methanol, chloroform (CHCl3), and n-hexane, were purchased from Merck (Darmstadt, Germany). A Milli-Q Plus system (Waters, Milford, MA) was used to obtain ultrapure water. Dulbecco's modified Eagle's medium (DMEM), trypsin/EDTA, 100× antibiotic–antimycotic, and Hoechst 33342 nuclei dye were purchased from Gibco, Invitrogen Corp. (Carlsbad, CA). Fetal bovine serum (FBS) was obtained from Biological Industry (Kibbutz Beit Haemek, Israel). A CellTiter 96® AQueuous One Solution kit was obtained from Promega (Fitchburg, WI).Synthesis of PNiPAAm-S-S-PXCL diblock copolymers
The polymerization reaction to create PNiPAAm21-S-S-PMCL25 proceeded as follows: 2-(2-pyridyldithio)-ethanol (Py-S-S-(CH2)2OH, 0.12 g, 0.64 mmol), serving as an initiator, and MCL (2.46 g, 19.20 mmol) were introduced into a flask, and then dissolved in 50 mL toluene under a dry nitrogen stream. Subsequently, 39 mg of SnOct2 (1.5 wt% based on the weight of Py-S-S-(CH2)2OH and MCL) was added to the flask. The flask was purged with nitrogen and refluxed for 24 h, and the solution was vacuum-concentrated under reduced pressure. The resulting product, Py-S-S-PMCL25, was dissolved in CHCl3, and then precipitated into excess n-hexane/diethyl ether (v/v 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) while stirring. The purified polymer was dried in vacuo at 50 °C for 24 h and analyzed. Fig. 1A shows the representative 1H nuclear magnetic resonance (NMR) spectrum of Py-S-S-PMCL25. The resonance peaks were assigned to the corresponding hydrogen atoms of Py-S-S-PMCL25. Subsequently, a mixture of Py-S-S-PMCL25 (0.93 g, 0.22 mmol, 1 equiv., Mn = 4210 g mol−1) and PNiPAAm21-SH (0.56 g, 0.22 mmol, Mn = 2540 g mol−1) was stirred in THF (10 mL) at room temperature. HfCl4·2THF (28.1 mg, 0.11 mmol) was added, and the mixture was stirred for 24 h at room temperature under nitrogen. The solution was then precipitated into excess n-hexane/diethyl ether (v/v 5:1) while stirring. Subsequently, the precipitated polymer was purified through dialysis (MW cutoff, 3.5 KDa) against THF for 2 days. The purified polymer, PNiPAAm21-S-S-PMCL25, was dried in vacuo at 50 °C for 24 h and analyzed. Fig. 1C and 2A show the representative 1H NMR and IR spectra of PNiPAAm21-S-S-PMCL25.
:1) while stirring. The purified polymer was dried in vacuo at 50 °C for 24 h and analyzed. Fig. 1A shows the representative 1H nuclear magnetic resonance (NMR) spectrum of Py-S-S-PMCL25. The resonance peaks were assigned to the corresponding hydrogen atoms of Py-S-S-PMCL25. Subsequently, a mixture of Py-S-S-PMCL25 (0.93 g, 0.22 mmol, 1 equiv., Mn = 4210 g mol−1) and PNiPAAm21-SH (0.56 g, 0.22 mmol, Mn = 2540 g mol−1) was stirred in THF (10 mL) at room temperature. HfCl4·2THF (28.1 mg, 0.11 mmol) was added, and the mixture was stirred for 24 h at room temperature under nitrogen. The solution was then precipitated into excess n-hexane/diethyl ether (v/v 5:1) while stirring. Subsequently, the precipitated polymer was purified through dialysis (MW cutoff, 3.5 KDa) against THF for 2 days. The purified polymer, PNiPAAm21-S-S-PMCL25, was dried in vacuo at 50 °C for 24 h and analyzed. Fig. 1C and 2A show the representative 1H NMR and IR spectra of PNiPAAm21-S-S-PMCL25.
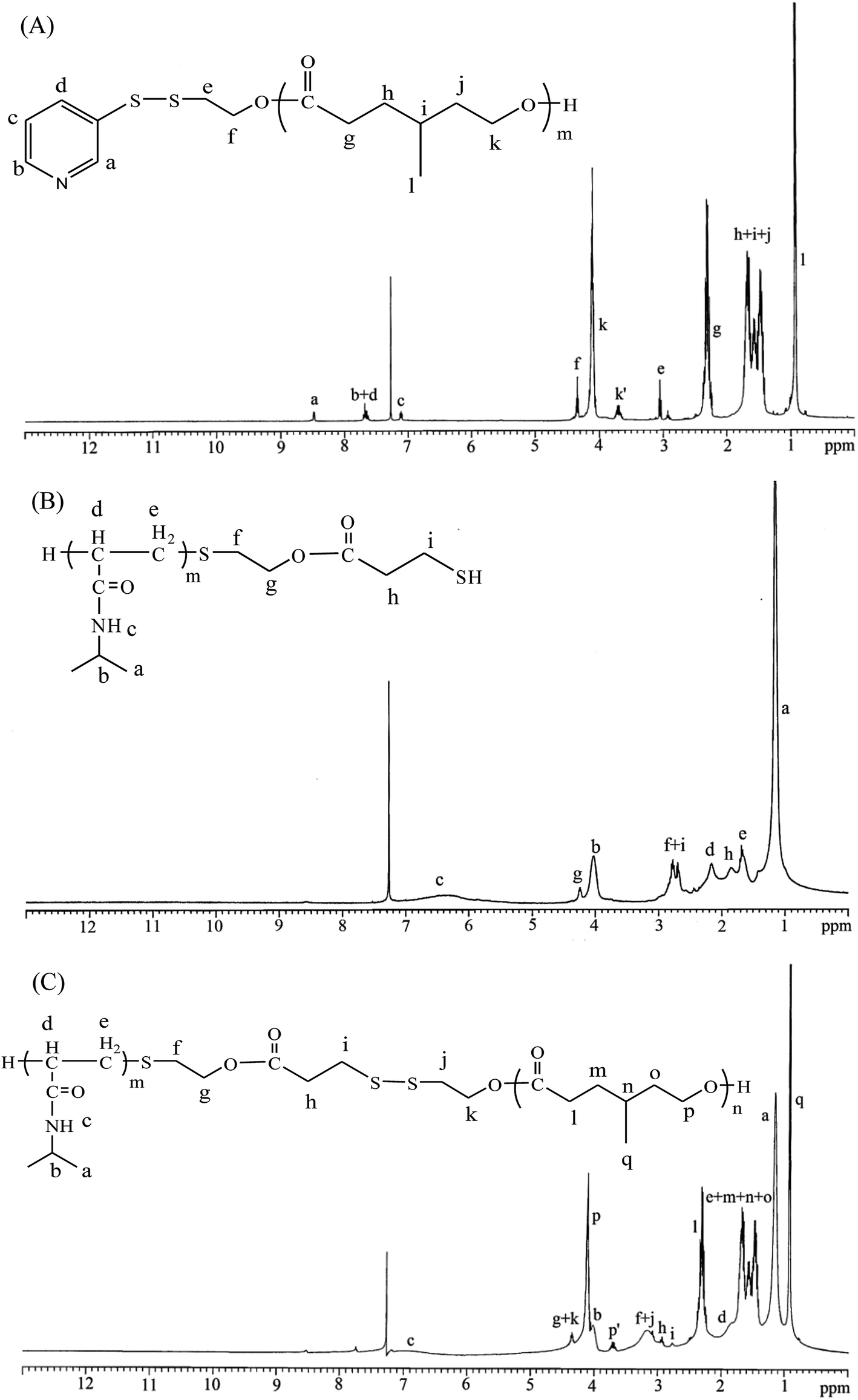 | ||
| Fig. 1 1H NMR spectra of (A) Py-S-S-PMCL25, (B) PNiPAAm21-SH, and (C) PNiPAAm21-S-S-PMCL25. | ||
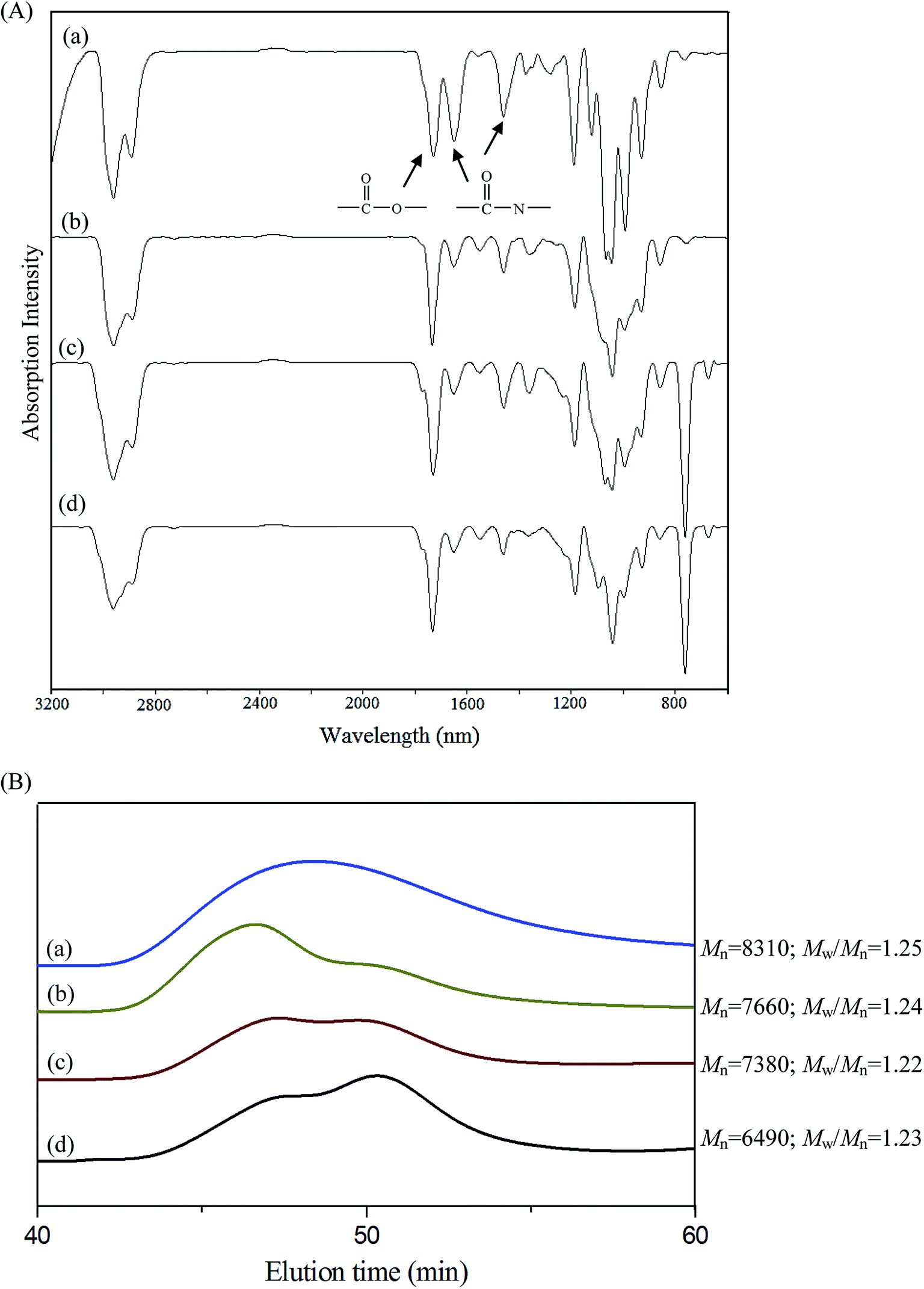 | ||
| Fig. 2 (A) FT-IR spectra and (B) GPC curves change of PNiPAAm21-S-S-PMCL25 micelle treated with various stimuli conditions for 10 min: (a) without, (b) with DTT, (c) with HIFU, (d) with combined DTT and HIFU (DTT: 10 mM; HIFU power output: 100% amplitude). | ||
Characterization
1H NMR spectra were recorded from CDCl3 solution on a Bruker AM 400 spectrometer. IR spectra were measured using a Bruker TENSOR 27 Fourier transform infrared (FT-IR) spectrophotometer (Bruker, Germany). Samples were placed on sodium chloride plates or pressed into potassium bromide pellets, and the number- and weight-average molecular weights (Mn and Mw, respectively) of the polymers were determined through gel permeation chromatography (GPC). GPC was conducted using an HPLC system equipped with a model PU-2031 refractive-index detector (Jasco, Tokyo, Japan) and Jordi Gel poly(divinyl benzene) columns with pore sizes of 100, 500, and 1000 Å. THF was used as an eluent at a flow rate of 0.5 mL min−1. Poly(ethylene glycol) standards of low dispersity (Polymer Sciences) were used to generate a calibration curve. Data were recorded and manipulated using a Windows-based software package (Scientific Information Service). Dynamic light scattering (DLS) experiments were carried out on the Zetasizer Nano ZS (Malvern, UK) spectrometer with an argon laser operating at 632.8 nm and a fixed scattering angle of 90° at 20 °C.LCST measurements
The optical transmittance of the aqueous polymer solution (1 mg mL−1) at various temperatures was measured at 500 nm by using a UV-vis spectrophotometer (Jasco V-550, Japan). The temperature of the sample cells was regulated using a temperature controller (Jasco ETC-505T, Japan). The heating rate was set at 0.1 °C min−1. The LCST values for the polymer solutions were determined at temperatures which the optical transmittance was 50%.Self-assembly behavior of polymers
