
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA microfluidic model of the blood–brain barrier to study permeabilization by pulsed electric fields†
M. Bonakdar *,
P. M. Graybill and
R. V. Davalos
*,
P. M. Graybill and
R. V. Davalos
Bioelectromechanical Systems Laboratory, Department of Biomedical Engineering and Mechanics, Virginia Polytechnic Institute and State University, Blacksburg, VA, USA. E-mail: mohammad@vt.edu
First published on 5th September 2017
Abstract
Pulsed electric fields interact with the blood–brain barrier (BBB) and have been shown to increase the BBB permeability under some pulsing regimes. Pulsed electric fields may enhance drug delivery to the brain by disrupting the integrity of the BBB and allowing otherwise impermeable drugs to reach target areas. Microfluidic, in vitro models offer an alternative platform for exploring the impact of pulsed electric fields on the BBB because they create physiologically relevant microenvironments and eliminate the confounding variables of animal studies. We developed a microfluidic platform for real-time measurement of BBB permeability pre- and post-treatment with pulsed electric fields. Permeability is measured optically by the diffusion of fluorescent tracers across a monolayer of human cerebral microcapillary endothelial cells (hCMECs) cultured on a permeable membrane. We found that this device is able to capture real-time permeability of hCMEC monolayers for both reversible and irreversible electroporation pulsing regimes. Furthermore, preliminary testing of deep brain stimulation pulsing regimes reveals possible impacts on BBB integrity. This device will enable future studies of pulsed electric field regimes for improved understanding of BBB permeabilization.
Introduction
Cerebral microvessels strictly regulate the transfer of substances between the blood and the brain tissue. This regulation by cerebral micro-vessels is called the blood–brain barrier (BBB), and is due to intercellular tight junctions (TJs) that form between brain capillary endothelial cells. In cerebral capillaries, TJ proteins are expressed 50–100 times more than in peripheral microvessels.1 TJs are formed by an intricate complex of transmembrane proteins (claudin and occludin) with cytoplasmic accessory proteins (ZO-1 and -2, cingulin, AF-6, and 7H6).2 By linking to the actin cytoskeleton, these proteins form a strong cell–cell connection.3 Brain endothelial cells, which form the endothelium of cerebral microvessels, are responsible for about 75–80% of the BBB resistance to substances, and other cells such as astrocytes and pericytes provide the remainder of the resistance.4The integrity of the BBB is essential for the health and proper functioning of brain tissue. BBB breakdown and increased permeability are observed in some diseases and injuries associated with the central nervous system (CNS) such as stroke, traumatic head injury, brain edema, Alzheimer's disease, AIDS, brain cancer and meningitis.5 The integrity of the BBB, while critical to brain health, limits the success rate of new therapies by hampering drug transport to the brain. There are two major pathways for the transfer of substances across the BBB which are categorized as paracellular and transcellular.5 Transport across the intercellular TJs, the paracellular pathway, restricts transport to small hydrophilic molecules of less than 500 Da in molecular weight.5 Transport through the cell membrane and intracellular space, the transcellular pathway, is facilitated by special carriers or passive diffusion for lipophilic molecules.5 Most CNS drugs are large, lipophilic molecules that cannot pass through the TJs between the endothelial cells6 or diffuse across the endothelial cell membrane due to their large size. These limitations highlight the importance of developing techniques to permeabilize the BBB temporarily for drug delivery purposes.
Advanced drug delivery methods are being widely investigated to enhance transport across the BBB including focused ultrasound,7–9 osmotic disruption,10 drug delivery vehicles,11–14 and pulsed electric fields (PEFs).15–17 Although all these techniques have inherent advantages and disadvantages, PEFs may offer advantages over other techniques due to their synergistic potential for treating a variety of CNS disorders. Several types of PEFs have clinical significance for treating brain conditions, including electrochemotherapy,18,19 tumour treating fields (TTFields),20,21 deep-brain stimulation (DBS),22,23 and irreversible electroporation.24–26 Recent studies show that pulsed electric fields (PEFs) can temporarily or permanently disrupt the BBB through either the paracellular or transcellular pathway.27 The type and extent of BBB disruption generally depends on the PEF's parameters such as amplitude, polarity, duration and frequency. Low frequency (∼1 Hz), high amplitude (∼500 V cm−1) PEFs applied for a short duration (1 min) can electroporate the constituent endothelial cells, opening the transcellular pathway. High frequency (∼200 Hz), low amplitude (∼25 V cm−1) PEFs applied for a long duration (>30 min) may disrupt tight junctions, increasing the permeability of the BBB through the paracellular pathway. To limit the scope of this study, we focus on BBB permeabilization by unipolar electroporation pulses as a case study for high-magnitude PEFs and DBS-type pulses as a preliminary case study of low-magnitude PEFs.
