
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe assembly of small molecule conjugate amphiphiles into a precise nanomedicine for colon cancer
Shihou Sheng†
a,
Yahong Chen†a,
Tao Zhanga,
Meng Dingb,
Yuanyu Wua,
Zhen Shena,
Gang Hanc and
Xu Wang *d
*d
aDepartment of Gastrointestinal Colorectal and Anal Surgery, China-Japan Union Hospital of Jilin University, Chang Chun 130000, China
bDepartment of Gastroenterology, China-Japan Union Hospital of Jilin University, Chang Chun 130000, China
cDepartment of Gastrointestinal Surgery, The Second Hospital of Jilin University, Chang Chun, 130041, China
dDepartment of Gastrointestinal Colorectal Surgery, The First Hospital of Jilin University, 130000, China. E-mail: e6e6e67@163.com
First published on 29th September 2017
Abstract
A small molecule conjugate based on a traditional Chinese medicine cantharidin (CTR), which is an anhydride, and the anticancer drug camptothecin (CPT) was designed. This small molecule conjugate is amphiphilic and was found to readily self-assemble into NPs. The NPs were systematically characterized and shown to release drugs in a pH dependent manner. Moreover, we found that cancer cells internalized the NPs via endocytosis. The ester bond could be opened by an esterase to release the two drugs in tumor cells with lower pH values. After that, these two drugs work synergistically to kill the cancer cells. Lastly, mice bearing subcutaneous HT29 colon cancer were i.v. injected with these NPs and great potential for inhibiting colon cancer was found.
Introduction
Over the last several decades, tremendous effort has been made on the development and application of engineered nanoparticles for cancer therapeutics to solve the key issues and overcome the drawbacks associated with conventional anticancer agents including poor solubility of drugs, short blood circulation time, low bioavailability, systemic toxicity and low therapeutic index.1–3 Although numerous so-called nanomedicines have been developed across the world with some of them in clinic trials, very few of them get approved and little benefit over their small molecule counterparts has been seen.4,5 The major reason for the status of nanomedicine lies in the complexity of constructing the systems, poor quality control with batch-to-batch variations as well as stability and reproducibility which makes them far from being a so-called precise medicine.4,6The current approach to nanomedicine involves loading of a certain drug, targeting ligands as well as imaging agents into carrier systems by either chemical conjugation or physical encapsulation.7–10 Moreover, electrostatic complexation can be also adopted for inversely charged molecules.11–14 These strategies eventually aims to build up a nanoparticle delivery system that can selectively deliver the drug payload to the tumor site and shrink the tumor burden.15–18 However, conventional way of preparing nanomedicine can inevitably bring the problems of difficult control over the drug loading, great batch-to-batch variations and poor reproducibility.19,20 In addition to these conventional strategies, developing small molecule amphiphiles in which there exist both hydrophilic molecules and hydrophobic molecules for nanomedicine based cancer therapy would be possible.21–23 Small molecule conjugate amphiphiles have been previously reported to conjugate two molecules with different water solubility together such as two anticancer drugs, one anticancer drug and one targeting or imaging agent.24–26 The benefits for this strategy is to precisely control the NPs formulation and the drug loading would be constant.27,28
Cantharidin (CTR), a very toxic compound, is isolated from Spanish fly or blistering beetle and other insects.29,30 CTR is considered to be a potent and specific inhibitor of protein phosphatases 1 (PP1) and 2A (PP2A).30 CTR has shown great activity on a variety of cancer cells including lung cancer, liver cancer, breast cancer, ovarian cancer as well as colon cancers.30 However, great side effects arise as exposure to CTR can cause severe chemical burns.16,31,32 Developing new methods for protecting CTR and site specifically deliver CTR into the tumor site is highly desirable. Notably, CTR is chemically an anhydride, which can be easily used to make drug conjugates with other molecules that possess hydroxyl/amine groups.
Camptothecin (CPT) is a cytotoxic quinolone alkaloid, which can inhibit the DNA enzyme topoisomerase 1 (topo 1).32–35 CPT takes it action by binding to topo 1 and DNA complex. This results in a ternary complex and thereby stabilizes it, which prevents DNA re-ligation and causes DNA damage, resulting in apoptosis.36 CPT also showed remarkable anticancer activity on a variety of cancers including bladder cancer, breast cancer, and colon cancer, etc.37 However, CPT is very hydrophobic and has extremely low water solubility and high adverse drug reaction.38 Because of these adverse effects, CPT analogues (topotecan/irinotecan) were developed.
Taking advantage of the anticancer effects and chemical properties of the two drugs as well as the drawbacks of them, we here showed the design of a small molecule conjugate making from CTR and CPT by one-step chemical conjugation (Scheme 1A). The as prepared small molecule conjugate (1, Scheme 1) has both hydrophilic cantharidin and hydrophobic CPT in its molecule hence 1 can self-assemble into NPs in water. We hypothesize that 1-NP can be endocytosed by cancer cells. The internalized 1-NP can be further degraded to release CTR and CPT inside the cells as the esterase39,40 and the acidic environment41 will possibly make 1 hydrolyze to CTR and CPT (Scheme 1B). To prove this mechanism, we established an subcutaneous colon cancer model to evaluate 1-NP both in vitro and in vivo (Scheme 1C).