Polymeric micelles of PNiPAAm-S-S-PXCL polymers were prepared through dialysis. Briefly, a solution of the PNiPAAm-S-S-PXCL polymer (30 mg) in DMF (5 mL) was placed in a dialysis bag with a molecular weight cutoff (MWCO) of 1.0 KDa and then dialyzed against deionized water at an ambient temperature for 24 h. The water was replaced at 2 h intervals.The critical micelle concentration (CMC) values of copolymers were investigated through the fluorescence probe method by using pyrene as the hydrophobic probe. Fluorescence spectra were recorded using a Hitachi F-4500 fluorescence spectrometer equipped with a 20 kW xenon discharge lamp. The slit width was 10 nm, and square quartz cells (1.0 × 1.0 cm) were used. First, the fluorescence spectra of pyrene were recorded in an aqueous solution at room temperature using the fluorescence spectrophotometer. Subsequently, sample solutions were prepared by adding known amounts of pyrene in acetone to a series of flasks. After the acetone had completely evaporated, various amounts of PNiPAAm-S-S-PXCL micelle solutions at concentrations, ranging from 0.0183 to 300 mg L−1, were added to each flask and mixed through vortexing. The pyrene concentration in the final solutions was 6.1 × 10−7 M. The flasks were allowed to stand overnight at room temperature to equilibrate the pyrene and micelles. For the measured fluorescence spectra, the excitation wavelength (λex) was 339 nm, and for the excitation spectra, the emission wavelength (λem) was 390 nm.
The average particle size, size distribution (size PD), and zeta potential of the micelles in the aqueous solution were measured using a dynamic light scattering (DLS). Measurements were recorded after the filtering of an aqueous micellar solution (C = 0.3 g L−1) by using a microfilter with an average pore size of 0.2 μm (Advantec MFS, Inc., Dublin, CA, USA).
The morphologies of the self-assembled micelles were observed using a transmission electron microscope (JEM 1200-EXII, Tokyo, Japan). Drops of the micelle solution (0.3 g L−1, not containing a staining agent) were placed on a carbon film-coated copper grid (400 mesh) and dried at room temperature. The micelles were observed at an accelerating voltage of 100 kV.
HIFU irradiation of polymer micelles
HIFU was generated using a commercially available ultrasound apparatus (Q125, Qsonica, Newtown, CT) that comprises three main components: an arbitrary waveform generator, a RF power amplifier (A150, Electronics & Innovation), and an acoustic lens transducer (H-101, Sonic Concept). The ultrasound amplitude can be adjusted in the range 20–100%, and the ultrasound frequency is 20 kHz. In all ultrasound irradiation experiments, the focal spot of the beams was the center of the micellar solution (10 mL) in a tube reactor immersed in a water tank. Following ultrasound irradiation, the tube reactor was removed from the water tank and the polymer solution was used for characterization.In vitro drug loading and release
IMC-loaded micelles were prepared in the following method. The anti-inflammatory drug IMC was served as a model drug. PNiPAAm21-S-S-PMCL25 (50-fold CMC value) and IMC (1:1 weight ratio to polymer) were dissolved in 6 mL of methylene chloride. The solution was then added dropwise to 150 mL of distilled water. The emulsion was stirred at an ambient temperature overnight to evaporate the methylene chloride. The unloaded IMC residue was removed through filtration using a Teflon filter (Whatman) with an average pore size of 0.45 μm, and the micelles were obtained through vacuum drying. A weighed amount of micelles was then disrupted by adding a 10-fold excess volume of DMF. The drug content was assayed spectrophotometrically at 320 nm by using a diode array UV-vis spectrophotometer. The experiments were conducted in triplicate. The results were presented as the average ± standard deviation. The following equations were used to calculate the drug-loading content and drug-entrapment efficiency:| Drug-loading content (%) = (weight of the drug in the micelles/weight of micelles) × 100 | (1) |
| Drug-entrapment efficiency (%) = (weight of the drug in the micelles/weight of the drug initially provided) × 100 | (2) |
The in vitro release of IMC in phosphate-buffered saline (PBS 0.01 M, pH 7.4) under various stimuli conditions was investigated through dialysis. Briefly, appropriate amounts of IMC-loaded micelles (110.2 mg) were suspended in 10 mL of PBS and DTT (10 mM) was added or ultrasonicated in a sonication bath for 10 min. The micellar solution was introduced to a dialysis membrane bag (MWCO = 3.5 kDa), and the bag was immersed in 50 mL of a PBS release medium. The medium was shaken (30 revolutions per min) at 25 °C or 40 °C. At predetermined intervals, 3 mL of released medium was taken out and the medium was replenished with an identical volume of fresh buffer solution to maintain a constant volume. The concentration of released IMC was determined using a UV-vis spectrophotometer at a wavelength of 320 nm. The rate of controlled drug release was measured on the basis of the cumulatively released weight of IMC by using the calibration curve for IMC. All release experiments were performed in triplicate.