Application of reversible electroporation in BBB permeabilization is shown in several studies.17,27–30 Electroporation must be reversible to avoid permanent damage to the BBB. We have recently shown in an in vitro model that 10 pulses of 380 V cm−1 can trigger the electroporation of adhered brain endothelial cells with minimal damage to the cells. When the electric field magnitude was increased to 450 V cm−1 for the same number of pulses, the permeabilization efficiency was increased but did not induce any cell death.31 Using an in vivo model Garcia et al. found that electroporation is predominantly reversible at electric fields less than 400 V cm−1 in the brain and reversibly disrupts the BBB.27 In a recent in vivo study Arena et al. found that sub-lethal BBB disruption could be achieved by 300 bursts of bipolar pulses with an amplitude of 250 V cm−1, where each burst consisted of 200 pulses with a duration of 850 ns.17 The BBB disruption seen in this study, however, may represent an increase in permeability through the paracellular pathway rather than by electroporation, since low amplitude pulses were used to treat the cells.17
In addition to the transcellular pathway, BBB disruption can be achieved through the paracellular pathway by disrupting the tight junctions between the endothelial cells. It has been shown that PEFs disrupt the cytoskeletal organization and delocalize junction-specific proteins such as VE-cadherin, which weaken the cell–cell integrity32 and enhance the paracellular permeability. In a recent study Lopez-Quintero et al. showed that PEFs of low amplitude (2.5 V cm−1) and high frequency (200 Hz), such as the ones used in deep brain stimulation, can increase the permeability of the BBB.33 Due to the low amplitude of these pulses, it is postulated that the permeabilization happens only through the paracellular pathway, since electroporation may not be induced. The disruption of the tight junctions and increased permeability of the BBB has also been reported after exposure to different types of electromagnetic fields34 including continuous wave electromagnetic fields,35 intense electromagnetic pulses36,37 and microwave radiation.38
So far the majority of the studies on BBB permeability have been conducted on animals. However, in vitro models can provide more freedom to explore parameters and offer quantitative data without the complications of animal studies.39 In vitro models exploit monolayer cultures of cerebral endothelial cells as the main constituent of the BBB, using different platforms such as transwells,40–42 micro-fabricated membranes,43 tubes,44 collagen matrices45 and microfluidic channels46–49 to monitor the integrity and permeability of the endothelial cells using tracers,50 trans-endothelial electrical resistance (TEER)40,46 or electrical impedance spectroscopy (EIS).48,51
In this study we developed a platform to investigate the disruptive effects of unipolar electroporation pulses and a single type of DBS pulsed electric field on the BBB in an accurate, quantitative manner using a microfluidic platform that allows real-time measurement of permeability.
Materials and methods
Device design and fabrication
A double layer microfluidic device with an embedded membrane is developed as a platform to model the BBB and study its permeabilization. The top layer contains a single channel, 3 cm long, 1 mm wide, and 250 μm high that contains the endothelial cell monolayer. The bottom channel contains an array of 6 channels, 800 μm wide and 80 μm high that are oriented perpendicular to the top channel. This orientation creates 6 intersection sites of the top and bottom channels, which are separated by the embedded membrane. We incorporate a microfluidic adaptor upstream of the double layer chip (connecting the syringe pump and peristaltic pump to the chip) to act as a bubble trap and to measure the fluorescent intensity of the top channel. The device schematic is shown in Fig. 1a. | ||
| Fig. 1 Schematic of the microfluidic device (a) and the experimental setup (b). Fluid lines for the top and bottom channels are shown in green and red, respectively. | ||
The double layer device is fabricated according to standard procedures for this application,52 but is briefly described here. Photolithography and deep reactive ion etching (DRIE) are first used to create negatively-patterned master molds of the microfluidic channels. The top and bottom channel layers of PMDS are patterned by replication molding using the master molds. The top layer is punched at four sites corresponding to the inlets and outlets of the top and bottom channels. A liquid PDMS glue is used for bonding, and is made by mixing PDMS and toluene with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight ratio. Prior to bonding, the PDMS layers are placed under vacuum for at least 15 minutes. A clean glass slide is spin-coated with the PDMS glue at 1600 rpm for 60 seconds. The top and bottom PDMS layers are taken from vacuum and immediately placed on the coated glass slide with the channels facing down. The PDMS layers remain on the slides for 1 minute to absorb a thin layer of glue. The PDMS layers are then peeled off the slide, and a 0.4 μm pore size polyester membrane cut from a transwell plate (Corning) is embedded between the layers before aligning and bonding the layers together. The assembled device is cured at 65 °C for 10 hours and then plasma bonded to a glass slide adjacent to the adaptor. Stainless steel needles 0.13 mm diameter (Kingli, China) are inserted through the sides of the device and into the inlet and the outlet of the top channel to enable the application of an electric potential.
:1 weight ratio. Prior to bonding, the PDMS layers are placed under vacuum for at least 15 minutes. A clean glass slide is spin-coated with the PDMS glue at 1600 rpm for 60 seconds. The top and bottom PDMS layers are taken from vacuum and immediately placed on the coated glass slide with the channels facing down. The PDMS layers remain on the slides for 1 minute to absorb a thin layer of glue. The PDMS layers are then peeled off the slide, and a 0.4 μm pore size polyester membrane cut from a transwell plate (Corning) is embedded between the layers before aligning and bonding the layers together. The assembled device is cured at 65 °C for 10 hours and then plasma bonded to a glass slide adjacent to the adaptor. Stainless steel needles 0.13 mm diameter (Kingli, China) are inserted through the sides of the device and into the inlet and the outlet of the top channel to enable the application of an electric potential.