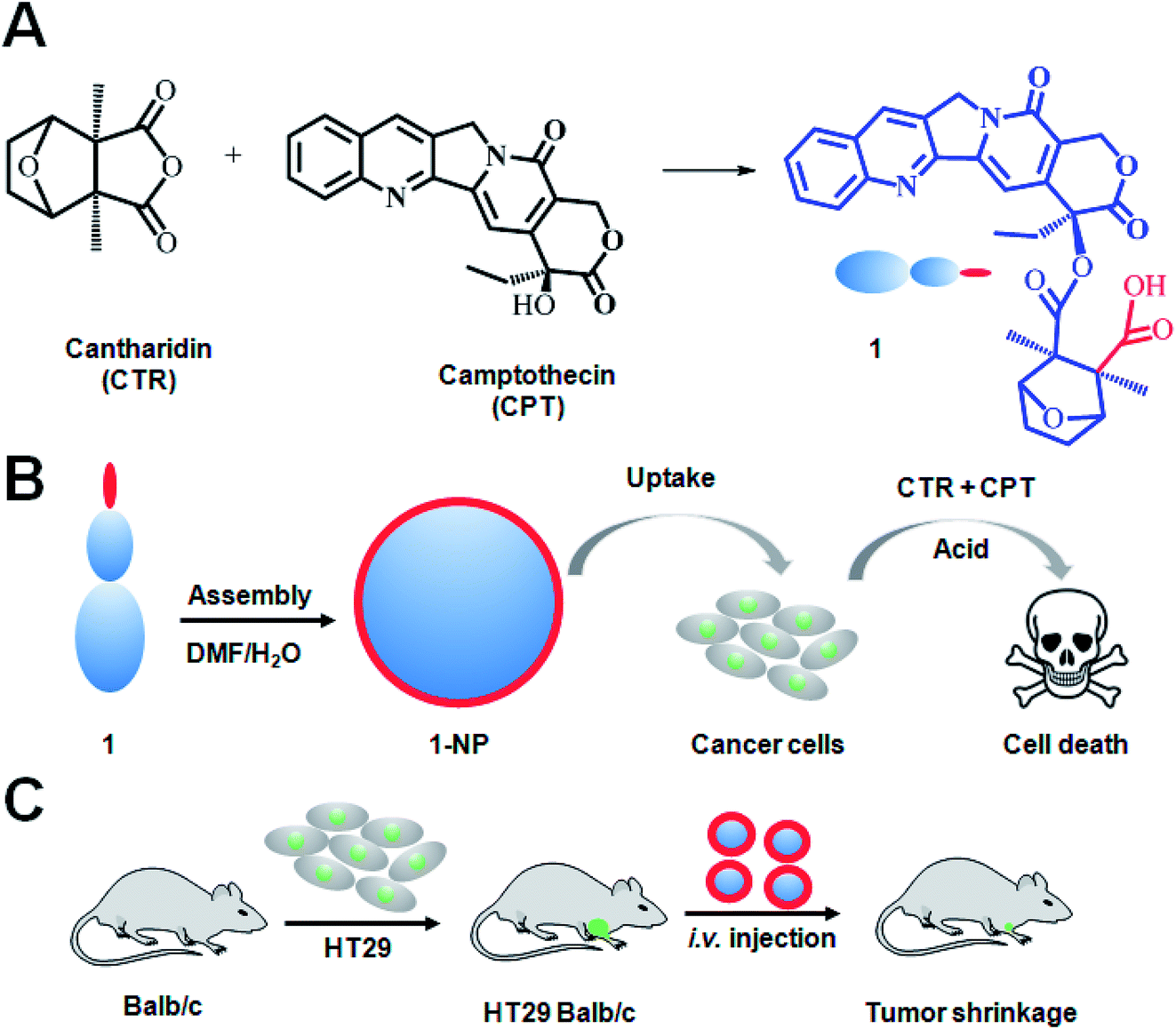 | ||
| Scheme 1 Design of small molecule conjugate amphiphiles for assembled 1-NPs for cancer therapy. Traditional Chinese medicine CTR was used conjugated with CPT to the small molecule conjugate 1 (A). 1 which has both hydrophilic CTR (carboxylic acid) and hydrophobic CPT can self-assemble into nanoparticles (1-NP). Due to the hydrophilicity, carboxylic acid groups are on the surface of nanoparticles. Cancer cells treated with 1-NPs will internalize the NPs via endocytosis. The ester bond could be opened to release the two drugs in tumor cells with lower pH values and esterase. These two drugs work synergistically to kill the cancer cells (B). To evaluate 1-NPs in vivo, mice model bearing subcutaneous HT29 cancer were i.v. injected with 1-NP (C). | ||
Results and discussion
1 was synthesized by the ring-opening reaction of CTR with CPT in the presence of pyridine. 1 was proved to be successfully synthesized and characterized by 1HNMR, ESI-MS and elemental analysis. The presence of both characteristic peaks in CTR and CPT in 1 indicated the successful conjugation of the two. Due to the presence of both hydrophilic and hydrophobic parts in the molecule, 1 was amphiphilic hence can be self-assemble into NPs. 1-NP was first characterized by DLS. As shown in Fig. 1A, 1-NP had a mean diameter of 122 nm and a polydispersity index at 0.107. Representative TEM images of the NPs shown in Fig. 1B indicated the spherical structures with a mean diameter at 68 nm. The TEM images clearly and directly showed the formation of NPs by assembling the small molecule conjugate. The driving force for the NPs formation could be the amphiphicity of the drug amphiphilies. The stability of NPs is a key factor for being a drug candidate. Here, the stability of 1-NP was monitored in PBS by DLS for one week. As shown in Fig. 1C, no evident change in size was found for these nanoparticles, denoting the stability of the NPs. As there are tremendous conjugates in 1-NP, release of them to exert their anticancer activities is necessary. To study this process, we incubated 1-NP in a dialysis bag (molecule weight cutoff, 500 Da) at three pH values (pH = 3.0, pH = 5.0 and pH = 7.4). It is well known in the tumor cells, the pH value is low.42 However, in the blood circulation, pH value is much higher at 7.4, indicating a neutral condition.42–44 Here we found 1-NP release the payload via an acid responsive manner. At acidic conditions (pH = 3.0), >80% drugs were released. However, ca. 20% and ca. 60% drugs were released at pH 7.4 and pH 5.0. This acid responsive release could be attributed to the hydrolysis of the ester bond between CTR and CPT. Moreover, this is beneficial for cancer cells for exerting the anticancer activity (Fig. 1D). | ||
| Fig. 1 Characterization of the small molecule conjugate amphiphiles assembled nanoparticles. Representative dynamic light scattering (DLS) (A) and TEM (B) of the as-prepared 1-NP. 1-NP was shown to be stable in PBS monitored by DLS for a week (C). The release of CPT at three different pH values was studied (D). | ||
To track the intracellular internalization of 1-NP, 1-NP was labeled by loading hydrophobic Nile red dye via encapsulation. The intracellular localization of 1-NP was studied by confocal laser scanning microscopy (CLSM). As shown in Fig. 2A, at 30 min, 1-NP was observed in the cells, indicating the NPs entering the cells in a rapid way. After 6 h incubation, the red fluorescence was very bright and full of the cell plasma, suggesting great amount of NPs were in the cells. We could clearly see here the time dependent uptake of NPs into the cells. To further quantify this process, cells were then treated with 1-NP with Nile red and the relative cell uptake was measured by flow cytometry. As shown in Fig. 2B, from 30 min to 6 h, we could find the great increase in fluorescence intensity, further indicating the time dependent uptake. This increase in uptake was almost 4-fold (Fig. 2C), indicating longer time point was beneficial for internalization of these NPs.