Cell culture
HeLa cells were maintained in DMEM/F12 1:1 medium containing 10% FBS and 1% antibiotic–antimycotic at 37 °C in a humidified atmosphere with 5% carbon dioxide (CO2).
In vitro cytotoxicity assay
The cytotoxicity of free DOX, the PNiPAAm21-S-S-PMCL25 polymer, and the DOX-loaded micelles of PNiPAAm21-S-S-PMCL25 was investigated through an MTS assay performed using a Promega CellTiter 96® AQueuous One Solution kit. The assay was performed according to manufacturer's instructions but with minor modifications. Briefly, HeLa cells were seeded in a 24-well plate (3 × 104 cells per well) overnight; they were then treated with various concentrations of free DOX, polymers and freeze-dried DOX-loaded micelles or a control DMSO vehicles added to a DMEM/F12 1:1 medium with 1% FBS in a humidified 37 °C incubator supplied with 5% CO2. After 48 h, the medium in each well was removed and replaced with 350 μL of warm PBS and 35 μL of CellTiter 96® AQueuous One Solution. The mixture was then incubated at 37 °C for 4 h. After incubation, 110 μL of supernatant from each well was moved to a 96-well plate and absorbance was measured at 485 nm using an ELISA reader (Hidex, Finland). All experiments were performed in triplicate.
Results and discussion
Synthesis of PNiPAAm-S-S-PXCL diblock copolymers
Scheme 1 presents a strategy for synthesizing a temperature-sensitive and redox/ultrasound dual-cleavable PNiPAAm-S-S-PXCL polymer containing a redox-cleavable disulfide linkage at the junction between the temperature-sensitive hydrophilic PNiPAAm and the degradable hydrophobic block PXCL through the ROP and nucleophilic substitution reactions. First, the hydroxyl group of Py-S-S-(CH2)2OH initiated the ROP of ε-CL or 4-substituted-ε-caprolactone (4-XCL, X = CH3, Ph) catalyzed by SnOct2 to yield the Py-S-S-PXCL pre-polymer. Subsequently, the coupling reaction of the PNiPAAm-SH and Py-S-S-PXCL was carefully performed at room temperature under an N2 atmosphere to minimize air oxidation and homodimerization, to yield the PNiPAAm-S-S-PXCL polymer. Copolymers with different compositions were prepared by altering the length of the PNiPAAm and PXCL segments. Table 1 lists the coupling results with moderate yield. The Mns of the block copolymers with different compositions ranged from 3870 to 10970 g mol−1, and the polydispersity index (Mw/Mn) ranged from 1.25 to 1.53. From the values reported in Table 1, it appears that a good correlation between theoretical (Mn,th) and experimental molar mass values Mn,NMR (determined by 1H NMR) is observed. Fig. 3 shows the representative GPC curves of PNiPAAm21-S-S-PMCL25 and Py-S-S-PMCL25. Compared with the peak in the GPC curves of Py-S-S-PMCL25, the GPC curves of PNiPAAm21-S-S-PMCL25 exhibited a monomodal distribution and revealed a shift in the peak towards the higher molecular weight region. 1H NMR spectroscopy and FT-IR results confirmed the effective coupling of PNiPAAm-SH with Py-S-S-PXCL to yield PNiPAAm-S-S-PXCL. The representative 1H NMR spectrum (Fig. 1C) revealed the presence of signals from the PNiPAAm, PXCL segments and the disulfide linkage moieties. The observed resonance peaks were assigned to the corresponding hydrogen atoms of the PNiPAAm block at δ = 6.50 (Hc, –NH), 4.01 (Hb, –CH(CH3)2), 1.19 (Ha, –CH(CH3)2), and 1.48–1.89 (Hd, He, –CH– and –CH2–) as well as to the corresponding hydrogen atoms of the –S–S– linkage moiety at δ = 4.35 (Hg+k). The PMCL block showed resonance peaks at δ = 4.12 (Hp, –CH2O–), 2.32 (Hl, –CH2–), 1.55–1.79 (Hm+n+o, –CH2– and –CH–), and 0.89 (Hq, –CH3). The IR spectrum of PNiPAAm21-S-S-PMCL25 indicated a typical carbonyl absorptions of an ester at 1720 cm−1, carbonyl absorption of an amide at 1634 and 1547 cm−1, and C–O absorption at 1050 cm−1 (Fig. 2A).
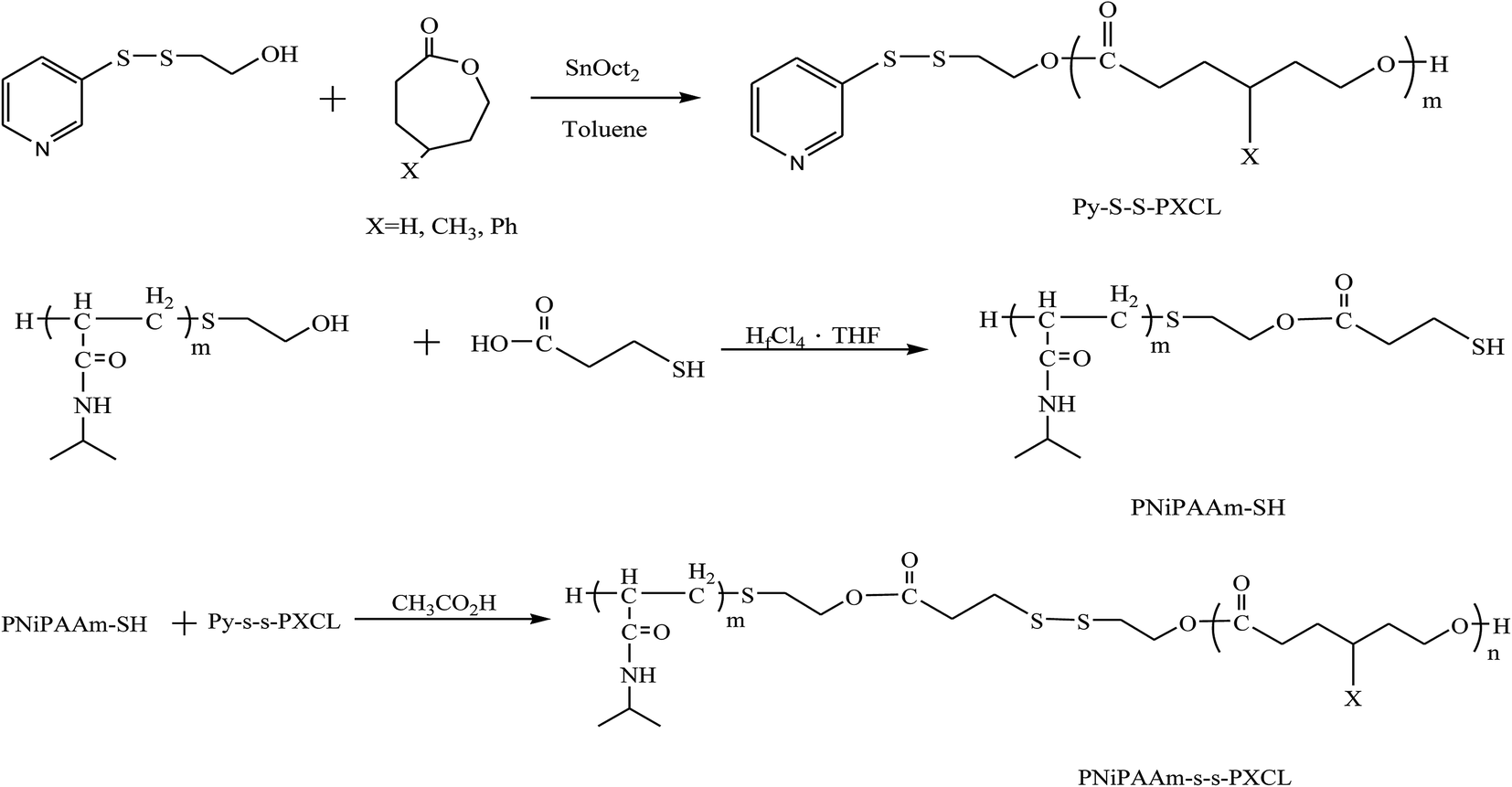 | ||
| Scheme 1 Synthesis temperature, ultrasound, and redox triplet-responsive PNiPAAm-S-S-PXCL copolymers. | ||
| Polymera | Isolated yield (%) | Mn,thb | Mn,NMRc | Mn,GPCd | PDId | LCSTe (°C) |
|---|---|---|---|---|---|---|
| a Abbreviations: PNiPAAm = poly(N-isopropyl acrylic amide); –S–S– = disulfide bond; PCL = poly(ε-caprolactone); PMCL = poly(4-methyl-ε-caprolactone); PBCL = poly(4-phenyl-ε-caprolactone).b Mn,th = Mn,PNiPAAm-SH + Mn,Py-S-S-PXCL (where Mn,PNiPAAm-SH is the number-average molecular weight of 2-thioethylcarbonyl-PNiPAAm, Mn,Py-S-S-PXCL is the number-average molecular weight of Py-S-S-PXCL).c Determine by 1H NMR spectroscopy of PNiPAAm-S-S-PXCL.d Determine by GPC relative to polyethylene glycol standards in THF.e LCST was determined by UV spectroscopy at 500 nm in 1 wt% aqueous polymer solution. The LCST values for the polymer solution were determined at temperature which the optical transmittance was 50%. The LCSTs were 41.9, 38.5, and 34.2 °C for PNiPAAm11-SH, PNiPAAm21-SH, and PNiPAAm43-SH, respectively. | ||||||
| PNiPAAm11-S-S-PCL17 | 84 | 3368 | 2960 | 3870 | 1.31 | 35.9 |
| PNiPAAm11-S-S-PMCL19 | 81 | 3862 | 3222 | 4480 | 1.37 | 33.8 |