Cell culture on chip
Human cerebral microvascular endothelial cells (hCMEC, EMD Millipore, Billerica, MA) were used for the BBB model in this study. Cells were cultured in culture flasks maintained with complete media at 37 °C in a water jacket incubator and were routinely passaged at 90% confluence. The complete culture medium for hCMEC cells consisted of EndoGRO basal medium supplemented with EndoGRO-LS supplement (0.2%), rh EGF (5 ng ml−1), L-glutamine (10 mM), hydrocortisone hemisuccinate (1 μg ml−1), heparin sulfate (0.75 U ml−1), ascorbic acid (50 μg ml−1) and FBS (5%), all from EMD Millipore. The cell culture protocol on chip is similar to an earlier study.31 Briefly, the microfluidic channel was sterilized with 70% ethanol, washed with PBS, and treated with 50 μg ml−1 bovine plasma fibronectin (Life Technologies) in PBS for one hour. The fibronectin solution was then removed and replaced with complete cell culture medium for another hour in the incubator. The cell suspension was then introduced into the culture channel at a concentration of 10 million cells per ml. The device was incubated for 1–2 hours to allow the cells adhere to the bottom of the channel. After 2 hours, pipet tips filled with complete media were placed at the channel outlets, providing nutrients for the duration of cell proliferation in static mode.After a confluent cell monolayer was formed in the channels, the chips were removed from the incubator and transferred to the inverted microscope for the duration of the permeabilization experiment. Due to the extended time required for the permeabilization experiments, a small incubator was designed and fabricated to fit on the microscope stage and accommodate the microfluidic chip during the experiment (Fig. 2). The small incubator was equipped with an indium tin oxide coated glass slide as a heater and was connected to a 5% CO2 balance air cylinder (Airgas, Radnor, PA).
 | ||
| Fig. 2 Stage-top incubator with the microfluidic device. | ||
On chip permeability measurement
Two fluorescent tracers of different molecular weight were used to measure the permeability: fluorescent sodium salt (376 Da) and FITC-dextran (70 kDa) (Sigma Aldrich, St Louis, MO). Each of the tracers were dissolved in serum-free media and circulated in the top (luminal) channel of the device with a peristaltic pump (Watson Marlow). A low conductivity buffer was injected through the bottom (abluminal) channel after passing through a bubble trap using a syringe pump (Harvard Apparatus). Fig. 1b shows the schematic of the experimental setup. Diffusion of the tracer molecules across the endothelial cell monolayer occurs across the membrane at the six intersection points between the top and bottom channels. The diffused concentration of fluorescent tracer molecules correlates with the permeability of the cell monolayer according to:
 | (1) |
 | (2) |
 | (3) |
Fluorescent microscopy is used to find the concentration of tracer molecules in the luminal and abluminal channels. The fluorescent light intensity varies linearly with the fluorescent molecule concentration,31 the exposure time of the microscope, and the height of the channel under observation (not shown here). Therefore, the fluorescent intensity has been used as a measure of the concentration when it is normalized by exposure time and channel height, which reduces eqn (3) to
 | (4) |
Taking into account the permeability across the membrane in the cell-free chip, Pm, the permeability of the cell layer, Pc, would be
 | (5) |
BBB disruption on chip
Two different sets of PEFs were used to stimulate the cells in the device. For each set of pulses, a separate pulse generator was used: (1) BTX pulse generator (Harvard Apparatus, Holliston, MA) was used to deliver unipolar electroporation pulses with high amplitude and 100 μs pulse width. (2) Low-amplitude, high-frequency pulses were delivered using a function generator (Agilent, Santa Clara, CA) and high voltage amplifier (Trek, Lockport, NY). Fig. S4 and S5 of the ESI† plot the waveforms of the PEFs tested. Due to the uniform cross section of the luminal channel, a uniform electric field is generated across the channel upon application of the potential. Table 1 summarizes the different treatments that were investigated in this study.| Treatment type | Voltage amplitude | Pulse # | Frequency | Pulse width |
|---|---|---|---|---|
| Electroporation pulses | 600 V | 10, 90 | 1 Hz | 100 μs |
| 1000 V | ||||