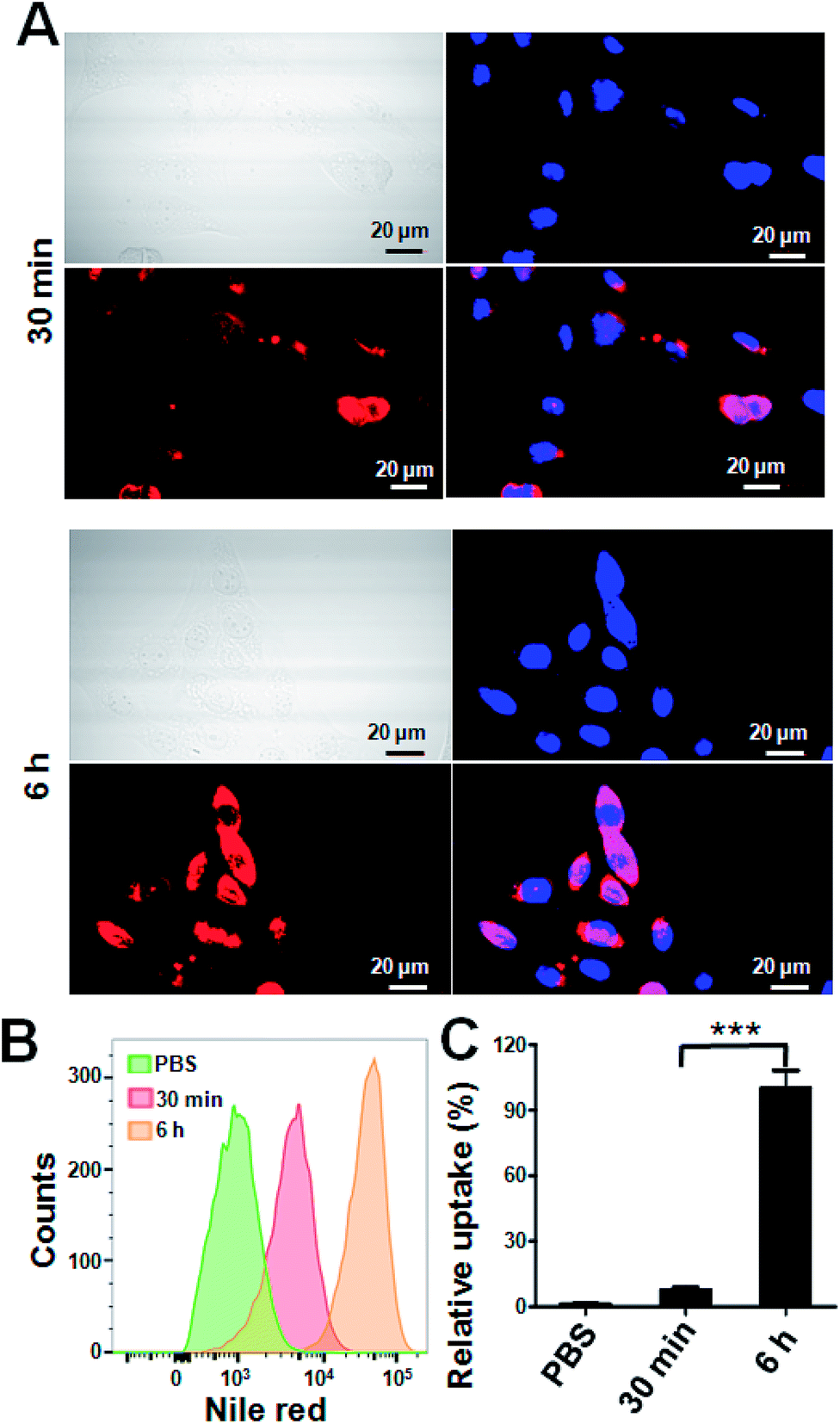 | ||
| Fig. 2 Intracellular uptake of 1-NP by CLSM and flow cytometry. Cells were treated with 1-NP with Nile red (1 μg ml−1) for 30 min and 6 h. Then cells were washed thoroughly by PBS and imaged by CLSM (Olympus, FLV1200). The cell nucleus was stained by DAPI. Scale bar = 20 μm (A). The cell uptake was also measured by flow cytometry. Cells were with 1-NP with Nile red (1 μg ml−1) for 30 min and 6 h. Then cells were washed thoroughly by PBS and measured by flow cytometry (B). The relative uptake shown here was normalized by 6 h (C). Data shown were mean values ± S.D. (n = 3). Statistic was indicated by ***p < 0.001. | ||
Both CTR and CPT possess very good anticancer activity.45 Simple chemical conjugate of the two compounds into 1 with an ester linkage may not hamper the anticancer effects. To prove this, two colon cancer cells, HT-29 and HCT-116 were treated with various drugs including CTR (Fig. 3A and B). It seemed that CPT had better anticancer activity on both HT-29 and HCT-116 cells than CTR did. Combination of CTR and CPT at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio resulted in greater potency on killing the cells as the curve shifts down. 1-NP was less potent in cell killing than mixture of free CTR and CPT at 1:1 ratio but better than both CPT and CTR used separately on both cell lines. The reason that 1-NP was less potent than mixture of CTR and CPT may be the direct action of both drugs on the cell lines. However, 1-NP was firstly internalized to the cells and the two drug should be hydrolyzed to release both CTR and CPT before they took their action. The half-inhibitory concentration (IC50) values were listed in Fig. 3C (table). IC50 values of 1-NP on HT-29 and HCT-116 were 0.146 and 0.216 μM. Moreover, the drug combination index (CI) at 50% inhibitory effect was calculated. When CI is above 1, equal to 1 or less than 1 means antagonistic, additive or synergistic separately. On both cell lines, the CI values for a mixture of CTR and CPT were less than 1. This was the same case for 1-NP. The results here meant synergy for both 1-NP and a combination of CTR and CPT at 1:1 ratio. To further show the potency and synergy of 1-NP, the growth inhibition assay on HT-29 cells was tested. Cells were treated with 1-NP and a mixture of CTR + CPT. On a period of 6 days, the absorbance at 570 nm by MTT assay at different days was measured and plotted in Fig. 3D. As the absorbance indicated the cell viabilities, lower absorbance suggested greater growth inhibition. A mixture of CTR and CPT completely inhibited the growth of cancer cells. Similarly, 1-NP showed the same inhibition effect. Colony formation assay is an in vitro cell survival assay, which is based on the ability of a single cell to grow into a colony.46 The colony is defined to consist of at least 50 cells. 1-NP showed a dose dependent inhibition effect on colony formation from 0.1 μM to 0.5 μM. Although a mixture of CTR and CPT at 0.1 μM inhibited the colony formation to ∼20%. At 0.25 μM and 0.5 μM, 1-NP showed better inhibition of colony formation ability than a mixture of CTR and CPT at 0.1 μM (Fig. 3E).