| PNiPAAm11-S-S-PBCL9 | 82 | 4763 | 3053 | 3890 | 1.51 | 39.8 |
| PNiPAAm43-S-S-PCL17 | 83 | 7502 | 7278 | 10300 |
1.43 | 33.4 |
| PNiPAAm43-S-S-PMCL19 | 74 | 7284 | 7779 | 10970 |
1.53 | 33.2 |
| PNiPAAm43-S-S-PMCL43 | 79 | 10612 |
5560 | 9290 | 1.34 | 32.8 |
| PNiPAAm21-S-S-PCL21 | 69 | 5013 | 4672 | 6410 | 1.47 | 36.2 |
| PNiPAAm21-S-S-PMCL25 | 67 | 7294 | 5625 | 8310 | 1.25 | 37.2 |
 | ||
| Fig. 3 GPC curves of (A) PNiPAAm21-S-S-PMCL25, and (B) Py-S-S-PMCL25. | ||
LCST behavior
We characterized the LCST behaviors of the PNiPAAm-S-S-PXCL copolymer solutions by measuring their cloud points. As illustrated in Fig. 4, the optical properties of the PNiPAAm-S-S-PXCL copolymers' transmittance changed (λ 500 nm) when heated. We determined the LCST at the temperature at which the optical transmittance was 50%. Table 1 presents the LCST results. The copolymerization of PNiPAAm with hydrophobic comonomers decreases the LCST, whereas the hydrophilic comonomers have the opposite effect.37 Fig. 4A indicates that when the hydrophilic block was fixed at PNiPAAm11-SH (LCST, 41.9 °C), the LCST values of the PNiPAAm11-S-S-PXCL copolymers decreased to 39.8 °C, 35.9 °C, and 33.8 °C for PNiPAAm11-S-S-PBCL9, PNiPAAm11-S-S-PCL17, and PNiPAAm11-S-S-PMCL19, respectively, which can be attributed to the increased hydrophobicity of the PXCL: hydrophobicity of PMCL19 > PCL17 > PBCL9. At constant hydrophobic block length PCL17, the LCST of the PNiPAAm-S-S-PCL17 copolymers decreased from 35.9 °C to 33.4 °C when the PNiPAAm segment length was increased from PNiPAAm11 to PNiPAAm43, which can be attributed to the reduced hydrophilicity of the PNiPAAm segment.38 The LCST of PNiPAAm21-S-S-PMCL25 is 37.2 °C, near body temperature (37.5 °C). The HIFU responsive behavior of PNiPAAm21-S-S-PMCL25 was first investigated and is shown Fig. 4B. No transition behavior in PNiPAAm21-S-S-PMCL25 was observed for treatment with HIFU irradiation (100% amplitude) for 10 min. The solution remained transparent with no decrease in transmittance, suggesting that the larger-sized aggregates had not formed. Under HIFU irradiation, the ultrasound-induced hydrolysis of the N-isopropyl amide units increase their polarity, like in the case of acrylic acid formation due to removal of N-isopropyl groups, the thermosensitive polymer becomes soluble in water and changes the hydrophilic–hydrophobic balance of the amphoteric polymer, thus leads to the disruption of the micelle.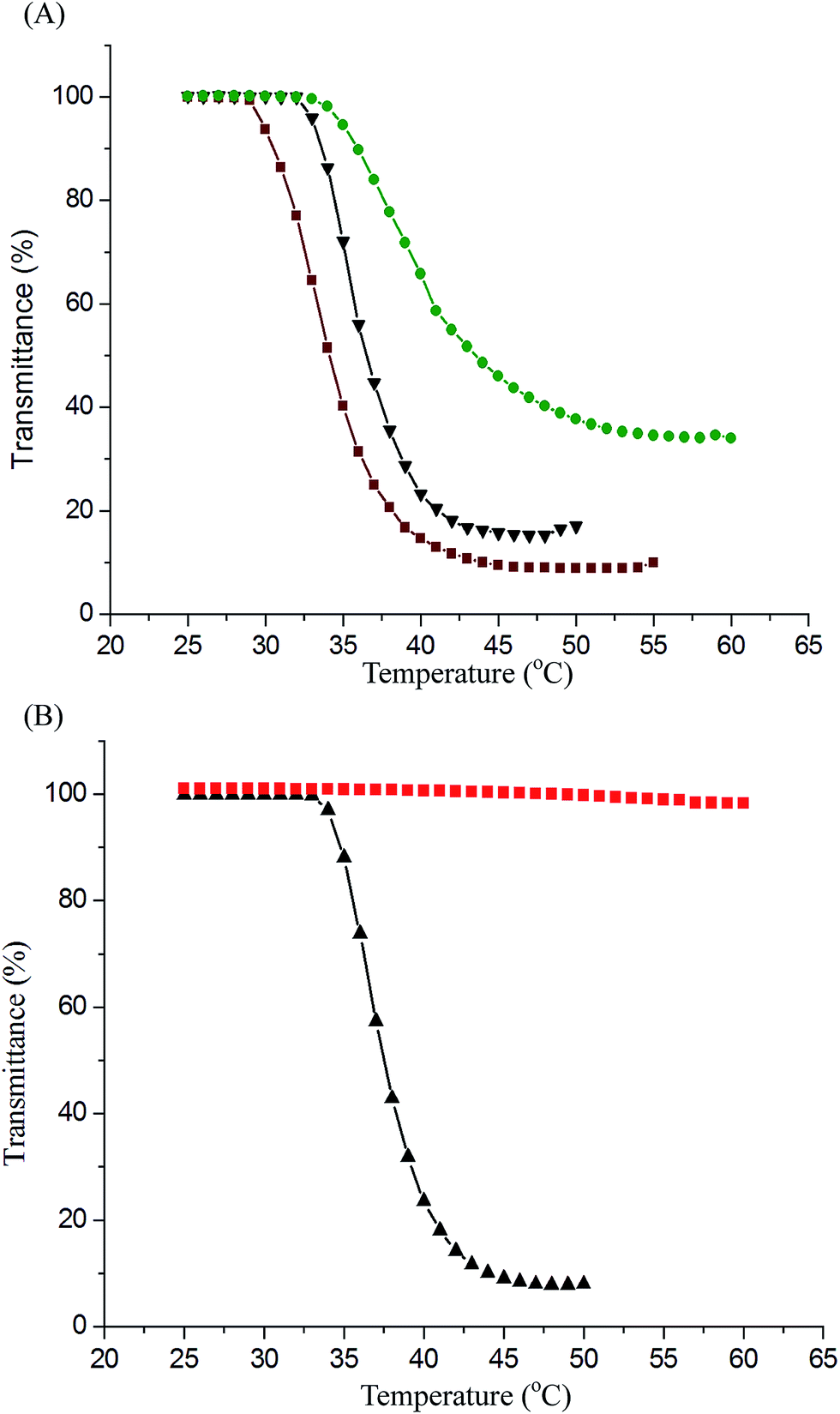 | ||
| Fig. 4 Phase-transition curves of PNiPAAm-S-S-PXCL in the 1 mg mL−1 PBS (0.01 M, pH 7.4): (A) PNiPAAm-S-S-PXCL with constant PNiPAAm11 segment: PNiPAAm11-S-S-PMCL19 (▼), PNiPAAm11-S-S-PCL17 (●), PNiPAAm11-S-S-PBCL18 (■), (B) PNiPAAm21-S-S-PMCL25 treatment before (▲), and after (■) with HIFU. | ||
Degradation of redox/HIFU-labile PNiPAAm-S-S-PMCL copolymer
FT-IR spectroscopy was applied to investigate the redox- and HIFU-triggered degradation of the polymer, by observing the spectra changes of a polymer solution under various conditions. Accordingly, 1 mg of PNiPAAm21-S-S-PMCL25 was dissolved in 1 mL of inhibitor-free THF and the solution was treated with DTT, HIFU, and combined DTT and HIFU, respectively. The experiment was performed at ambient temperature. The solutions before and after treatment were dried and characterized. The FT-IR results (Fig. 2A) show that the absorption of carbonyl groups in amide bonds (approximately 1634 and 1547 cm−1) decreases significantly after treatment with DTT or HIFU. The decreased tendency under combined stimuli is more prominent than either alone. Fig. 2B shows the change in GPC traces, and the Mn of PNiPAAm21-S-S-PMCL25 decreased from 8310 to 6490 g mol−1 after treatment with DTT and or HIFU in 10 min. A low molecular-weight new peak appears, indicating that the cleavage occurs. Ultrasound-induced chain scission occurs preferentially at the weak points. We thought that the disulfide bond in the copolymer is more easily broken under HIFU treatment because the –S–S– bond energy (approximately 54 kcal mol−1) is much lower than the C–O bond energy (approximately 85.5 kcal mol−1).29 However, the results went against our intuition. The DTT- and HIFU-induced changes in the molecular structure of our copolymer, as already stated, confirm the preferential cleavage of ester (C–O) and amide (C–N) bonds. In the same condition, a similar cleavage change was observed for PNiPAAm15-b-PMCL32 without the disulfide bond (spectra not shown) because of the existence of mechano-labile amide bonds. The result suggests that the hydrolysis of the amide bond is more easily induced through the ultrasound irradiation and DTT because of less hindrance in the side group.26 The disulfide (S–S) bond in the main chain is rigid and more difficult cleavage than the N-isopropyl amide side group. The solvodynamic shear or short-lived and localized hot spot produced through ultrasonic cavitation is responsible for the side-chain scission.Micelles of PNiPAAm-S-S-PXCL