| 2500 V | ||||
| High-frequency pulses | 7.5 V | Continuous | 200 Hz | 10 μs |
| 75 V |
Finite element modelling of fluid flow
The flow velocity distribution in the abluminal channel was determined by a three-dimensional finite element model constructed in Comsol Multiphysics (Comsol, Stockholm, Sweden). An arbitrary fluid velocity was given as the inlet boundary condition and atmospheric pressure was given as the outlet boundary condition. The fluid flow was solved under the laminar flow assumption. The flow rates in each of the six channels and the streamline pattern in the horizontal mid-plane were obtained.Results and discussion
Flow distribution in the channel
The bottom channel has a symmetric branching pattern to provide similar flow rates in all six channels, as done earlier for microfluidic gradient generators.53 As shown in Fig. 3a, the numerical modelling of the fluid flow shows that despite initial expectations, the fluid flow is not uniformly distributed in all of the six channels. The two middle channels have about 18% greater flow rate compared to the outer four channels. The numerical modelling also reveals that the streamlines of the six channels occupy different portions of the outlet channel after merging (Fig. 3b). The differences in flow rate and outflow regions must be considered during image analysis to determine the correct permeability of each channel. | ||
| Fig. 3 Numerical modelling of fluid flow in the bottom channel. (a) Fluid velocity and flow rate contributions in each of the channels. The scale bar shows an arbitrary velocity unit. (b) The streamlines of individual abluminal channels at the merging point determine the portion of the merged channel corresponding to each abluminal channel. | ||
Determination of the baseline permeability
The permeability of the cell-free device, Pm, is determined prior to running experiments involving cells. This control is needed in order to determine the contribution of the cell monolayer in the permeability experiments according to eqn (5). Fig. 4 shows the fluorescent images at the outlet of the bottom channel for each of the fluorescent tracers as well as the permeability values for each of the tracers. The comparison shows that the permeability of the sodium salt across the membrane is significantly higher than the 70 kDa FITC-dextran, which is expected due to the difference in the molecular weights of the two tracer molecules. | ||
| Fig. 4 (a) Merging of streamlines at the outlet channel in the bottom layer for sodium salt and 70 kDa FITC-dextran. (b) Membrane and cell monolayer permeability for two tracers. | ||
We also observed that increasing the molecular weight of the tracer resulted in the observed fluorescent tracks of each of the six channels becoming more distinct, which is due to a lower diffusion rate of the tracer.
Molecular weight dependence of permeability across the endothelial cell monolayer
Fig. 4b shows the diffusion results of the two fluorescent markers across the cell monolayer on the chip. As expected, 70 kDa FITC-dextran is significantly less permeable across the endothelial monolayer compared to sodium salt. After accounting for the membrane permeability, we found the permeability of the endothelial monolayer to be 5.99 ± 4.91 × 10−6 cm s−1 for sodium salt and 4.95 ± 2.37 × 10−7 cm s−1 for 70 kDa FITC-dextran.Effect of electroporation pulses on permeabilization of the BBB
Electroporation pulses were administered at 2500 V, 1000 V, and 600 V to generate electric field magnitudes of 833 V cm−1, 333 V cm−1, and 200 V cm−1 respectively within the device. Fig. 5 illustrates the prevailing trends for monolayer permeability to sodium salt for each electroporation treatment. Averaged results from all experiments of sodium salt and 70 kDa FITC-dextran are presented in the ESI.†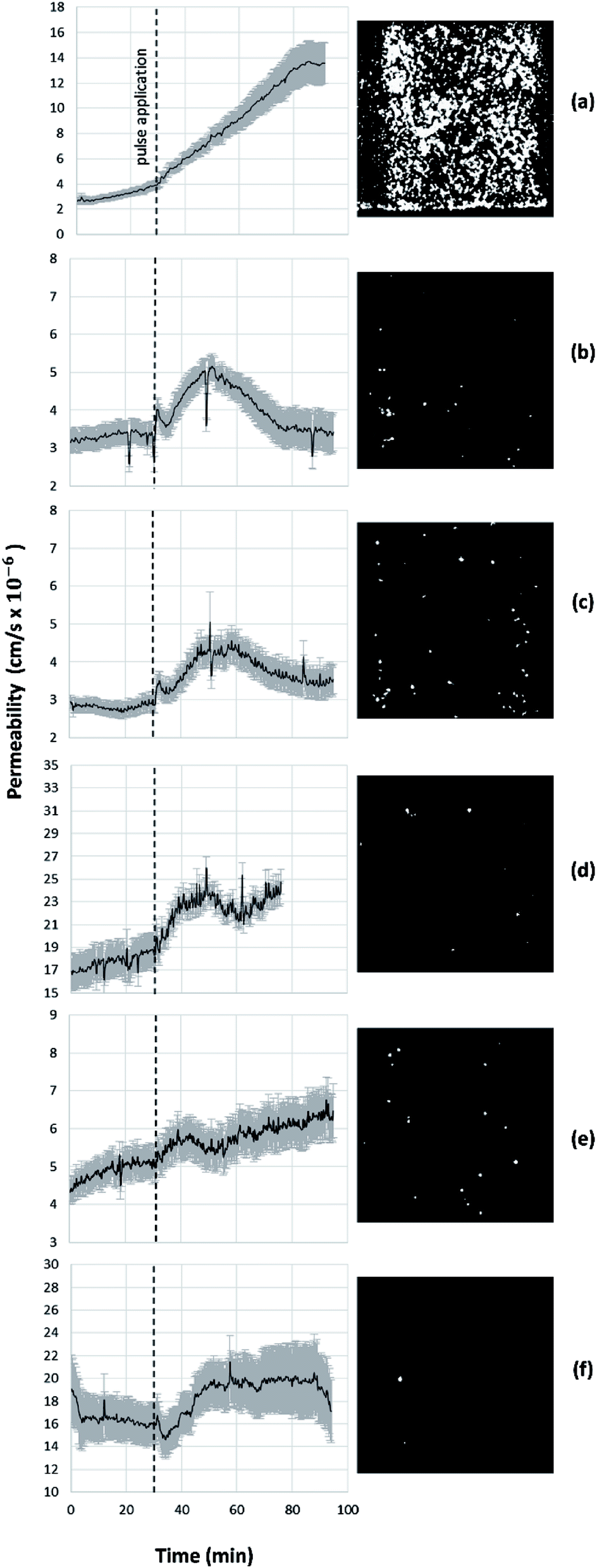 | ||
| Fig. 5 Permeabilization of the BBB to fluorescent sodium salt after application of (a) 90 pulses of 833 V cm−1, (b) 10 pulses of 833 V cm−1, (c) 90 pulses of 333 V cm−1, (d) 10 pulses of 333 V cm−1, (e) 90 pulses of 200 V cm−1 (f) 10 pulses of 200 V cm−1. The dashed line indicates pulse application. Images (right) show post-treatment staining for dead cells with propidium iodide. | ||