:1 ratio resulted in greater potency on killing the cells as the curve shifts down. 1-NP was less potent in cell killing than mixture of free CTR and CPT at 1:1 ratio but better than both CPT and CTR used separately on both cell lines. The reason that 1-NP was less potent than mixture of CTR and CPT may be the direct action of both drugs on the cell lines. However, 1-NP was firstly internalized to the cells and the two drug should be hydrolyzed to release both CTR and CPT before they took their action. The half-inhibitory concentration (IC50) values were listed in Fig. 3C (table). IC50 values of 1-NP on HT-29 and HCT-116 were 0.146 and 0.216 μM. Moreover, the drug combination index (CI) at 50% inhibitory effect was calculated. When CI is above 1, equal to 1 or less than 1 means antagonistic, additive or synergistic separately. On both cell lines, the CI values for a mixture of CTR and CPT were less than 1. This was the same case for 1-NP. The results here meant synergy for both 1-NP and a combination of CTR and CPT at 1:1 ratio. To further show the potency and synergy of 1-NP, the growth inhibition assay on HT-29 cells was tested. Cells were treated with 1-NP and a mixture of CTR + CPT. On a period of 6 days, the absorbance at 570 nm by MTT assay at different days was measured and plotted in Fig. 3D. As the absorbance indicated the cell viabilities, lower absorbance suggested greater growth inhibition. A mixture of CTR and CPT completely inhibited the growth of cancer cells. Similarly, 1-NP showed the same inhibition effect. Colony formation assay is an in vitro cell survival assay, which is based on the ability of a single cell to grow into a colony.46 The colony is defined to consist of at least 50 cells. 1-NP showed a dose dependent inhibition effect on colony formation from 0.1 μM to 0.5 μM. Although a mixture of CTR and CPT at 0.1 μM inhibited the colony formation to ∼20%. At 0.25 μM and 0.5 μM, 1-NP showed better inhibition of colony formation ability than a mixture of CTR and CPT at 0.1 μM (Fig. 3E).
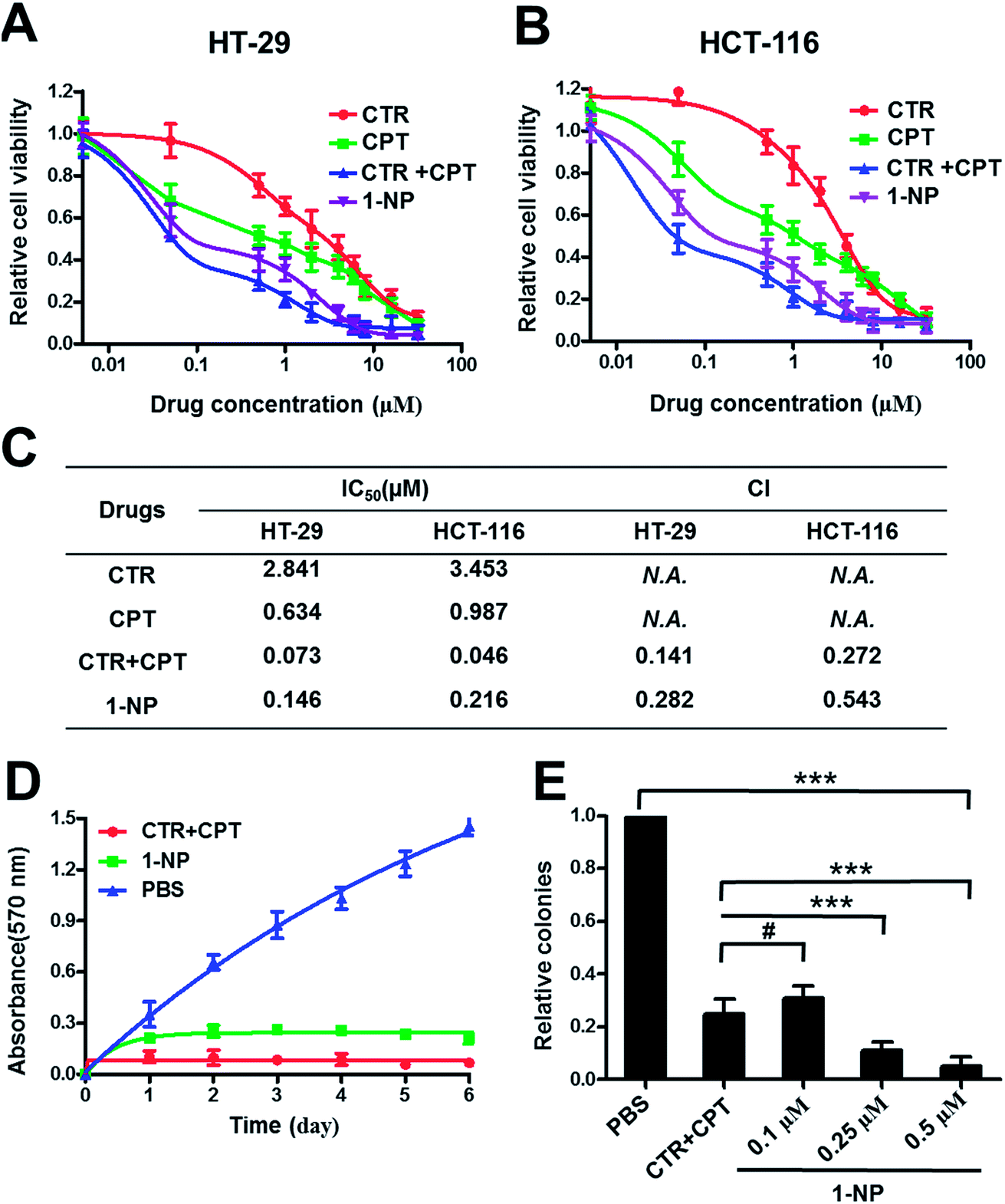 | ||
| Fig. 3 In vitro evaluation of the anticancer activity of 1-NP. 1-NP was screened on two colon cancer cells HT-29 and HCT-116 for 72 h drug incubation. CTR, CPT, CTR + CPT (mixture of them at 1:1 molar ratio) were also dosed to the HT-29 (A) and HCT-116 cells (B). The IC50 values were listed and the CI values were calculated (C). Growth inhibition assay (D) and colony formation assay (E) were further performed on HT-29 cells. Data shown were mean values ± S.D. (n = 3). Significance is defined by ***p < 0.001 and #p > 0.05. | ||
To unveil the mechanism of how 1-NP took the action, the cell apoptosis of 1-NP was measured (Fig. 4). CTR and CPT induced apoptosis rates at 10.8% and 12.8% respectively. However, a mixture of CTR and CPT resulted in a more profound apoptosis rate at 37.3%. 1-NP had a much similar apoptosis rate at 24.5%. This result coordinated well with the potency screened by MTT assay.