The amphiphilic nature of PNiPAAm-S-S-PXCL polymers, which consist of a hydrophilic PNiPAAm segment and a hydrophobic PXCL segment, enables the formation of micelles in water. The CMC of a block copolymer is an essential parameter for evaluating the self-assembly behavior of block copolymers in aqueous solution. In this study, the characteristics of the PNiPAAm-S-S-PXCL micelles in the aqueous phase were investigated using fluorescence techniques. The CMC of PNiPAAm-S-S-PXCL in the aqueous phase was determined using pyrene as a probe molecule. Pyrene is preferentially located in the hydrophobic core, leading to the change of the surrounding environment from polar to non-polar. The fluorescence intensity of the excitation spectrum of pyrene increased with the PNiPAAm21-S-S-PXCL25 polymer concentration (Fig. 5A). In the pyrene excitation spectrum, a red-shift of the (0, 0) band of pyrene from 336 to 341 nm was observed during partitioning into the micellar hydrophobic core. Fig. 5B illustrates the intensity ratios I341/I336 of pyrene excitation spectra versus the logarithm of PNiPAAm21-S-S-PXCL25 polymers concentrations. The CMC was determined on the basis of the intersection of straight-line segments drawn through the points of the lowest polymer concentrations, which lie on a nearly horizontal line, and the points of the rapidly rising region of the plot. Table 2 lists the CMC values of the various PNiPAAm-S-S-PXCL polymers. The PNiPAAm-S-S-PXCL polymers formed micelles in the aqueous phase, with their CMC values ranging from 1.3 to 26.6 mg L−1. The PNiPAAm-S-S-PXCL polymers exhibited low CMC values which are consistent with the high stability characteristic of polymer micelles. Declining CMC values were observed with increasing hydrophilicity of the hydrophilic segment or increasing hydrophobicity of the hydrophobic segment. At a fixed length of the hydrophobic block (PMCL19), the CMC values of the PNiPAAm-S-S-PMCL19 series of polymers increased from 3.3 to 26.6 mg L−1 when the hydrophilic PNiPAAm chain length increased from PNiPAAm11 to PNiPAAm43. The result is attributable to the lower hydrophilicity of PNiPAAm43 than that of PNiPAAm11. The flexibility and steric hindrance of the hydrophobic segment increased and the CMC values decreased. Polymeric micelles exhibiting lower CMC values are generally more stable in drug delivery applications because they remain stable in a dilute form in the blood after being administered.39 | ||
| Fig. 5 (A) Excitation spectra of PNiPAAm21-S-S-PMCL25 copolymer monitored at λem = 390 nm, (B) plot of I341/I336 intensity ratio (from pyrene excitation spectra; pyrene concentration = 6.1 × 10−7 M) versus the logarithm of concentration (logC) for PNiPAAm21-S-S-PMCL25 copolymer micelle, (C) DLS curves of PNiPAAm21-S-S-PMCL25 copolymer micelle, (D) storage stability of PNiPAAm21-S-S-PMCL25 micelles determined by average size and zeta potential. Each value represents the mean ± SD (n = 3). | ||
| Polymer | CMC (mg L−1) | Drug loading contenta (%) | Drug entrapment efficiencya (%) | Micelle sizeb (nm) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Blank | PD | Zeta potential (mv) | With IMC | PD | Zeta potential (mv) | ||||
| a Feed weight ratio IMC/polymer = 1/1.b Micelle size and particle size distribution (PD) determined by DLS. | |||||||||
| PNiPAAm11-S-S-PCL17 | 15.2 | 11.7 ± 1.1 | 23.3 ± 2.2 | 109.2 ± 20.5 | 0.08 | −15.7 | |||
| PNiPAAm11-S-S-PMCL19 | 3.3 | 23.4 ± 0.7 | 46.9 ± 1.4 | 117.5 ± 29.2 | 0.05 | −15.7 | 132.0 ± 48.6 | 0.11 | −16.3 |
| PNiPAAm11-S-S-PBCL9 | 1.3 | 37.0 ± 0.6 | 73.9 ± 1.1 | 140.6 ± 45.3 | 0.04 | −14.7 | 168.9 ± 40.8 | 0.08 | −30.5 |
| PNiPAAm43-S-S-PCL17 | 22.2 | 22.2 ± 0.7 | 44.4 ± 1.4 | 112.1 ± 33.3 | 0.06 | −7.0 | |||
| PNiPAAm43-S-S-PMCL19 | 26.6 | 29.0 ± 0.2 | 57.9 ± 0.3 | 124.6 ± 34.6 | 0.08 | −9.6 | 145.3 ± 56.1 | 0.12 | −11.6 |
| PNiPAAm43-S-S-PMCL43 | 25.3 | 41.0 ± 1.1 | 81.9 ± 2.2 | 127.6 ± 34.3 | 0.07 | −10.8 | 156.6 ± 62.9 | 0.19 | −14.0 |
| PNiPAAm21-S-S-PCL21 | 9.6 | 28.0 ± 0.2 | 56.0 ± 0.4 | 118.6 ± 30.7 | 0.05 | −34.5 | |||
| PNiPAAm21-S-S-PMCL25 | 8.5 | 37.5 ± 0.3 | 73.0 ± 1.4 | 146.2 ± 36.8 | 0.16 | −27.7 | 158.5 ± 49.6 | 0.13 | −5.1 |
The mean hydrodynamic diameters of blank micelles and those incorporating IMC, ranged from 109.2 to 146.2 nm and from 132.0 to 168.9 nm, respectively, and with acceptable size distribution (PD ≤ 0.19). Fig. 5C shows that the diameter of the original blank PNiPAAm21-S-S-PMCL25 micelles is 146 nm, with a narrow size distribution.
The stability of the PNiPAAm21-S-S-PMCL25 micelles was examined by incubating the nanosystems at 25 °C for 2 weeks. Average diameter and zeta potential were used as the indicators to evaluate the micellar stability. As illustrated in Fig. 5D, the size and zeta potential of PNiPAAm21-S-S-PMCL25 micelles showed no significant change during the 2 weeks. This suggests that the micelles are stable for at least 2 weeks; thus further application can be promised.