For high-magnitude pulses of 833 V cm−1, the results clearly demonstrate irreversible electroporation of the endothelial monolayer after 90 pulses but reversible electroporation of the monolayer after 10 pulses for sodium salt (Fig. 5) and 70 kDa FITC-dextran (ESI†). In the case of irreversible electroporation at 833 V cm−1 and 90 pulses, permeability rapidly increases immediately after pulsing and does not recover, indicative of cell death as verified by staining. We find that approximately 55 minutes after pulsing, the permeability value begins to stabilize and approach a steady value over 300% higher than pre-treatment, as shown in Fig. 5a. For reversible electroporation at 833 V cm−1 and 10 pulses, the maximum permeability (>130% baseline) occurs 15–20 minutes post-treatment, and remarkably is followed by a complete return to pre-treatment levels approximately 50 minutes post-treatment. The rate-of-change in permeability after treatment in Fig. 5b and c shows that the endothelial monolayer permeabilizes more rapidly than it recovers. For experiments with 70 kDa FITC-dextran at the same treatment conditions (833 V cm−1, 10 pulses), the duration of increased permeability post-treatment is shorter than that for sodium salt (see ESI†), indicating that the smaller molecules are able to pass through the monolayer for a longer time following the treatment.
Lower-magnitude pulsing reveals reversible electroporation of the endothelial monolayer and high cell viability post-treatment. Treatment with 333 V cm−1 and 90 pulses induces reversible electroporation of similar time-scale but lower magnitude than that of 833 V cm−1 and 10 pulses. However, unlike treatment at 833 V cm−1 and 10 pulses, permeability levels do not return to pre-treatment levels, likely indicating irreversible electroporation of a small percentage of cells, as supported by the greater prevalence of dead cells in the live-dead stain in Fig. 5c. Results for 333 V cm−1 and 10 pulses and 200 V cm−1 and 90 pulses show reversible electroporation and high monolayer viability post-treatment. It is apparent that the monolayer reversibly permeabilizes immediately following treatment. Averaged results for sodium salt show a sustained increase in permeability of approximately 16% after treatment at 333 V cm−1 for 10 pulses and approximately 14% after treatment at 200 V cm−1 for 90 pulses.
In a previous study conducted on mouse brain endothelial cells, it was found that regardless of the EF amplitude, increasing the pulse number beyond 10 pulses significantly reduces the chance of cell recovery.31 This finding is in agreement with the results of the current study that indicate an increased permeability of the cell monolayer after exposure to 90 pulses. On the other hand, in the same study, minimal cell electroporation was observed after treatment with a maximum of 10 pulses at electric fields below 300 V cm−1. Surprisingly, the current study shows that permeability increases with 10 pulses of even 200 V cm−1. Knowing that this treatment falls below the threshold for electroporation as stated before, we postulate that the permeability increases through the paracellular pathway due to the deformation of the cells and opening of the TJs. Therefore, BBB disruption could be achieved at a threshold lower than that of electroporation. The exact mechanism of paracellular permeabilization with EF is not known, however the possibility of cytoskeletal reorganization and cell shrinkage was raised in a previous study.32
DBS-relevant pulses and permeability of the BBB
Fig. 6 shows the results of permeabilization in response to high frequency low amplitude pulsing similar to DBS treatment pulses. Our preliminary results suggest that the permeability of hCMEC monolayers to sodium salt increases due to DBS-relevant pulse conditions at 2.5 V cm−1. However, at this EF magnitude no increase is observed for the larger molecules of 70 kDa FITC-dextran. The permeabilization process was also investigated when increasing the pulse amplitude to 25 V cm−1. For this pulsing condition the permeability of both tracers increased, probably due to larger openings in the cell monolayer. | ||
| Fig. 6 Permeabilization of the BBB upon continuous application of deep brain stimulation-relevant pulses. For fluorescent sodium salt at (a) 25 V cm−1 and (b) 2.5 V cm−1 and 70 kDa FITC-dextran at (c) 25 V cm−1 and (d) 2.5 V cm−1. | ||
Given the fact that the two amplitudes of 2.5 and 25 V cm−1 are far below the threshold for cell electroporation, it is expected that the increased permeability is through the paracellular pathway because of TJ opening. Further experimentation is needed to fully address this issue.