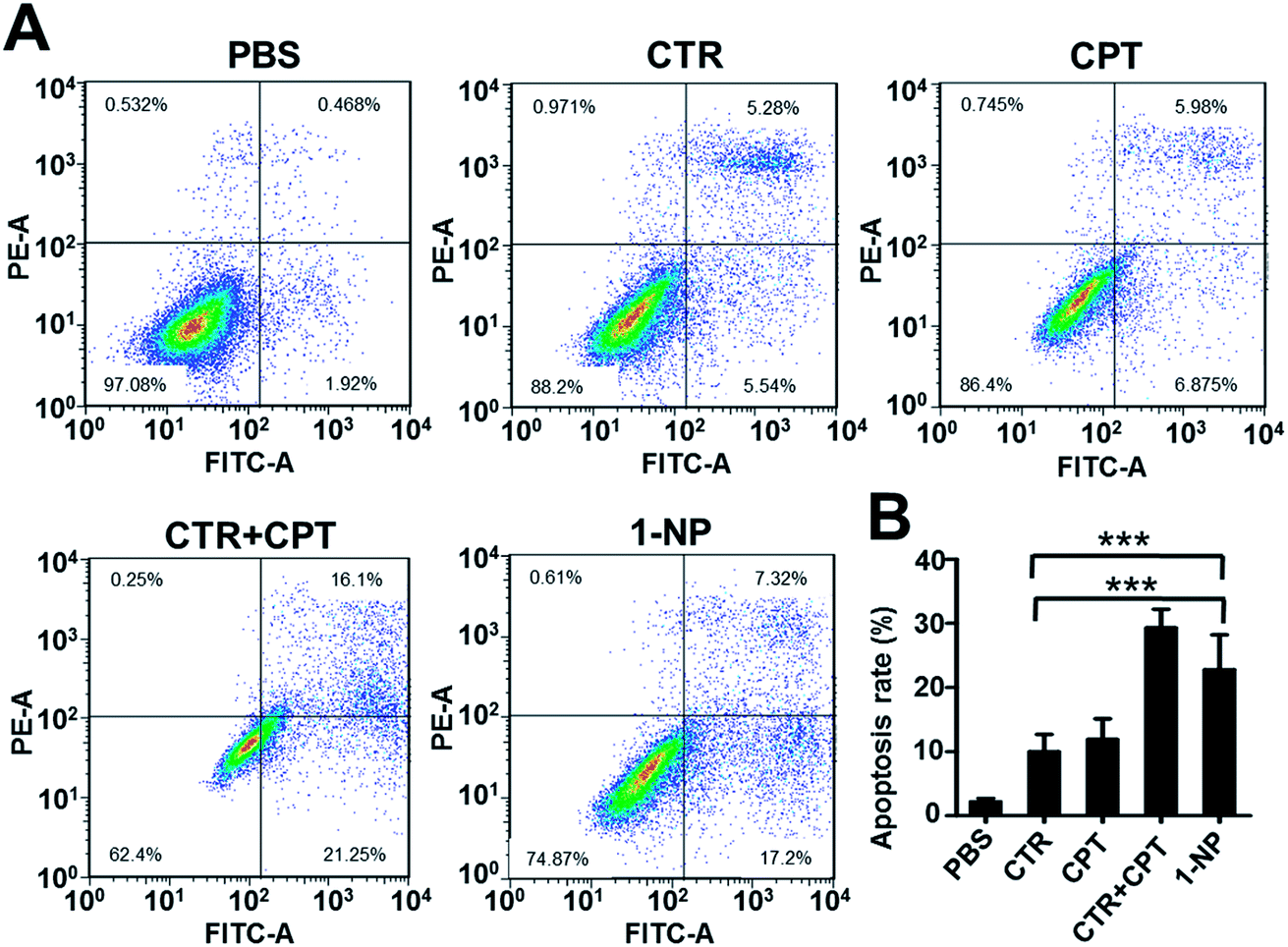 | ||
| Fig. 4 1-NP exhibited anticancer effect by inducing apoptosis. Cells were seeded in 6-well plate at a density of 5 × 105 cells per well overnight. The cells were then treated with CTR (5 μM), CPT (5 μM) and CTR + CPT (5 μM) as well as 1-NP (5 μM) with PBS as a control for 48 h. The representative apoptosis images were shown in (A) and statistic results were shown in (B). Data shown were mean values ± S.D. (n = 3). Significance is defined by ***p < 0.001. | ||
At last, we established an in vivo HT-29 colon cancer model to evaluate the efficacy of these drugs. Balb/c mice with subcutaneous HT29 cancer were treated with CTR, CPT, CTR + CPT and 1-NP for 3 times. The tumor volume and body weight change were plotted against time. As shown in Fig. 5A, the tumor of the PBS treated group grew very rapidly till 6000 mm3 in 30 days. CTR and CPT could inhibit the tumor growth to a certain extent. As cantharidin is too toxic and the solubility is too low, its disodium salt (disodium cantharidin) was used here instead of CTR itself.30 It seems CPT (4 mg kg−1) had better anticancer effect on HT29 cancer than CTR at 3 mg kg−1 as indicated by the tumor volume was ∼2100 mm3 for the former drug treated group and ∼3200 mm3 for the latter at the end of study. Notably, a mixture of CPT (4 mg kg−1) and CTR (3 mg kg−1) which is approximately at 1:1 molar ratio caused all the mice to death in the first 9 days. This great toxicity from the combination of the two free drugs indicated the impossible combination of the free drugs at this dose. However, 1-NP almost completely stopped the tumor growth and showed the best antitumor activity. To evaluate the systemic toxicity, the body weight change of the mice were plotted in Fig. 5B. Mice treated with most of the drugs did not show great body weight change except for the free drug combination group (CPT at 4 mg kg−1 plus CTR at 3 mg kg−1), indicating uncontrollable systemic toxicity and all the mice died in 9 days. To give insight into to what extent the tumor growth was inhibited, the tumors of the mice were collected at the end of study and weighed in Fig. 5C. As the mice treated with a mixture of CTR and CPT died, we did not collected the tumor for comparison here. The non-treated mice had tumor mass at 5.3 g; however, these were 1.69 g, 3.26 g and 0.45 g for CPT, CTR and 1-NP. 1-NP treated mice had less than one tenth of the tumor in PBS treated mice. This was also ca. one fourth and one seventh of the mice treated with CPT and CTR. Together, 1-NP displayed best anticancer activity in vivo with limited systemic toxicity.