The micelles incorporating IMC were larger than the blank micelles because of the incorporation of the hydrophobic drug; however, the micelle size remained <200 nm for all formations. A suitable size for nanoparticles (<200 nm in diameter) is beneficial for prolonged blood circulation and passive targeting through the enhanced permeability and retention (EPR) effect.40 The mean diameter of the micelles increased with the length of the hydrophilic segment, which can be attributed to the increased hydrated diameter engendered by the longer length of the hydrophilic PNiPAAm block. However, at a fixed hydrophilic block length (PNiPAAm43), the micelles size increased with the length of the hydrophobic segment (PMCL). These responses indicated that micelle size is dependent on the polymer composition (i.e., the length of the hydrophobic segment or the hydrophilicity of the hydrophilic moiety in the chain). Fig. 6 shows the spherical morphology of the PNiPAAm21-S-S-PMCL25 micelles. When the drug was incorporated, the micelle size slightly increased (Fig. 6B). The average diameter of the micelle was smaller than that recorded through DLS; this is mainly because TEM revealed the actual core dimensions of the micelles in the dry state, whereas DLS revealed the average dimensions of the micelles in aqueous solution.
 | ||
| Fig. 6 TEM photograph of the micelles formed by PNiPAAm21-S-S-PMCL25: (A) blank, (B) loading IMC; TEM image change of the PNiPAAm21-S-S-PMCL25 micelles treat with different stimuli for 10 min: (C) with 10 mM DTT, (D) with HIFU irradiation (output power: 100% amplitude), (E) combined DTT and HIFU; and (F) with 10 mM DTT for 24 h. Scale bar 0.2 μm. | ||
Temperature-sensitive and redox/HIFU-cleavable behaviors of micelles
The temperature-sensitive and redox and or HIFU degradation of the micelles were evaluated by monitoring changes in NR fluorescence at 25 °C or 40 °C, with the reducing agent DTT (10 mM) treatment, under HIFU irradiation (power output 100% amplitude), or using a combination of the two stimuli condition, depending on the reduction in the NR fluorescence intensity at 610 nm. Because of the micelle disruption, the encapsulated NR is released into water from the micelle core, which can lead to fluorescence quenching, resulting in a decrease in fluorescence intensity.41 After NR and PNiPAAm21-S-S-PMCL25 were dissolved in THF (1 mg mL−1, NR to polymer ratio = 1:3), water was added to induce micelle formation and concomitant NR encapsulation by the micelle core. Subsequently, THF was removed through evaporation, and the nonsolubilized NR was filtered through microfiltration (0.2 μm pore filter). The final micelle concentration was adjusted to 0.2 mg mL−1. Fig. 7A and B show the fluorescence emission spectra of NR loaded in the PNiPAAm11-S-S-PMCL25 and PNiPAAm15-b-PMCL32 without disulfide bond micelles before and after treatment under HIFU irradiation with different amplitudes (0–100%) for 10 min. As the amplitude of HIFU increased, the intensity of the fluorescence emission spectra of NR significant decreased with and without disulfide bond linkage polymers. A similar reduced trend in normalized fluorescence emission intensity was observed for PNiPAAm11-S-S-PMCL25 and PNiPAAm15-b-PMCL32 micelles after treatment with HIFU (Fig. 7C). Fluorescence emission intensity reduces quickly as HIFU amplitude increases, which should be attributed to the relatively strong cavitation effect at a higher acoustic intensity. Fig. 7D shows the fluorescence emission spectra recorded at different ultrasonic irradiation times. After treatment with HIFU irradiation, the intensity of the fluorescence emission spectra of NR rapidly decreased as the treatment time increased. To more accurately compare the kinetics of the apparent release of NR with different stimuli conditions, Fig. 7E displays normalized fluorescence plotted against time under different treatment conditions: without DTT and HIFU irradiation, only with DTT, only with HIFU irradiation, and with a combination of both stimuli. In the absence of DTT and HIFU irradiation, the emission intensity remained constant over time, indicating the stable encapsulation of the dye in a hydrophobic environment. By contrast, when expose to HIFU irradiation for 10 min, the emission intensity decreased to 40%. Over the same period (10 min), DTT treatment decreased emission intensity to 80%. However, in the case of both HIFU irradiation and DTT, the emission intensity rapidly decreased to 11% over 10 min. These results indicate that micelles disintegrate more rapidly under HIFU irradiation than exposed to DTT.
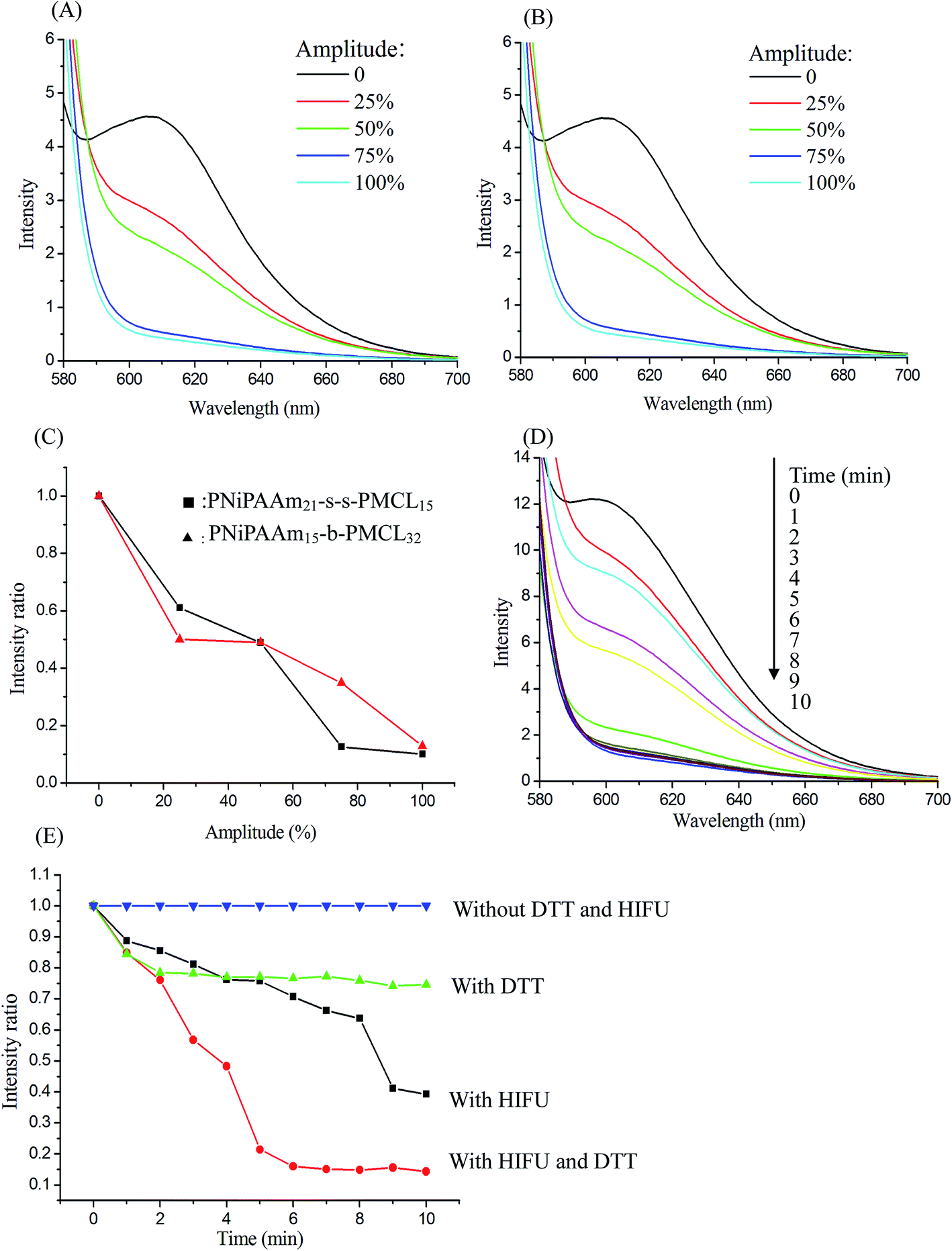 | ||
| Fig. 7 Fluorescence spectra of NR-loaded PNiPAAm21-S-S-PMCL25 (A) and PNiPAAm15-b-PMCL32 (B) micelles under HIFU treatment in 10 min at different amplitude power, (C) normalized fluorescence emission intensity vs. amplitude of the NR-loaded micelle with HIFU treatment, (D) fluorescence emission intensity of NR-loaded PNiPAAm21-S-S-PMCL25 micelles decay under different irradiation time (HIFU power output: 100% amplitude), (E) normalized fluorescence emission intensity vs. time of the NR-loaded micelle under various stimuli treatment in 10 min (DTT: 10 mM; HIFU power output: 100% amplitude). | ||
The disruption of the BCP micelles using a reducing agent and or ultrasonic was also confirmed by TEM. The morphological changes of the micelles were observed after treatment with different stimuli. The TEM images in Fig. 6C–F provided more insight into the effect of ultrasonic or redox-cleavage both alone and combined on the micelles of PNiPAAm21-S-S-PMCL25. Fig. 6C depicts the morphology of the micelles after treatment with DTT for 10 min. The TEM micrographs indicate distinct morphologies with some aggregation because of redox-induced cleavage of some disulfide bonds and consequently the aggregation and precipitation of hydrophobic PXCL blocks. However, after HIFU irradiation, disintegrated small nanoparticles (no aggregate) were observed (Fig. 6D). Because the micelle was disrupted quickly by HIFU, suggesting the hydrophobic blocks of the copolymers are degraded into numerous different small molecules. When both HIFU irradiation and DTT were stimulated, the spherical micelle disintegrated, and smaller irregularly shaped nanoparticle were formed (Fig. 6E). Similar, as the treatment time with DTT was extended to 24 h, the spherical micelle disintegrated, and smaller irregularly shaped nanoparticle were observed (Fig. 6F). Therefore, the TEM micelle characterization results revealed that altering the external environment with a reducing agent and ultrasonic engenders the dissociation of the micelles.