Although future studies must be conducted before making definitive conclusions, our results for high-frequency, low-amplitude pulses of 25 V cm−1 suggest an increase in BBB permeability during DBS pulsing. Results for 2.5 V cm−1 are within the range of experimental noise, but in general do not seem to have as dramatic an increase on BBB permeability. In addition to disruption with PEFs, the current platform is also capable of investigating other types of physical disruptions such as osmotic disruption. Mannitol, which is known to increase BBB permeability, was briefly studied at concentrations up to 1 M for very short durations and resulted in the disruption of the monolayer integrity as expected (results shown in ESI†).
The permeability diagrams seem to be noisy in some cases. We suspect that the noise is caused by slight changes in the outflow velocity due to bubble formation upstream or an inconsistent delivery rate from the syringe pump or the peristaltic pump because the noise correlates between all six channels during experimentation.
Application of BBB model to future research
Many PEF regimes are applicable to BBB research and represent testable conditions with this device. This study investigated the effect of unipolar electroporation pulses and a single case of DBS pulses on BBB permeability. Future studies could expand this scope to explore other BBB-relevant pulsing regimes. For example, high-frequency irreversible electroporation (H-FIRE) uses bursts of bipolar pulses of 1–5 μs to electroporate cells and promises several advantages over traditional IRE treatments.54,55 The capability of H-FIRE pulses to permeabilize the BBB has also been explored.17 Furthermore, the DBS pulsing which was used in this study is a special case of high frequency signals which may span a wide range of parameters such as shape, amplitude, frequency, etc. Each of these parameters may affect the permeability of the BBB differently. Future research on other pulse regimes may prove valuable for optimizing treatments by minimizing or maximizing BBB permeability.Increasing the permeability of the BBB by PEFs may allow otherwise impenetrable chemotherapies to penetrate the brain for improved tumor treatments. In this study, we used two fluorescent tracers of different molecular weights to examine the effect of molecular weight on BBB permeability following PEF treatment. Molecular weight, among other factors such as lipophilicity and molecular charge, has a profound impact on a substance's ability to cross the BBB. Many commonly administered chemotherapeutic agents have molecular weights similar to that of fluorescent sodium salt (doxorubicin: 543.5 Da, cisplatin: 300.0 Da, paclitaxel: 853.9 Da), and thus may show a similar increase of BBB permeability to these agents after treatment by PEFs. The investigation of BBB permeability to chemotherapeutic agents is left for future studies.
Conclusions
A platform was developed enabling real-time monitoring of permeability across cell monolayers with fluorescent microscopy. This platform was used to study the effects of unipolar electroporation PEFs on permeabilization of the BBB. It was found that this device captures the reversible and irreversible effects of electroporation pulses on the endothelial monolayer permeability. Furthermore, our preliminary results for a low-amplitude, high-frequency pulsing regime suggest that this device can resolve the effects of DBS pulses. The increased permeability of the BBB model at sub-electroporation pulses suggests that permeabilization can occur through the paracellular pathway by opening the TJs. This microfluidic platform will be valuable for future studies of permeability across cell monolayers, including osmotic permeability studies, high-frequency irreversible electroporation (H-FIRE) studies, and further DBS studies. As PEFs become increasingly important to medical treatments, this microfluidic, in vitro model will be valuable for studying the permeabilization of the BBB.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by NIH (R01CA213423), NSF (CAREER CBET-1055913), and the National Cancer Institute's Cancer Center grant P30CA012197 issued to the Wake Forest Baptist Comprehensive Cancer Center. P. M. G. is supported by Virginia Tech's BIOTRANS IGEP.References
- N. J. Abbott, J. Anat., 2002, 200, 523–534 CrossRef.
- Y. Chen and L. Liu, Adv. Drug Delivery Rev., 2012, 64, 640–665 CrossRef CAS PubMed.
- B. T. Hawkins and T. P. Davis, Pharmacol. Rev., 2005, 57, 173–185 CrossRef CAS PubMed.
- G. Li, M. J. Simon, L. M. Cancel, Z. D. Shi, X. Ji, J. M. Tarbell, B. Morrison 3rd and B. M. Fu, Ann. Biomed. Eng., 2010, 38, 2499–2511 CrossRef PubMed.
- B. M. Fu, Curr. Pharm. Biotechnol., 2012, 13, 1346–1359 CAS.
- S. Santaguida, D. Janigro, M. Hossain, E. Oby, E. Rapp and L. Cucullo, Brain Res., 2006, 1109, 1–13 CrossRef CAS PubMed.
- K. Hynynen, N. McDannold, N. A. Sheikov, F. A. Jolesz and N. Vykhodtseva, NeuroImage, 2005, 24, 12–20 CrossRef PubMed.
- C. Poon, D. McMahon and K. Hynynen, Neuropharmacology, 2017, 120, 20–37 CrossRef CAS PubMed.
- L. H. Treat, N. McDannold, N. Vykhodtseva, Y. Zhang, K. Tam and K. Hynynen, Int. J. Cancer, 2007, 121, 901–907 CrossRef CAS PubMed.
- R. A. KrolI, E. A. Neuwelt and E. A. Neuwelt, Neurosurgery, 1998, 42, 1083–1099 CrossRef.
- E. C. Unger, T. Porter, W. Culp, R. Labell, T. Matsunaga and R. Zutshi, Adv. Drug Delivery Rev., 2004, 56, 1291–1314 CrossRef CAS PubMed.
- A. Kumari, S. K. Yadav and S. C. Yadav, Colloids Surf., B, 2010, 75, 1–18 CrossRef CAS PubMed.
- W. M. Pardridge, Nat. Rev. Drug Discovery, 2002, 1, 131–139 CrossRef CAS PubMed.
- X. Li, J. Tsibouklis, T. Weng, B. Zhang, G. Yin, G. Feng, Y. Cui, I. N. Savina, L. I. Mikhalovska and S. R. Sandeman, J. Drug Targeting, 2017, 25, 17–28 CrossRef CAS PubMed.
- S. V. Lopez-Quintero, A. Datta, R. Amaya, M. Elwassif, M. Bikson and J. M. Tarbell, J. Neural Eng., 2010, 7, 016005 CrossRef CAS PubMed.
- P. A. Garcia, J. H. Rossmeisl Jr, J. L. Robertson, J. D. Olson, A. J. Johnson, T. L. Ellis and R. V. Davalos, PLoS One, 2012, 7, e50482 CAS.
- C. B. Arena, P. A. Garcia, M. B. Sano, J. D. Olson, T. Rogers-Cotrone, J. H. Rossmeisl Jr and R. V. Davalos, Technology, 2014, 2, 206–213 CrossRef.
- L. G. Salford, B. Persson, A. Brun, C. Ceberg, P. C. Kongstad and L. M. Mir, Biochem. Biophys. Res. Commun., 1993, 194, 938–943 CrossRef CAS PubMed.
- B. Agerholm-Larsen, H. K. Iversen, P. Ibsen, J. M. Moller, F. Mahmood, K. S. Jensen and J. Gehl, Cancer Res., 2011, 71, 3753–3762 CrossRef CAS PubMed.
- E. D. Kirson, Z. Gurvich, R. Schneiderman, E. Dekel, A. Itzhaki, Y. Wasserman, R. Schatzberger and Y. Palti, Cancer Res., 2004, 64, 3288–3295 CrossRef CAS PubMed.
- K. D. Swanson, E. Lok and E. T. Wong, Curr. Neurol. Neurosci. Rep., 2016, 16, 8 CrossRef PubMed.
- J. S. Perlmutter and J. W. Mink, Annu. Rev. Neurosci., 2006, 29, 229–257 CrossRef CAS PubMed.
- H. S. Mayberg, A. M. Lozano, V. Voon, H. E. McNeely, D. Seminowicz, C. Hamani, J. M. Schwalb and S. H. Kennedy, Neuron, 2005, 45, 651–660 CrossRef CAS PubMed.
- P. A. Garcia, J. H. Rossmeisl, R. E. Neal, T. L. Ellis, J. D. Olson, N. Henao-Guerrero, J. Robertson and R. V. Davalos, J. Membr. Biol., 2010, 236, 127–136 CrossRef CAS PubMed.
- J. H. Rossmeisl Jr, P. A. Garcia, T. E. Pancotto, J. L. Robertson, N. Henao-Guerrero, R. E. Neal, T. L. Ellis and R. V. Davalos, J. Neurosurg., 2015, 123, 1008–1025 CrossRef PubMed.
- P. Garcia, T. Pancotto, J. Rossmeisl Jr, N. Henao-Guerrero, N. Gustafson, G. Daniel, J. Robertson, T. Ellis and R. Davalos, Technol. Cancer Res. Treat., 2011, 10, 73–83 CrossRef CAS PubMed.
- P. A. Garcia, J. H. Rossmeisl Jr, J. L. Robertson, J. D. Olson, A. J. Johnson, T. L. Ellis and R. V. Davalos, PLoS One, 2012, 7, e50482 CAS.
- M. Hjouj, D. Last, D. Guez, D. Daniels, S. Sharabi, J. Lavee, B. Rubinsky and Y. Mardor, PLoS One, 2012, 7, e42817 CAS.
- S. Sharabi, D. Last, D. Guez, D. Daniels, M. I. Hjouj, S. Salomon, E. Maor and Y. Mardor, Bioelectrochemistry, 2014, 99, 30–39 CrossRef CAS PubMed.