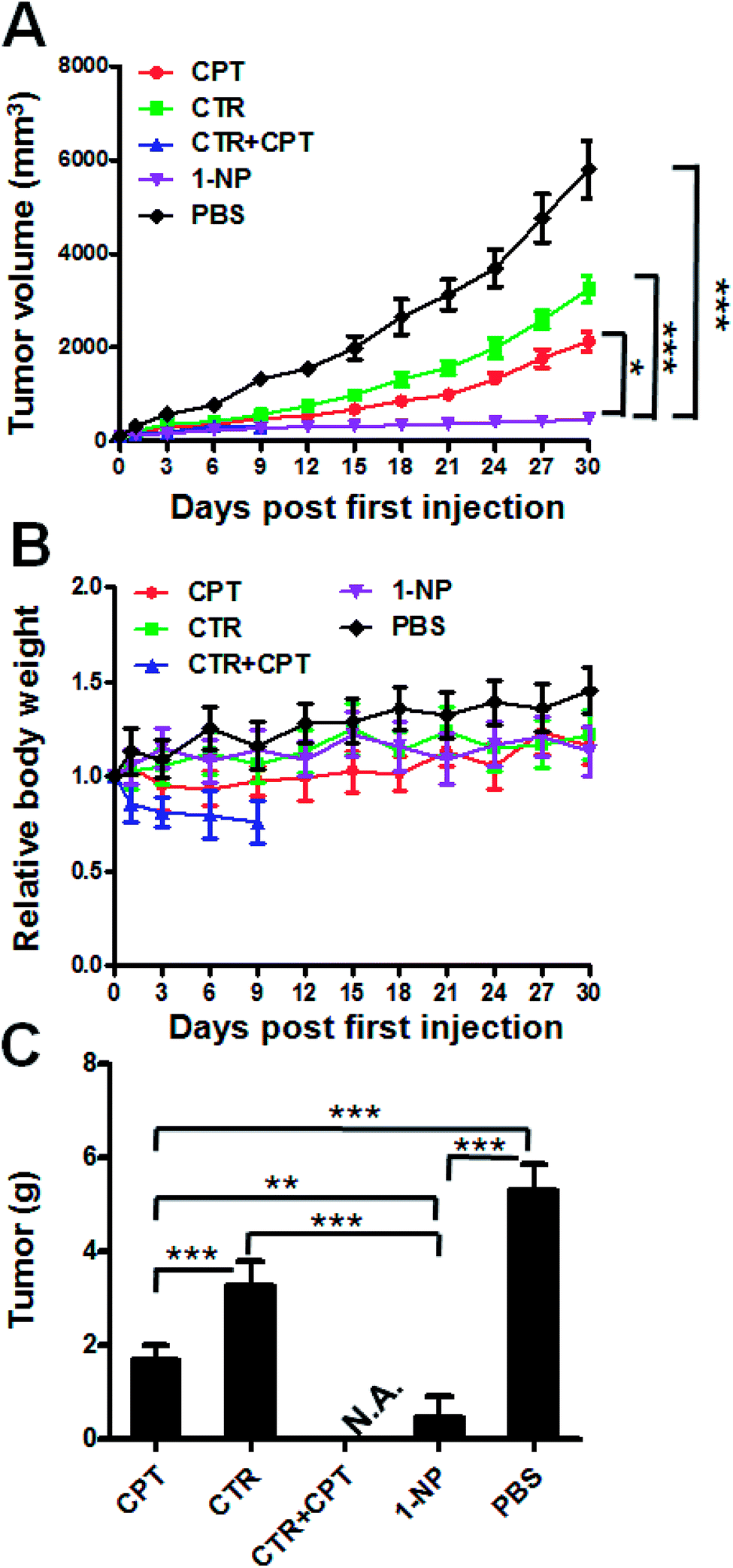 | ||
| Fig. 5 In vivo evaluation of 1-NP on a subcutaneous colon cancer model. Balb/c nude mice bearing HT-29 tumor were i.v. injected at day 0, 3, 6 with CTR (3 mg kg−1), CPT (4 mg kg−1), CTR (3 mg kg−1) + CPT (4 mg kg−1) and 1-NP (6.4 mg kg−1) with PBS as a control. The first treatment date was set as day 0. The tumor volume (A) and body weight (B) of the mice were monitored. At the end of the study, the mice were killed and the tumor were collected and weighed (C). Significance is defined as ***p < 0.001, **p < 0.01, *p < 0.05. | ||
Conclusions
We have shown here the construction a novel drug–drug conjugate by integrating CTR and CPT in one molecule. This conjugate 1 was synthesized by ring-opening of CTR in the presence of base. As conjugate 1 is amphiphilic, it can self-assemble into NPs in aqueous solution. 1-NP was shown to release the drug payload via a acid responsive manner. Moreover, 1-NPs were internalized into the HT-29 colon cancer cells by CLSM and flow cytometry. We then studied the in vitro anticancer activity of 1-NP as compared to the free drugs and their combination. 1-NP were shown to be potent to kill the cells and resulted in synergy of the two free drugs. Moreover, in vivo tumor shrinkage by 1-NP was observed. Future further evaluation of 1-NP for potential clinic use was envisaged.Materials and methods
CTR was purchased from Nanjing Zelang Co., Ltd. CPT and Nile red were purchased from Sigma-Aldrich. Pyridine and all other chemicals were of reagent grade and used as received.Synthesis of small molecule conjugate 1
CPT (0.352 g) was dissolved in dry DMF in a flask, to which CTR (0.196 g) and 10 μl pyridine was added. The reaction mixture was kept stirring at 60 °C overnight. Afterwards, DMF was removed by vacuum evaporator. The products were collected and sonicated in water and dried under vacuum. Conjugate 1 (1H-NMR in d6-DMSO: δ 0.90 (t, 3H), 1.47 (s, 6H), 1.96 (m, 4H), 2.15 (m, 2H), 4.7 (d, 2H), 5.3 (s, 2H), 5.48 (s, 2H), 7.13 (s, 1H), 7.72 (t, 3H), 7.87 (t, 1H), 8.16 (t, 2H), 8.7 (s, 1H); ESI, negative model, M − H, m/z = 543.3; exper. C: 65.68%; H: 5.88%; N: 5.11%; found, C: 65.24%; H: 5.76%; N: 5.19%).Self-assemble of 1 into 1-NPs
Small molecule conjugate 1 which was previously dissolved in DMF and then it was injected into warm PBS solution (60 °C). DMF was dialyzed to be removed and the NPs were filtered by 200 μm cellulose filter before DLS (Nano Zeta sizer 90, Malvern) and TEM (JEOL-1011) measurement.Drug release
Drug release from 1-NP was performed by using a dialysis method. 5 ml solution of 1-NP at a known CPT concentration (1 mM) was put into a dialysis bag (molecular cut-off at 500 Da). To monitor the drug release, buffered solutions at different pH values (pH = 3.0, pH = 5.0 and pH = 7.4) were used. The dialysis bag was dialyzed against 20 ml of buffered solution. At preferable time point, 300 μl of solution was collected for UV-Vis absorbance measurement of CPT at 360 nm. The cumulative CPT release was calculated.Cell lines
HT-29 and HCT-116 cell lines were purchased from Wuhan University. The two cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS), 0.03% L-glutamine and 1% penicillin/streptomycin in 5% CO2 at 37 °C.NP uptake by confocal laser scanning and flow cytometry
HT-29 cells were seed onto a glass in 6-wells plate at a density of 2 × 105 cells per well in 2 ml culture media overnight before use. Then cells were then treated with 1-NP with Nile red for 30 min and 6 h at a dye concentration of 1 μg ml−1 Nile red respectively. Then the cells were washed by three times with cold PBS, and fixed with 4% formaldehyde. Cell nucleus was stained by DAPI (2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride). Cells were then thoroughly washed by PBS for three times and observed using confocal laser scanning microscope (CLSM, Olympus FV1200).HT-29 cells were seed in 6-wells plate at a density of 6 × 105 cells per well in 2 ml culture media overnight before use. Then cells were treated with 1-NP with Nile red at a dye concentration of 1 μg ml−1 for 30 min and 6 h. After treatment, the cells were thoroughly washed and analyzed on a BD FACSCalibur™ flow cytometer (BD Biosciences).