Evaluation of drug-loading content, drug-entrapment efficiency and stimuli-responsive release of IMC
The drug-loading content and drug-entrapment efficiency of the polymeric micelles were determined using UV-vis absorption spectroscopy. IMC, a common, hydrophobic, non-steroidal, and anti-inflammatory drug, was selected as a model drug to investigate drug-loading in the hydrophobic core. The intensity of IMC at maximum absorbance wavelength (320 nm) was proportional to its concentration. After IMC was released and the polymer precipitate was removed, the amount of loaded IMC was determined on the basis of the absorbance at 320 nm. Table 2 lists the calculated drug-loading content and drug-entrapment efficiency values. At a constant feed weight ratio (WIMC/Wcopolymer 1:1), the maximum drug-loading content and drug-entrapment efficiency were 41.0% and 81.9%, respectively. The drug-loading content and drug-entrapment efficiency increased with the length of the hydrophilic or hydrophobic segment.
To compared the influence of stimuli triggers on drug delivery, the IMC release from the triple stimuli-responsive PNIPAAm21-S-S-PMCL25 micelles in the PBS (pH 7.4) was investigated through dialysis under various conditions for 25 h: (i) in the absence of DTT and HIFU at 25 °C or 40 °C; (ii) only with DTT (10 mM) at 40 °C; (iii) combined HIFU and DTT at 40 °C. Fig. 8 illustrates the release profiles of IMC from the IMC-loaded micelles of PNiPAAm21-S-S-PMCL25. Environmental factors, such as temperature, reducing agent and ultrasonic, significantly affected the release of IMC from the micelles. At a fixed pH of 7.4, the release rate of IMC at 40 °C was much faster than that at 25 °C. The release rate increased slightly from 52% to 57% when increased temperature from 25 °C to 40 °C during a 25 h period, which could be ascribed to the micelle constriction when the temperature was higher than the LCST (37.2 °C). At a temperature 40 °C, the IMC release rate further accelerated when in the presence of DTT (10 mM), with approximately 65% of the encapsulated IMC being released sustainably during a 25 h period. This reductive–responsive release behavior can be attributed to the easier cleavage of the N-isopropyl-amide and disulfide bonds in a DTT solution and disruption of the micelle structure under reductive conditions. Because of the combined stimuli (DTT and HIFU), IMC release amount was increased to 93% over 25 h. Compared with a redox stimulus, HIFU significantly enhances the release rate from the micelles. This is due to the amide bond of the PNiPAAm was hydrolyzed under HIFU stimulus, caused the hydrophobic–hydrophilic balance of the copolymer be changed irreversibly, leading to the disruption of micelles and release of encapsulated cargos. The release rate of the control PNiPAAm15-b-PMCL32 micelle without a disulfide bond was also investigated under the same condition. The release rate of the PNiPAAm15-b-PMCL32 (71%) is slower than the PNiPAAm21-S-S-PMCL25 (93%) due to the lack redox responsiveness. These results suggest that HIFU could be a promising trigger for the cargo release from PNiPAmm21-S-S-PMCL25 copolymer micelle containing the mechanophore. Possible morphologies and structure changes of the micelles under the stimuli of temperature, HIFU, and reducing agent are presented in Scheme 2.
 | ||
| Fig. 8 Cumulative IMC release profiles from PNiPAAm21-S-S-PMCL25 micelles in PBS (0.01 M, pH 7.4) with different conditions: (▲) at 25 °C, (■) at 40 °C, (●) at 40 °C with DTT, (◆) after HIFU irradiation for 10 min, then at 40 °C with DTT, and (▼) PNiPAAm15-b-PMCL32 without reductive disulfide linkage at 40 °C with HIFU and DTT (DTT: 10 mM; HIFU output power: 100% amplitude). Each value represents the mean ± SD (n = 3). | ||
 | ||
| Scheme 2 Schematic illustration of the micellization of the PNiPAAm-S-S-PXCL and possible morphology changes under the stimuli of temperature, HIFU, and reducing agent. | ||
In vitro cytotoxicities of the polymer and DOX-loaded micelles
The MTS assay was used to evaluate the in vitro cytotoxicities of the PNiPAAm21-S-S-PMCL25 polymer, DOX-loaded micelles, and free DOX, by incubating HeLa cells with various polymer concentrations or DOX dosages. Fig. 9A illustrates the relative cell viability of cells treated with various concentrations of PNiPAAm21-S-S-PMCL25. Compared with the control, the measured cell viability was nearly 80% during the polymer concentration 10 to 1000 μg mL−1. The nanoparticles exhibited slight toxicity against the HeLa cells. In addition, Fig. 9B presents the in vitro cytotoxicities of the DOX-loaded micelles and free DOX at various DOX dosages (0.125–10 μg mL−1). The viabilities of HeLa cells were decreased with increasing DOX concentrations. The IC50 values represent the DOX dosage that is necessary for inhibitory concentration to produce 50% cell death. The DOX-loaded PNiPAAm21-S-S-PMCL25 micelles effectively inhibited the proliferation of the HeLa cells, with IC50 of 2.33 μg mL−1. The DOX-loaded micelles possessed slightly higher IC50 values than did free DOX (1.25 μg mL−1). This could be because of the longer time required for DOX to be released from micelles to tumor cells via an endocytosis mechanism, while free DOX entered into the cells by a passive diffusion process.42 Compared with DOX-loaded PNiPAAm15-b-PMCL32 without a disulfide bond linkage, PNiPAAm21-S-S-PMCL25 micelles could quickly release the encapsulated DOX upon redox stimuli.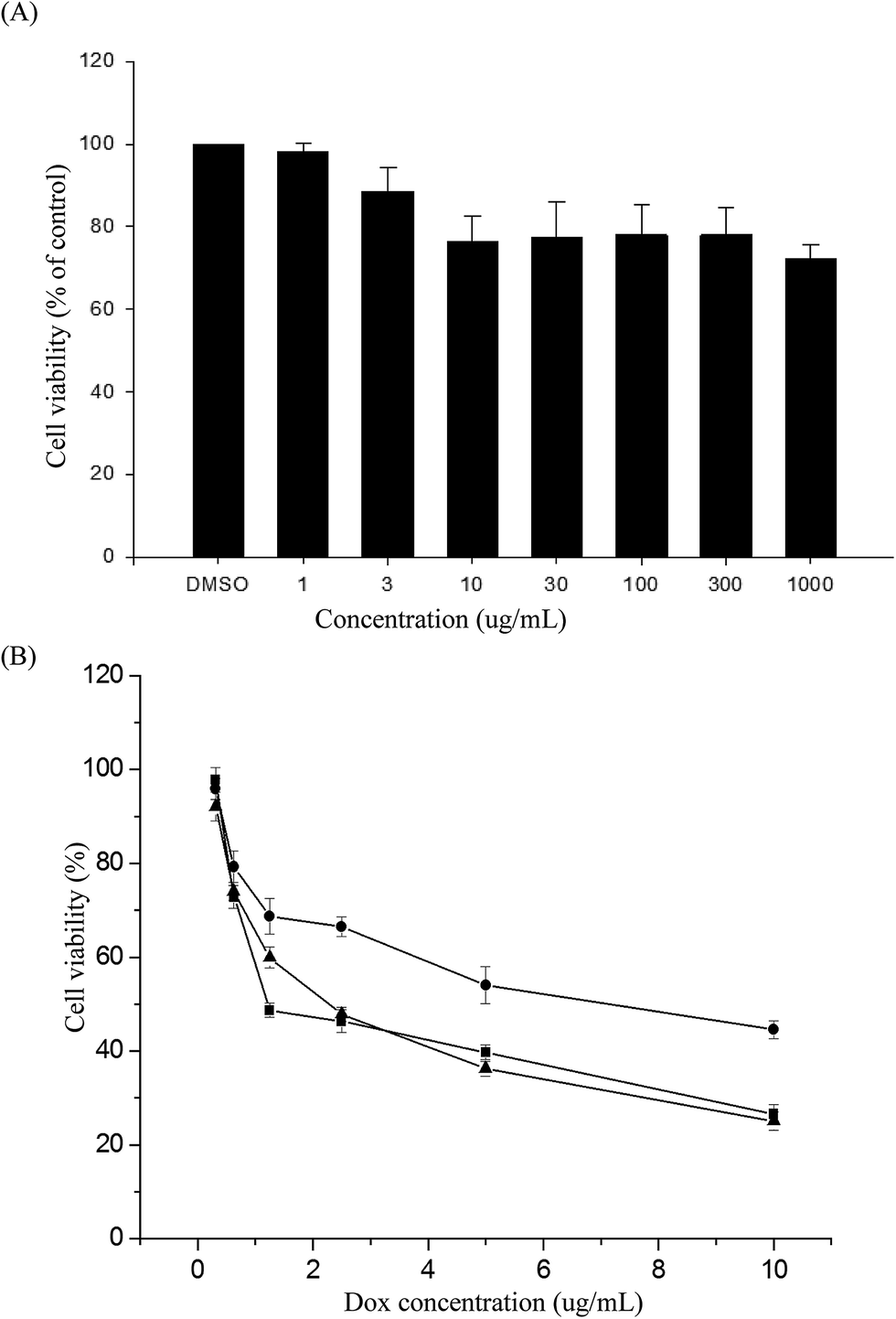 | ||
| Fig. 9 Cell viabilities of HeLa cells treated (A) with various concentration of PNiPAAm21-S-S-PMCL25, (B) with dox-loaded PNiPAAm21-S-S-PMCL25 (▲), free DOX (■), and DOX-loaded PNiPAAm15-b-PMCL32 without reductive disulfide linkage (●) for 48 h. Each value represents the mean ± SD (n = 3). | ||
Conclusions
Redox- and ultrasonic-triggered thermo-responsive PNiPAAm-S-S-PXCL BCPs with a disulfide bond linkage junction between the temperature-sensitive hydrophilic and biodegradable hydrophobic blocks were successfully synthesized using ROP and nucleophilic substitution reactions. The copolymers formed micelles in aqueous solution, had hydrodynamic sizes of <200 nm, and were spherical. Under the combined stimulation of reducing agent and ultrasonic, the micellar nanoparticles could be dissociated; therefore, the loaded molecules could be released from the assemblies more efficiently than that under only one stimulus and under HIFU-induced release faster than redox-induced release. Cell viabilities were evaluated in response to the concentration of the polymer (1–1000 μg mL−1), and the nanoparticles exhibited slight toxicity against HeLa cells at these concentrations. The DOX-loaded PNiPAAm21-S-S-PMCL25 micelles effectively inhibited the proliferation of the HeLa cells, with a half-maximal inhibitory concentration (IC50) of 2.33 μg mL−1. Therefore, the study results indicate that temperature, reducing agent, and ultrasonic triple-sensitive PNiPAAm21-S-S-PMCL25 BCPs can be used for triggered drug delivery.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was supported by grants from Ministry of Science and Technology (MOST105-2221-E-182-080) and Chang Gung Memorial Hospital (CMRPD5F0012). The manuscript was edited by Wallace Academic Editing.References
- X. Hu, Y. Zhang, Z. Xie, X. Jing, A. Bellotti and Z. Gu, Biomacromolecules, 2017, 18, 649–673 CrossRef CAS PubMed.