- S. Sharabi, B. Kos, D. Last, D. Guez, D. Daniels, S. Harnof, Y. Mardor and D. Miklavcic, Radiol. Oncol., 2016, 50, 28–38 CAS.
- M. Bonakdar, E. M. Wasson, Y. W. Lee and R. V. Davalos, Biophys. J., 2016, 110, 503–513 CrossRef CAS PubMed.
- C. Kanthou, S. Kranjc, G. Sersa, G. Tozer, A. Zupanic and M. Cemazar, Mol. Cancer Ther., 2006, 5, 3145–3152 CrossRef CAS PubMed.
- S. V. Lopez-Quintero, A. Datta, R. Amaya, M. Elwassif, M. Bikson and J. M. Tarbell, J. Neural Eng., 2010, 7, 16005 CrossRef CAS PubMed.
- R. Stam, Brain Res. Rev., 2010, 65, 80–97 CrossRef PubMed.
- A. H. Frey and S. R. Feld, Ann. N. Y. Acad. Sci., 1975, 247, 433–439 CrossRef CAS PubMed.
- G. Ding, K. Li, X. Wang, Y. Zhou, L. Qiu, J. Tan, S. Xu and G. Guo, Environ. Sci. Technol., 2009, 22, 255–258 Search PubMed.
- J. X. Zhou, G. R. Ding, J. Zhang, Y. C. Zhou, Y. J. Zhang and G. Z. Guo, Biomed. Environ. Sci., 2013, 26, 128–137 CAS.
- L. G. Salford, A. Brun, K. Sturesson, J. L. Eberhardt and B. R. Persson, Microsc. Res. Tech., 1994, 27, 535–542 CrossRef CAS PubMed.
- J. Bicker, G. Alves, A. Fortuna and A. Falcão, Eur. J. Pharm. Biopharm., 2014, 87, 409–432 CrossRef CAS PubMed.
- M. P. Chan, S. Morisawa, A. Nakayama, Y. Kawamoto and M. Yoneda, Environ. Toxicol., 2006, 21, 223–235 CrossRef CAS PubMed.
- S. Nakagawa, M. A. Deli, H. Kawaguchi, T. Shimizudani, T. Shimono, A. Kittel, K. Tanaka and M. Niwa, Neurochem. Int., 2009, 54, 253–263 CrossRef CAS PubMed.
- J. Barar and Y. Omidi, J. Biol. Sci., 2008, 8, 556–562 CrossRef.
- S. H. Ma, L. A. Lepak, R. J. Hussain, W. Shain and M. L. Shuler, Lab Chip, 2005, 5, 74–85 RSC.
- L. Cucullo, M. S. McAllister, K. Kight, L. Krizanac-Bengez, M. Marroni, M. R. Mayberg, K. A. Stanness and D. Janigro, Brain Res., 2002, 951, 243–254 CrossRef CAS PubMed.
- J. A. Kim, H. N. Kim, S.-K. Im, S. Chung, J. Y. Kang and N. Choi, Biomicrofluidics, 2015, 9, 024115 CrossRef PubMed.
- R. Booth and H. Kim, Lab Chip, 2012, 12, 1784–1792 RSC.
- F. R. Walter, S. Valkai, A. Kincses, A. Petneházi, T. Czeller, S. Veszelka, P. Ormos, M. A. Deli and A. Dér, Sens. Actuators, B, 2016, 222, 1209–1219 CrossRef CAS.
- L. M. Griep, F. Wolbers, B. de Wagenaar, P. M. ter Braak, B. B. Weksler, I. A. Romero, P. O. Couraud, I. Vermes, A. D. van der Meer and A. van den Berg, Biomed. Microdevices, 2013, 15, 145–150 CrossRef CAS PubMed.
- B. Prabhakarpandian, M. C. Shen, J. B. Nichols, I. R. Mills, M. Sidoryk-Wegrzynowicz, M. Aschner and K. Pant, Lab Chip, 2013, 13, 1093–1101 RSC.
- S. Nag, The Blood-Brain Barrier: Biology and Research Protocols, 2003, pp. 133–144 Search PubMed.
- D. H. Jo, R. Lee, J. H. Kim, H. O. Jun, T. G. Lee and J. H. Kim, Sci. Rep., 2015, 5, 1–9 Search PubMed.
- B.-h. Chueh, D. Huh, C. R. Kyrtsos, T. Houssin, N. Futai and S. Takayama, Anal. Chem., 2007, 79, 3504–3508 CrossRef CAS PubMed.
- S. K. Dertinger, D. T. Chiu, N. L. Jeon and G. M. Whitesides, Anal. Chem., 2001, 73, 1240–1246 CrossRef CAS.
- C. B. Arena, M. B. Sano, J. H. Rossmeisl, J. L. Caldwell, P. A. Garcia, M. N. Rylander and R. V. Davalos, Biomedical engineering online, 2011, 10, 102 CrossRef PubMed.
- C. B. Arena, M. B. Sano, M. N. Rylander and R. V. Davalos, IEEE Trans. Biomed. Eng., 2011, 58, 1474–1482 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07603g |
| This journal is © The Royal Society of Chemistry 2017 |