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay
HT-29 and HCT-116 cells were harvested in a logarithmic growth phase and seeded on a 96-well plate in 100 μL media at a density of 2000 cells per well overnight. The cells were then treated with CPT, CTR, a mixture of CPT and CTR at a molar ratio at 1:1, and 1-NP. The concentration range of each drug was 0.005 μM to 32 μM. The drug incubation lasted for 72 h. At the end of incubation, 10 μl of 5 mg ml−1 MTT solution in PBS was added into each well and incubated for another 4 h. 100 μl of acidified sodium dodecyl sulfate (SDS) solution was added into each well and incubated for another 12 h. Finally, a microplate reader at a wavelength at 570 nm read the plate.
Growth inhibition
HT-29 cells were seeded in 96 well plates at 2000 cells per well overnight in 100 μl of culture media. Then the cells were treated with a mixture of CPT and CTR at 1:1 ratio, and 1-NP with PBS treated cells as control. The concentration of each drug was set at 1 μM. At a period of 6 days (one plate per day), MTT assay was performed on one plate. The absorbance at 570 nm of each drug was plotted against the incubate time.
Colony formation
HT-29 cells were seeded in six well plates at 2000 cells per well overnight in 10 ml of culture media. Then the cells were treated with a mixture of CPT and CTR at 1:1 ratio, and 1-NP with PBS treated cells as control. A mixture of CPT and CTR at 1:1 ratio (0.1 μM CPT and 0.1 μM CTR) and 1-NP (0.1 μM) were used to treated the HT-29 cells. Moreover, 1-NP at two more concentrations at 0.25 and 0.5 μM were used to treat the cells. After two weeks, the culture media was discarded and then cells were washed, fixed. Crystal violet (0.1%) was used to stain the colonies formed as previously described.
Apoptosis
HT-29 cells were seeded in 6 well plates at a density of 5 × 105 cells per well overnight. Cells were then treated with CPT, CTR, a mixture of CPT and CTR and 1-NP. The concentration for each drug is 5 μM for 48 h. Then PBS was used to wash the cells for three times, trypsinized, and stained by Annexin-V/FITC apoptosis kit. The apoptosis rate was measured by flow cytometry.Animal use
All the animals in use were applied to the animal welfare and guidelines in Jilin University, Changchun, China. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of “Animal use of the China-Japan Union Hospital” and approved by the Animal Ethics Committee of “China-Japan Union Hospital animal protocols and guidelines for the welfare and ethics of the experimental animals”.Antitumor efficacy evaluation
Balb/c nude mice was used to establish the HT-29 subcutaneous model. HT-29 cancer cells at 1 × 107 per mice were subcutaneously injected to the mice right flank. When the tumor grows up to ∼150 mm3, treatment was started. Mice were randomly grouped into 5 groups with 5 mice in each group (n = 5). They were treated with PBS (control), CPT (4 mg kg−1), CTR (disodium cantharidin, 3 mg kg−1), a mixture of CPT (4 mg kg−1) and CTR (disodium cantharidin, 3 mg kg−1) and 1-NP (6.4 mg kg−1, roughly equal to one molar ratio of CPT at a dose of 4 mg kg−1 plus disodium cantharidin at a dose of 3 mg kg−1). Mice were treated at day 0, day 3 and 6. The tumor size was measured by caliper, and body weight was monitored every three days. The tumor size was calculated by equation:47| V = 0.5 × L × W2 |
Statistical analyses
The data were expressed as mean ± standard deviation (S.D.). Statistical analysis was performed using Students' test. Significance is defined as by #p > 0.05, *p < 0.05, **p < 0.01 and ***p < 0.001.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the science foundation from Health Department of Jilin Province (2013ZC014).References
- T. Sun, Y. S. Zhang, B. Pang, D. C. Hyun, M. Yang and Y. Xia, Angew. Chem., Int. Ed., 2014, 53, 12320–12364 CAS
.