- M. Wei, Y. Gao, X. Li and M. J. Serpe, Polym. Chem., 2017, 8, 127–143 RSC.
- Z. Su and X. Jiang, Polymer, 2016, 93, 221–239 CrossRef CAS.
- X. Wang, G. Jiang, X. Li, B. Tang, Z. Wei and C. Mai, Polym. Chem., 2013, 4, 4574–4577 RSC.
- W. Yuan, T. Shen, J. Wang and H. Zou, Polym. Chem., 2014, 5, 3968–3971 RSC.
- L. Wang, J. Luan, L. Du, L. Li, Z. Liu and R. Zhuo, RSC Adv., 2016, 6, 111161–111169 RSC.
- S. Tan, Z. Lu, J. Zhao, J. Zhang, M. Wu, Q. Wu and J. Yang, Polym. Chem., 2016, 7, 4106–4111 RSC.
- F. Jiang, S. Chen, Z. Cao and G. Wang, Polymer, 2016, 83, 85–91 CrossRef CAS.
- Y. Y. Xiao, X. L. Gong, Y. Kang, Z. C. Jiang, S. Zhang and B. J. Li, Chem. Commun., 2016, 52, 10609–10612 RSC.
- F. Coumes, A. Malfait, M. Bria, J. Lyskawa, P. Woisel and D. Fournier, Polym. Chem., 2016, 7, 4682–4692 RSC.
- G. Vancoillie, W. L. A. Brooks, M. A. Mees, B. S. Sumerlin and R. Hoogenboom, Polym. Chem., 2016, 7, 6725–6734 RSC.
- Y. Zhang, S. Chen, M. Pang and W. Zhang, Polym. Chem., 2016, 7, 6880–6884 RSC.
- K. Jia, Y. Cheng, X. Liu, X. Li and J. Dong, RSC Adv., 2015, 5, 640–642 RSC.
- X. Chang, C. Ma, G. Shan, Y. Bao and P. Pan, Polymer, 2016, 90, 122–131 CrossRef CAS.
- Z. Tao, K. Peng, Y. Fan, Y. Liu and H. Yang, Polym. Chem., 2016, 7, 1405–1412 RSC.
- Y. Lu, H. Zou, H. Yuan, S. Gu, W. Yuan and M. Li, Eur. Polym. J., 2017, 91, 396–407 CrossRef CAS.
- A. Seeboth, D. Lötzsch, R. Ruhmann and O. Muehling, Chem. Rev., 2014, 114, 3037–3068 CrossRef CAS PubMed.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- H. Ren, D. Chen, Y. Shi, H. Yu and Z. Fu, Polymer, 2016, 97, 533–542 CrossRef CAS.
- Z. Cao, H. Wu, J. Dong and G. Wang, Macromolecules, 2014, 47, 8777–8783 CrossRef CAS.
- X. Liu, D. Hu, Z. Jiang, J. Zhuang, Y. Xu and X. Guo, Macromolecules, 2016, 49, 6186–6192 CrossRef CAS.
- T. Yildirim, L. Yildirim, R. Yañez-Macias, S. Stumpf, C. Fritzsche, S. Hoeppener, C. Cuerrero-Sanchez, S. Schubert and U. S. Schubert, Polym. Chem., 2017, 8, 1328–1340 RSC.
- H. Xia, Y. Zhao and R. Tong, Adv. Exp. Med. Biol., 2016, 880, 365–384 CrossRef CAS PubMed.
- T. Manouras and M. Vamvakaki, Polym. Chem., 2017, 8, 74–96 RSC.
- J. Xuan, M. Pelletier, H. Xia and Y. Zhao, Macromol. Chem. Phys., 2011, 212, 498–506 CrossRef CAS.
- J. Xuan, O. Boissière, Y. Zhao, B. Yan, L. Trembly, S. Lacelle, H. Xia and Y. Zhao, Langmuir, 2012, 28, 15463–16468 CrossRef PubMed.
- R. Deckers, A. Paradissis, C. Oerlemans, M. Talelli, G. Storm, W. E. Hennink and J. F. W. Nijsen, Langmuir, 2013, 29, 9483–9490 CrossRef CAS PubMed.
- R. Tong, H. Xia and X. Lu, J. Mater. Chem. B, 2013, 1, 886–894 RSC.
- R. Tong, X. Liu and H. Xia, Chem. Commun., 2014, 50, 3575–3578 RSC.
- Y. Li, R. Tong, H. Xia, H. Zhang and J. Xuan, Chem. Commun., 2010, 46, 7739–7741 RSC.
- R. S. Lee, S. W. Wang, Y. C. Li and J. Y. Fang, RSC Adv., 2015, 5, 497–512 RSC.
- Y. K. Lin, S. W. Wang, Y. C. Yu and R. S. Lee, Int. J. Polym. Mater. Polym. Biomater., 2017 DOI:10.1080/00914037.2017.1291514.
- R. S. Lee and W. H. Chen, React. Funct. Polym., 2011, 71, 455–462 CrossRef CAS.
- K. Y. Peng, S. W. Wang and R. S. Lee, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2769–2781 CrossRef CAS.
- J. Y. Fang, Y. K. Lin, S. W. Wang, Y. C. Yu and R. S. Lee, RSC Adv., 2016, 6, 107669–107682 RSC.
- H. Huang, S. Wu, Z. Xie, F. Meng, X. Jing and Y. Huang, Biomacromolecules, 2012, 13, 3004–3012 CrossRef PubMed.
- M. C. Alf, T. A. Hatton and K. K. Gleason, Polymer, 2011, 52, 4429–4434 CrossRef CAS.
- Y. Tachibana, M. Kurisawa, H. Uyama and S. Kobayashi, Biomacromolecules, 2003, 4, 1132–1134 CrossRef CAS PubMed.
- R. Ranjan and W. J. Brittain, Macromol. Rapid Commun., 2007, 28, 2084–2089 CrossRef CAS.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS PubMed.
- A. P. Goodwin, J. L. Mynar, Y. Ma, G. R. Fleming and J. M. J. Frechet, J. Am. Chem. Soc., 2005, 127, 9952–9953 CrossRef CAS PubMed.
- J. Hu, J. He, D. Cao, M. Zhang and P. Ni, Polym. Chem., 2015, 6, 3205–3216 RSC.
| This journal is © The Royal Society of Chemistry 2017 |