- S. Parveen, R. Misra and S. K. Sahoo, Nanomedicine, 2012, 8, 147–166 CrossRef CAS PubMed
- R. Singh and J. W. Lillard Jr, Exp. Mol. Pathol., 2009, 86, 215–223 CrossRef CAS PubMed
- V. J. Venditto and F. C. Szoka, Adv. Drug Delivery Rev., 2013, 65, 80–88 CrossRef CAS PubMed
- M. Ferrari, Trends Biotechnol., 2010, 28, 181–188 CrossRef CAS PubMed
- I. Linkov, F. K. Satterstrom and L. M. Corey, Nanomedicine, 2008, 4, 167–171 CrossRef CAS PubMed
- D. Peer, J. M. Karp, S. Hong, O. C. FaroKHzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed
- Q. Yang, Y. Yang, L. Li, W. Sun, X. Zhu and Y. Huang, ACS Appl. Mater. Interfaces, 2015, 7, 6661–6673 CAS
- S. M. Moghimi, A. C. Hunter and J. C. Murray, FASEB J., 2005, 19, 311–330 CrossRef CAS PubMed
- J. W. Yoo, D. J. Irvine, D. E. Discher and S. Mitragotri, Nat. Rev. Drug Discovery, 2011, 10, 521–535 CrossRef CAS PubMed
- P. M. Biesheuvel and M. A. Cohen Stuart, Langmuir, 2004, 20, 2785–2791 CrossRef CAS PubMed
- M. Dominska and D. M. Dykxhoorn, J. Cell Sci., 2010, 123, 1183–1189 CrossRef CAS PubMed
- Y. Q. Niu, T. He, J. Song, S. P. Chen, X. Y. Liu, Z. G. Chen, Y. J. Yu and S. G. Chen, Chem. Commun., 2017, 53, 7541–7544 RSC
- J. Li, X. Feng, B. Liu, Y. Yu, L. Sun, T. Liu, Y. Wang, J. Ding and X. Chen, Acta Biomater., 2017, 61, 21–40 CrossRef CAS PubMed
- J. D. Byrne, T. Betancourt and L. Brannon-Peppas, Adv. Drug Delivery Rev., 2008, 60, 1615–1626 CrossRef CAS PubMed
- S. Wang, L. Shang, L. Li, Y. Yu, C. Chi, K. Wang, J. Zhang, R. Shi, H. Shen, G. I. Waterhouse, S. Liu, J. Tian, T. Zhang and H. Liu, Adv. Mater., 2016, 28, 8379–8387 CrossRef CAS PubMed
- A. Kumari, S. K. Yadav and S. C. Yadav, Colloids Surf., B, 2010, 75, 1–18 CrossRef CAS PubMed
- L. D. Zhang, Y. J. Yu, C. Joubert, G. Bruder, Y. Liu, C. C. Chang, M. Simon, S. G. Walker and M. Rafailovich, Polymers, 2016, 8 Search PubMed
- N. Kamaly, Z. Xiao, P. M. Valencia, A. F. Radovic-Moreno and O. C. Farokhzad, Chem. Soc. Rev., 2012, 41, 2971–3010 RSC
- D. J. Crommelin and A. T. Florence, Int. J. Pharm., 2013, 454, 496–511 CrossRef CAS PubMed
- J. Xie, G. Liu, H. S. Eden, H. Ai and X. Chen, Acc. Chem. Res., 2011, 44, 883–892 CrossRef CAS PubMed
- J. Zhang and P. X. Ma, Adv. Drug Delivery Rev., 2013, 65, 1215–1233 CrossRef CAS PubMed
- G. Yu, K. Jie and F. Huang, Chem. Rev., 2015, 115, 7240–7303 CrossRef CAS PubMed
- R. C. Huxford, J. Della Rocca and W. B. Lin, Curr. Opin. Chem. Biol., 2010, 14, 262–268 CrossRef CAS PubMed
- Y. Yu, T. Wu, A. Johnson-Buck, L. Li and X. Su, Biosens. Bioelectron., 2016, 82, 248–254 CrossRef CAS PubMed
- Z. Tao, X. Dang, X. Huang, M. D. Muzumdar, E. S. Xu, N. M. Bardhan, H. Song, R. Qi, Y. Yu, T. Li, W. Wei, J. Wyckoff, M. J. Birrer, A. M. Belcher and P. P. Ghoroghchian, Biomaterials, 2017, 134, 202–215 CrossRef CAS PubMed
- Z. Cheng, A. Al Zaki, J. Z. Hui, V. R. Muzykantov and A. Tsourkas, Science, 2012, 338, 903–910 CrossRef CAS PubMed
- X. Gao, L. Yang, J. A. Petros, F. F. Marshall, J. W. Simons and S. Nie, Curr. Opin. Biotechnol., 2005, 16, 63–72 CrossRef CAS PubMed
- A. T. Dossey, Nat. Prod. Rep., 2010, 27, 1737–1757 RSC
- C. E. P. Galvis, L. Y. V. Mendez and V. V. Kouznetsov, Chem. Biol. Drug Des., 2013, 82, 477–499 Search PubMed
- A. Nel, T. Xia, L. Madler and N. Li, Science, 2006, 311, 622–627 CrossRef CAS PubMed
- C. F. He, S. H. Wang, Y. J. Yu, H. Y. Shen, Y. Zhao, H. L. Gao, H. Wang, L. L. Li and H. Y. Liu, Cancer Biol. Med., 2016, 13, 299–312 CrossRef PubMed
- Y. Pommier, E. Leo, H. L. Zhang and C. Marchand, Chem. Biol., 2010, 17, 421–433 CrossRef CAS PubMed
- Z. Topcu, J. Clin. Pharm. Ther., 2001, 26, 405–416 CrossRef CAS PubMed
- Y. Pommier, ACS Chem. Biol., 2013, 8, 82–95 CrossRef CAS PubMed
- B. L. Staker, M. D. Feese, M. Cushman, Y. Pommier, D. Zembower, L. Stewart and A. B. Burgin, J. Med. Chem., 2005, 48, 2336–2345 CrossRef CAS PubMed
- M. C. Prewett, A. T. Hooper, R. Bassi, L. M. Ellis, H. W. Waksal and D. J. Hicklin, Clin. Cancer Res., 2002, 8, 994–1003 CAS
- Y. Sadzuka, H. Takabe and T. Sonobe, J. Controlled Release, 2005, 108, 453–459 CrossRef CAS PubMed
- Y. Q. Zheng, B. C. Yu, K. L. Ji, Z. X. Pan, V. Chittavong and B. H. Wang, Angew. Chem., Int. Ed., 2016, 55, 4514–4518 CrossRef CAS PubMed
- Z. X. Zhou, W. J. Murdoch and Y. Q. Shen, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 507–515 CrossRef CAS
- J. Lu, E. Choi, F. Tamanoi and J. I. Zink, Small, 2008, 4, 421–426 CrossRef CAS PubMed
- E. S. Lee, K. Na and Y. H. Bae, J. Controlled Release, 2003, 91, 103–113 CrossRef CAS PubMed
- Y. Yu, Q. Zhang, J. Buscaglia, Y. Wang, C.-C. Chang, Y. Liu, Y. Guo, Z. Yang, K. Levon and M. H. Rafailovich, Analyst, 2016, 141, 4424 RSC
- Y. Yu, Q. Zhang, C. C. Chang, Y. Liu, Z. Yang, Y. Guo, Y. Wang, D. K. Galanakis, K. Levon and M. Rafailovich, Analyst, 2016, 141, 5607–5617 RSC
- V. Srivastava, A. S. Negi, J. K. Kumar, M. M. Gupta and S. P. S. Khanuja, Bioorg. Med. Chem., 2005, 13, 5892–5908 CrossRef CAS PubMed
- N. A. Franken, H. M. Rodermond, J. Stap, J. Haveman and C. van Bree, Nat. Protoc., 2006, 1, 2315–2319 CrossRef CAS PubMed
- C. Delloye-Bourgeois, E. Brambilla, M. M. Coissieux, C. Guenebeaud, R. Pedeux, V. Firlej, F. Cabon, C. Brambilla, P. Mehlen and A. Bernet, J. Natl. Cancer Inst., 2009, 101, 237–247 CrossRef CAS PubMed
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |