
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlasma pharmacokinetics and brain distribution kinetics of lapachol in rats using LC-MS and microdialysis techniques
Lu Bai†
a,
Ying Han†b,
Pingxiang Xua,
Binbin Xiaac,
Yuming Zhaoa,
Xiaorong Lia and
Ming Xue *a
*a
aDepartment of Pharmacology, Beijing Laboratory for Biomedical Detection Technology and Instrument, School of Basic Medical Sciences, Capital Medical University, Beijing 100069, China. E-mail: xuem@ccmu.edu.cn
bCollege of Medical Sciences, Inner Mongolia University for Nationalities, Huhehaote, Inner Mongolia 028000, China
cDepartment of Pharmacy, Beijing Luhe Hospital Affiliated to Capital Medical University, Beijing 101149, China
First published on 20th November 2017
Abstract
The objective of this paper was to investigate the plasma pharmacokinetics and brain distribution kinetics of lapachol in rats. A sensitive and specific high-performance liquid chromatography-tandem mass spectrometry method was developed and validated for quantification of the bioactive naphthoquinone lapachol in rat plasma and brain dialysates after oral administration. The analytes were determined using the negative electrospray ionization mode in multiple reaction monitoring (MRM). The chromatographic separation was on a ZORBAX SB-C18 column coupled with a C18 guard column using a mobile phase composed of acetonitrile–water containing 0.1% formic acid at a flow rate of 0.5 mL min−1. The methods were sensitive with good linearity and no endogenous material interferences. The inter- and intra-day precision and accuracy of lapachol in plasma and the brain were lower than 12%. The methods were successfully applied to the quantification and pharmacokinetic study of lapachol in rats. The results indicated that the disposition profile of lapachol fitted to first order elimination and the two-compartment open model. Lapachol could pass through the blood brain barrier and went through enterohepatic circulation in rats with extending in vivo exposure time after oral administration. In summary, these findings provide an important pharmacological foundation for developing a novel drug and the clinical use of lapachol.
Introduction
Lapachol is a natural naphthoquinone compound obtained from several vegetal species, mainly bignoniaceae (Tabebuia sp.), synthesized for the first time by Vasanth et al.1,2 Extensive pharmacological activities have been described for lapachol, such as antibiotic,3,4 antifungal,5 anti-inflammatory,4 antiprotozoal,6,7 antipsoriatic,8 antiviral,9 immunomodulatory and interceptive actions.10,11 Xu et al. found that C6 glioma could been inhibited by lapachol both in vivo and vitro,12 suggesting that lapachol could be further used in malignant glioma therapy. Eyong et al., who investigated some neurological activities of lapachol, found that lapachol displayed significant anxiolytic and antidepressant effects at different doses tested.13 These results indicated that lapachol might be able to cross the blood brain barrier (BBB) to exert an effect on the brain.Although lapachol has been studied extensively in the biological activities and pharmacodynamics, little is known about the pharmacokinetic and target tissue distribution profile of lapachol in vivo.14 It is particular to the case of central nervous system-acting drugs, which must pass through the BBB before they can exert their effects.15 Detection of drug concentration, both in plasma and target tissue brain, is crucial to understand the time course of the neuroprotective activity of lapachol. In addition, the pharmacokinetic profile of lapachol will help with the determination of dosage regimen to ensure efficient clinical application, especially in the brain disease.
In the present study, we developed accurate, sensitive and specific LC-ESI-MS/MS methods combined with microdialysis technique to investigate the plasma pharmacokinetics and brain distribution kinetics of lapachol in rats. The methods were validated and could be successfully applied for determination of lapachol in pharmacokinetic studies. Our results provided important information for developing a novel drug and for better use of lapachol in clinic practice.
Experimental
Chemicals and reagents
The experiment compound lapachol [2-hydroxy-3-(3′-methyl-2′-butenyl-)-1,4-naphthoquinone] and the internal standard (IS) isoliquiritigenin were purchased from Sigma-Aldrich and the purity was greater than 99%. Acetonitrile was of HPLC grade and purchased from Fisher Scientific (Fair lawn, NJ, USA). Formic acid was of HPLC grade and purchased from Dikma Reagent Company (Beijing, China). Water used in the experiment was triply distilled. All other chemicals, reagents and solvents used were of analytical grade. The artificial cerebrospinal fluid (ACSF) buffer (124 mM NaCl, 5 mM KCl, 1.25 mM NaH2PO4, 2 mM MgSO4, 2 mM CaCl2, 26 mM NaHCO3, 10 mM D-glucose) was prepared weekly, filtered, degassed to obtain a pH of 7.1–7.4, and used as the perfusate.Apparatus and conditions
The LC-MS/MS system consisted of a HPLC system (Agilent Technologies, Palo Alto, CA, USA) including a G1379A vacuum degasser, a HP G1312A binary pump, G1313A auto-sampler and triple quadrupole mass spectrometer equipped with electrospray source (Series 6410, Agilent Technologies). The LC separation was performed on a ZORBAX SB-C18 column (150 mm × 2.1 mm i.d., 5 μm; Agilent, USA) coupled with a C18 guard column. All chromatography was performed at ambient temperature. The mobile phase was composed of acetonitrile and water containing 0.1% formic acid. The gradient elution was changed linearly from 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 (v/v) 0.1% formic acid aqueous solution-acetonitrile to 30:70 at a flow-rate of 0.5 mL min−1; the running time of samples was 12 min.
:40 (v/v) 0.1% formic acid aqueous solution-acetonitrile to 30:70 at a flow-rate of 0.5 mL min−1; the running time of samples was 12 min.
Mass spectrometric measurements were performed on a triple quadrupole mass spectrometer equipped with electrospray source working in the negative ion mode. Nitrogen was used as a sheath gas, mass spectral analysis were performed under the optimizing and automatic conditions, using a sheath flow rate of 10 L min−1, the ion spray voltage of 4 kV, and a heated capillary temperature of 350 °C. The optimized fragmentation transitions for the multiple reactions monitoring (MRM) were the m/z 241 → 186 for lapachol and the m/z 255 → 119 for the IS.
Preparation of standard and quality control (QC) samples
Stock solutions of lapachol (1 mg mL−1), and the IS (1 mg mL−1) were prepared in methanol, respectively. Calibration standards were prepared by diluting the stock solution with methanol, to get the concentrations of 100, 50, 10, 5, 1 and 0.5 μg mL−1 for analysis of lapachol in plasma and 300, 100, 50, 10, 5, 1, 0.5 and 0.1 ng mL−1 for analysis of brain dialysis samples. The stock solution of the IS was also diluted to the final concentrations of 5 μg mL−1 and 5 ng mL−1 for the analysis of plasma and dialysis samples respectively. All solutions were kept at −20 °C until analysis. Three levels of QC samples (0.5, 5 and 50 μg mL−1 for plasma analysis and 0.1, 10 and 100 ng mL−1 for brain dialysis analysis) were prepared in the same methods as the calibration standards.Experimental animals and blood samples collection
Adult male Sprague-Dawley (SD) rats (250 ± 10 g) were obtained from the Laboratory Animals Center of Capital Medical University (LAC, CMU, Beijing, China). Blank plasma was obtained from the healthy rats. After aliquot, the blood controls were stored at −80 °C and thawed at room temperature for calibration curves and quality control (QC) samples. The experiments were carried out in accordance with the current guidelines for the Care and Use of Laboratory Animals as adopted by the National Health Ministry of China.16,17 The employed protocols were approved by the Animal Center of Capital Medical University (Beijing, China).Six rats were anesthetized with chloral hydrate (10%) by intraperitoneal injection (0.3 mL/100 g), standard silicone tubing (0.20 inch i.d. × 0.37 inch OD, Helix Medical, USA) were implanted in the jugular vein for blood sampling before experiment. Each rat was housed individually in a rat cage and allowed to recover for two days.18,19 The rats were fasted for 12 h before administration, but had free access to water. All rats were orally administered with lapachol suspension (dissolved in normal saline coupled with 1% Tween-80) at a dose of 34 mg kg−1. The blood (150 μL) was obtained from the jugular cannula and collected in heparin pre-treated polypropylene centrifuge tubes before administration and post-dosing at 0.25, 0.5, 1, 3, 5, 7, 9, 12, 24 and 36 h, respectively. The plasma was separated by centrifuged at 7000 rpm for 10 min immediately and stored at −80 °C until further analysis.
Microdialysis sampling and brain distribution kinetics
Four rats were anesthetized with chloral hydrate (10%, 0.3 mL/100 g, i.p. route) and remained anesthesia throughout the experimental period. The rats were immobilized on a stereotaxic frame (Anhui, China) for brain microdialysis and the body temperature was maintained at 37 °C by heating light. After the skull was surgically exposed, a small hole was drilled on the skull based on stereotaxic coordinates. The intra-cerebral guide cannula was implanted into the striatum (coordinates: AP, −1.2 mm; LAT, −2 mm; VERT, 4 mm) according to the Paxinos and Watson atlas and secured by dental cement.20 The position of the probes in the cerebral cortex was confirmed by standard histological procedures at the end of the studies.After two days of recovery, the rats were anesthetized with chloral hydrate again, a microdialysis probe was implanted into the striatum through the guide cannula and the rats were placed in a freely moving system to ensure that the animals were awake and active during the sampling period. The microdialysis probe was perfused with ACSF at a flow rate of 1.5 μL min−1. Before sample collection, the probe was allowed to equilibrate for two hours. Then the rats were orally administered with lapachol suspension (136 mg kg−1), the collection times of the microdialysate were 20, 40, 60, 100 and 140 min, and 3, 5, 7, 9, 11, 19, 27 and 37 h. 30 μL of each sample microdialysate was collected for 37 h, and stored at −80 °C until analysis.
Microdialysis probe in vivo relative recovery (RRin vivo) was calibrated by measurements of probe in vitro recovery (RRin vitro), in vitro loss (RLin vitro) and in vivo loss (RLin vivo).20 RRin vitro was determined by placing the microdialysis membrane in 100 ng mL−1 lapachol solution and perfusing ACSF at 1.5 L min−1 by an infusion pump (CMA/100). After 20 min, the dialysate was collected and analyzed by LC-MS. ACSF containing lapachol (100 ng mL−1) passed through the microdialysis probe, which was placed in a vial containing ACSF; the dialysate was collected and analyzed by LC-MS to get RLin vitro. In the similar manner, RLin vivo was determined by perfusing the lapachol solution (100 ng mL−1) into a microdialysis probe inserted into rat brain. The probe relative recovery (RR) and relative loss (RL) were calculated according to eqn (1), in which Cd was the concentration of lapachol in the microdialysate; Cs was the concentration in the sample, and Cp was the concentration in the perfusate. The probe in vivo relative recovery (RRin vivo) was defined by the following eqn (2). The concentrations of lapachol detected in the microdialysate (Cm) were corrected to concentrations (Cf) in the brain as the eqn (3)
| RR = (Cd/Cs) × 100%; RL = (Cp − Cd)/Cp × 100% | (1) |
| RRin vivo/RLin vivo = RRin vitro/RLin vitro | (2) |
| Cf = Cm/RRin vivo | (3) |
Preparation of samples
Plasma samples (0.1 mL) were spiked with 100 μL of methanol and the IS (5 μg mL−1) stock solution, respectively. Then the mixture was extracted with 400 μL of ethyl acetate by vortexing for 2 min. After centrifugation at 12000g for 10 min, the supernatant was transferred into a clean test tube and evaporated to dryness under a flow of nitrogen gas at 35 °C. The residue was reconstituted in 100 μL of methanol and a 10 μL aliquot was injected into the LC-MS/MS system for analysis.
20 μL of brain samples from microdialysis were spike with 20 μL of methanol and the IS (5 ng mL−1) stock solution. The procedure was the same as that described above. The residue was dissolved in 20 μL the methanol, and aliquots (5 μL) were injected into the LC-MS/MS system.
Method validation
The methods were validated according to the Food and Drug Administration (FDA) guidelines for its specificity, linearity, the lower limit of quantification (LLOQ), accuracy, precision, stability, extraction recovery and matrix effect.To evaluate the selectivity, six independent samples of rat blank plasma and blank brain dialysate were analyzed by comparing with the blank samples spiked with the analytes, and a random rat sample after administration of a single dose of lapachol for excluding endogenous material interference.
To evaluate the linearity of LC-MS methods, the calibration curves of rat plasma and brain dialysate were determined in six replicates on six separate days, respectively. The calibration graphs were constructed using a linear regression of the test compound/IS peak area ratio (y) to the nominal plasma concentration of the test compound (x, μg mL−1). The concentration of each sample was determined using the equation of linear regression obtained from the calibration curve. The LLOQ was considered as the final concentration that produced at least a signal-to-noise ratio of 10. In addition, the LLOQ should be quantified reliably with acceptable accuracy and precision (<15%).
The precision and accuracy of methods were assessed by performing replicate analysis of the QC samples at low, middle and high concentrations against the calibration standards. Six replicates of the QC samples at each concentration were evaluated on the same day for intra-day precision, while repeated analysis at each concentration of the QC samples six times per day over six consecutive days for inter-day precision. The accuracy and precision are expressed in terms of the relative percent error (RE) and relative standard deviation (R.S.D.), respectively.
To evaluate the extraction recovery and matrix effect, six different blank rat plasma and microdialysis samples were extracted and spiked reference QC solutions. The corresponding peak areas divided by those of QC samples (n = 6) to get extraction recovery, and then compared with those of neat standard solutions for matrix effects (n = 6). The extraction recovery and matrix effect of the IS was determined in similar method.
Six replicates of QC samples were used to assess the stability of analytes in the plasma and brain dialysate. The long-term stability was determined after storage of the QC samples at −80 °C for 30 days. To get auto-sampler stability, QC samples were kept at ambient for 24 h and then analyzed. The freeze–thaw stability was evaluated after three cycles of freeze–thaw (−80 °C) on consecutive days.
Results and discussions
Method development
Sample pre-treatment is a critical step for determination of the test drug in biological sample by minimizing the matrix effect that occurs within LC-MS detection and increasing the relative recovery. Currently, protein precipitation, solid phase extraction and liquid–liquid extraction was three of the most useful preparation methods employed in biological samples.21 After several trials, liquid–liquid extraction was found to be more suitable for the determination of lapachol in rat plasma and brain dialysate with ethyl acetate. An internal standard is necessary for the determination of the analytes in biological samples. In initial stage of the experiment, several compounds were compared and screened. Finally, isoliquiritigenin, a kind of flavonoid compound, was found to be optimal for the IS due to its better chromatographic resolution and reduction of interference within the plasma and dialysate matrix.Some instrument parameters were investigated and optimized to maximize the mass spectra responses to lapachol and the IS. Negative ion mode was found to provide better sensitivity for detecting both the lapachol and the IS. The capillary temperature and sheath flow rate were also optimized to obtain the deprotonated molecules of the analytes. The fragment energy was optimized to achieve the maximum response of the fragment ion peaks. The precursor-product ion pairs used for the multiple reaction monitoring were m/z 241 → 186 for the test compound lapachol and m/z 255 → 119 for the IS, shown in Fig. 1.
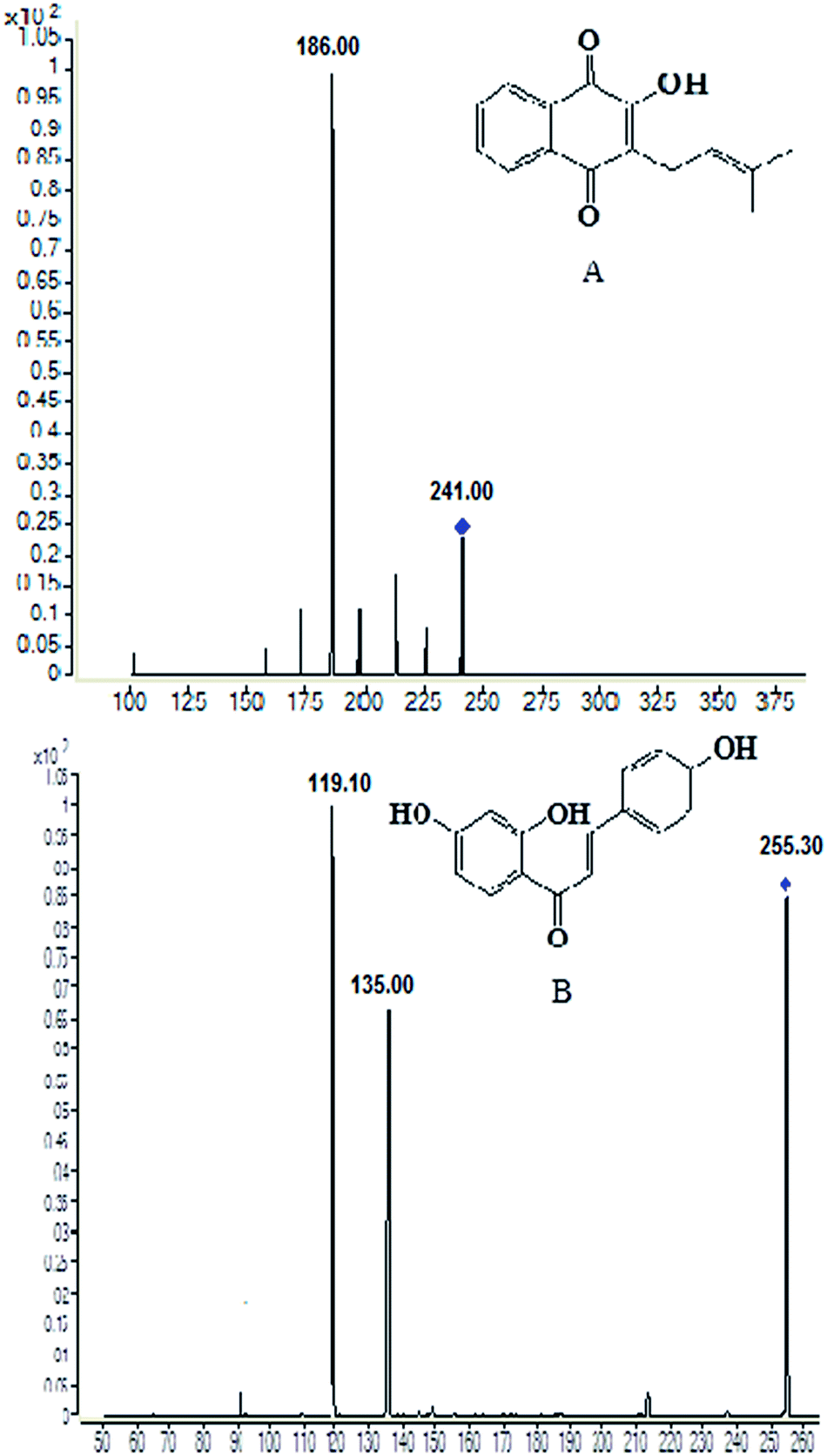 | ||
| Fig. 1 Chemical structures and production scan mass spectra of lapachol (A) and the internal standard isoliquiritigenin (B). | ||
Selectivity
The results for the selectivity of samples were shown in Fig. 2. The retention times were 8.7 min for lapachol and 5.5 min for the IS. There were no endogenous substance peaks and drug metabolite peaks interfered with the analytes and the IS in the plasma and brain dialysate at the retention times.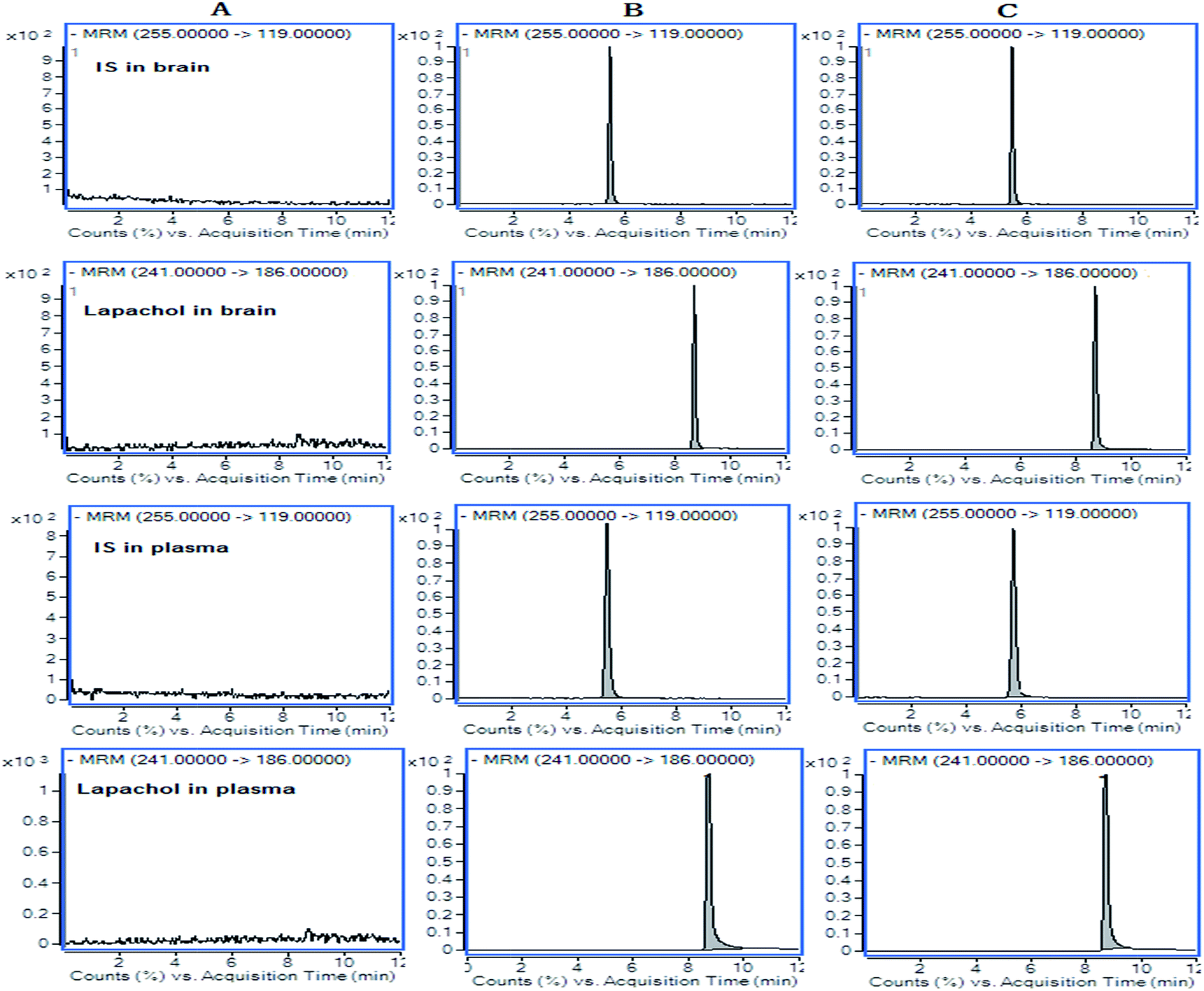 | ||
| Fig. 2 Representative LC-MS/MS chromatograms for lapachol and IS. (A) Blank samples in rat plasma and dialysate; (B) a spiked dialysate sample with IS and 50 ng mL−1 of lapachol and a spiked plasma sample with IS and 1 μg mL−1 of lapachol; (C) random dialysate sample (95 ng mL−1) at 100 min after administration of lapachol and random plasma sample (0.73 μg mL−1) at 36 h after administration of lapachol. | ||
Calibration curve and sensitivity
The calibration curves for quantification of lapachol in the plasma and dialysate showed good linear relationships, with the ranges 0.5–100 μg mL−1 and 0.1–300 ng mL−1 respectively. The mean equations for the calibration curves for lapachol were y = 0.4321 + 0.224x (r2 = 0.997) and y = 0.1516 + 0.0994x (r2 = 0.995), respectively in rat plasma and brain. The assay proved to be linear and acceptable.Sensitivity was evaluated by determining LLOQ that could be reliably and reproducibly measured at least six replicates. The LLOQ data was 0.5 ng mL−1 for lapachol in rat plasma and 0.1 ng mL−1 in the brain dialysate.
Precision and accuracy
The precision and accuracy of the methods were determined in rat plasma and dialysate by performing replicate analyses of QC samples. The intra-day and inter-day precision and accuracy of the methods were presented in Table 1. The precision (R.S.D.%) was less than 10% and the accuracy (RE) was less than 5% both in the plasma and brain. According to FDA guidance for the bioanalytical method validation, the data indicated that the precision and accuracy were acceptable.| Matrix | Spiked | Intra-day | Inter-day | ||||
|---|---|---|---|---|---|---|---|
| Measured mean ± SD | Precision (RSD%) | Accuracy (RE%) | Measured mean ± SD | Precision (RSD%) | Accuracy (RE%) | ||
| Plasma (μg mL−1) | 0.5 | 0.51 ± 0.01 | 2.49 | 1.32 | 0.5 ± 0.01 | 2.76 | 0.98 |
| 5 | 5.03 ± 0.08 | 1.51 | 0.7 | 5.1 ± 0.09 | 1.74 | 2.23 | |
| 50 | 50.5 ± 1.4 | 2.76 | 1.7 | 50.4 ± 2 | 3.93 | 0.70 | |
| Brain (ng mL−1) | 0.1 | 0.10 ± 0.004 | 3.94 | 4.88 | 0.1 ± 0.01 | 9.09 | 4.38 |
| 10 | 10.3 ± 0.19 | 1.86 | 2.85 | 10.3 ± 0.18 | 1.71 | 3.08 | |
| 100 | 100 ± 0.71 | 0.70 | 0.63 | 100.±1.46 | 1.45 | 0.41 | |
Extraction recovery and matrix effect
The extraction recovery of the QC samples was presented in Table 2. The average extraction recovery of the internal standard was 97.2% and 88.1% in the plasma and brain dialysate, respectively. The data indicated that the extraction recovery of lapachol and IS over the concentration range was acceptable.| Matrix | Spiked | Recovery | Matrix effect | ||
|---|---|---|---|---|---|
| Mean ± SD (%) | R.S.D.% | Mean ± SD (%) | R.S.D.% | ||
| Plasma (μg mL−1) | 0.5 | 84.79 ± 5.19 | 6.09 | 91.01 ± 7.93 | 8.72 |
| 5 | 87.04 ± 2.49 | 2.86 | 94.78 ± 3.71 | 3.91 | |
| 50 | 97.67 ± 3.16 | 3.24 | 96.86 ± 9.95 | 10.27 | |
| Brain (ng mL−1) | 0.1 | 80.87 ± 10.98 | 13.58 | 86.58 ± 10.58 | 12.23 |
| 10 | 88.71 ± 6.25 | 7.05 | 90.85 ± 8.15 | 8.97 | |
| 100 | 85.09 ± 6.89 | 8.09 | 93.15 ± 8.55 | 9.17 | |
The matrix effect was monitored by post-extraction spike method. The results of lapachol were in the range of 86.58–96.86, shown in Table 2, the matrix effect of IS were 95.1% and 92%, indicating that no co-eluting endogenous substance existed in the rat plasma and brain dialysate affected the reproducibility and the accuracy of the assay.
Stability
The stability of lapachol in the blood and brain dialysate under different conditions was shown in the Table 3, which indicated that the samples were all stable for 24 h of storage at room temperature, three freeze–thaw cycles and storage at −80 °C for 30 days. The concentrations of the analytes in rat plasma and brain dialysate were not significantly affected by ambient, frozen storage and freeze–thaw conditions.| Matrix | Spiked | Storage for 30 days at −80 °C | Three freeze–thaw cycles | Room temperature for 24 h | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Measured mean ± SD | RSD (%) | RE (%) | Measured mean ± SD | RSD (%) | RE (%) | Measured mean ± SD | RSD (%) | RE (%) | ||
| Plasma (μg mL−1) | 0.5 | 0.51 ± 0.01 | 2.69 | 1.63 | 0.52 ± 0.02 | 3.51 | 3.23 | 0.52 ± 0.02 | 3 | 3.58 |
| 5 | 5.11 ± 0.11 | 1.89 | 1.97 | 5.12 ± 0.14 | 2.73 | 2.56 | 5.17 ± 0.02 | 0.3 | 3.38 | |
| 50 | 50.38 ± 1.07 | 2.13 | 0.77 | 49.5 ± 1.08 | 2.17 | −1.05 | 50.81 ± 0.96 | 1.9 | 1.62 | |
| Brain (ng mL−1) | 0.1 | 0.11 ± 0.01 | 7.01 | 9.39 | 0.10 ± 0.01 | 9.71 | 3.66 | 0.11 ± 0.01 | 11.2 | 6.03 |
| 10 | 10.33 ± 0.52 | 5.05 | 3.26 | 10.6 ± 0.37 | 3.5 | 6.12 | 10.33 ± 0.45 | 4.4 | 3.32 | |
| 100 | 101.2 ± 3.25 | 3.21 | 1.25 | 101.4 ± 3.2 | 3.15 | 1.44 | 98.67 ± 3.68 | 3.7 | −1.33 | |
Plasma pharmacokinetics and brain distribution kinetics
The data was calculated and treated by the software DAS version 2.0 (Chinese Pharmacological Society, Beijing, China). The main pharmacokinetic parameters of lapachol in plasma were presented in Table 4. The concentration–time profile of lapachol in rats fitted to a first order elimination, two-compartment open model. The plasma concentration–time profile of lapachol in rat was shown in Fig. 3. Lapachol appeared in blood rapidly after oral administration, reached the maximum concentration at 0.52 h, and was eliminated from blood with elimination half-life time (7.62 h), which indicated that lapachol was fast absorbed, but distributed and eliminated slowly. A weak double peak in the plasma concentration–time profile indicated that lapachol could be via enterohepatic circulation in rats after oral administration, which corresponded with the double peak in the distribution of lapachol in rat brain dialysates. The distribution kinetic parameters of lapachol in rat brain were also calculated by the ware DAS 2.0. The main kinetic parameters were presented in Table 5. The brain tissue concentration–time profile of lapachol was shown in Fig. 4. Lapachol was distributed slowly in rat brain to achieve the maximum concentration after 5.85 h, but eliminated faster than in the plasma with elimination half-life time, hinting that there was possibility of some efflux transports on the BBB systems which could accelerate the elimination of lapachol in rat brain.22| Parameters | Unit | Mean ± S.D. |
|---|---|---|
| t1/2α | h | 2.38 ± 1.02 |
| t1/2β | h | 7.62 ± 3.40 |
| t1/2Ka | h | 0.07 ± 0.06 |
| Vd | L kg−1 | 0.75 ± 0.08 |
| AUC | (μg mL−1)h | 343 ± 88.3 |
| CL(s) | L h−1 kg−1 | 0.10 ± 0.02 |
| tmax | h | 0.52 ± 0.21 |
| Cmax | μg mL−1 | 43.2 ± 7.04 |
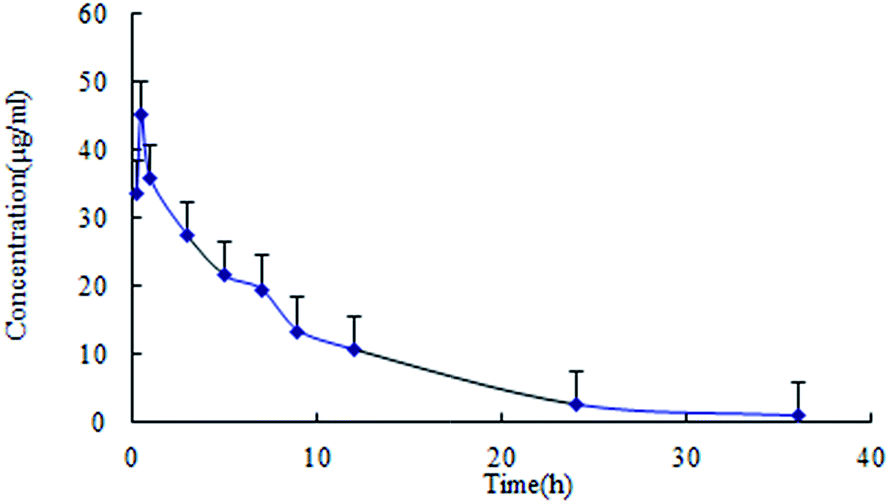 | ||
| Fig. 3 Plasma pharmacokinetic profile of lapachol following single oral administration of lapachol to rats (n = 6). | ||
| Parameters | Unit | Mean ± S.D. |
|---|---|---|
| t1/2α | h | 0.12 ± 0.05 |
| t1/2β | h | 4.78 ± 0.29 |
| t1/2Ka | h | 3.17 ± 0.48 |
| Vd | L kg−1 | 2.09 ± 2.54 |
| AUC | (ng mL−1)h | 3866 ± 683 |
| CL(s) | L h−1 kg−1 | 0.04 ± 0.01 |
| tmax | h | 5.85 ± 0.48 |
| Cmax | ng mL−1 | 252 ± 41.9 |
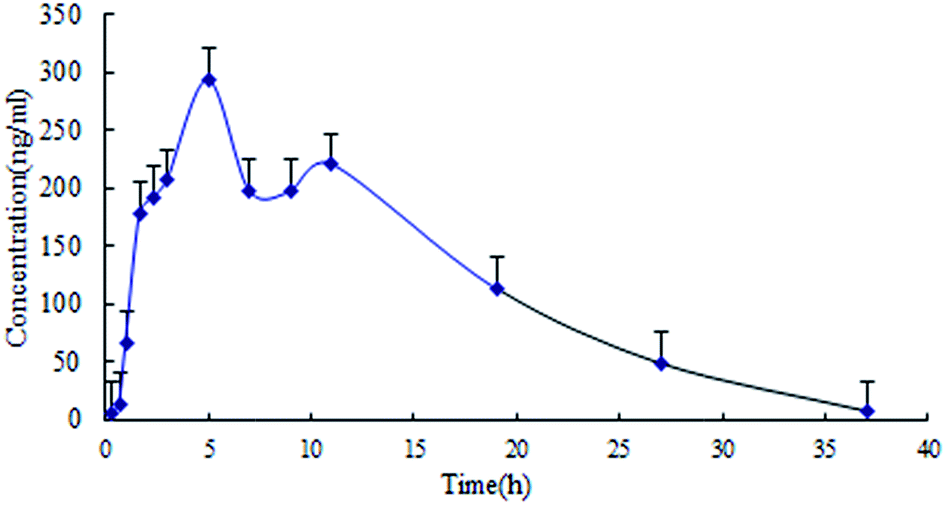 | ||
| Fig. 4 Brain distribution kinetic profile of lapachol in rat cerebrospinal fluid following single oral administration of lapachol to rats (n = 4). | ||
The blood brain barrier is a very important natural barrier between the blood and brain to maintain the in vivo homeostasis of brain parenchymal microenvironment. The drugs affected the brain should firstly penetrate the BBB so that the effects could be exerted. The BBB is a key regulatory interface that could prevent drug delivery to the central nerve system.23 Whether drugs can penetrate the BBB and distribute in the brain tissues in definite level are very crucial for discovering and developing the effective cerebral drugs. To date, the main interest in lapachol has been focused on its pharmacological activity, there is no articles published about the drug metabolism and pharmacokinetics. In this study, a specific, rapid and effective LC-MS/MS method has been developed for determination of lapachol both in rat plasma and brain tissues. All the validation data, such as the precision, accuracy, matrix effect and stability was within the required limit.24–27 This method has been successfully used to study the plasma pharmacokinetics and brain distribution kinetics of test drug lapachol in rats, which combined with the microdialysis technique that is a effective method for simultaneously collecting the bio-samples to monitor the local tissue drug concentrations continuously in the awake and unrestrained animals, with the advantages of real-time, live and dynamics.
Our results indicated that the in vivo disposition profile of lapachol fitted to first order elimination and two-compartment open model in rat plasma. After an oral administration, lapachol underwent an entero-hepatic circulation in rats that could extend the in vivo exposure time and tissue action duration. Our data also showed that lapachol could pass through the BBB and the elimination half-life was 4.78 h, suggesting that lapachol was eliminated faster in the brain than that in the plasma. The AUC ratio between the brain and plasma was 0.028% for lapachol, strongly suggesting that there were involvement of active efflux across the BBB.28 Nonetheless, the drug concentration levels in rat plasma and brain tissues were relatively high and could reach to the effective therapeutic concentrations.12 Lapachol have some pharmacological marked effects in brains such as anti-glioma, anxiolytic and antidepressant,12,13 and the definite distribution concentrations in brain tissues might be the prerequisite for the effective application of lapachol in clinical practice. For achievement of satisfactory effects, it is necessary to further improve the penetration index via changing the dosage form of lapachol such as nanoparticle.
Conclusion
In this paper, for the first time, the plasma pharmacokinetics and brain distribution kinetics of lapachol was studied. A LC-MS/MS method has been developed for determination of lapachol in rat plasma and dialysates combined with the microdialysis technique. The performance criteria for the precision and accuracy, recovery and matrix effect, sensitivity, linearity and stability have been assessed and were within the FDA recommended guidelines. The results indicated that this method could be successfully used for the pharmacokinetic studies of lapachol in rats. Accordingly, these findings provide an important pharmacological foundation for developing a novel drug and the effective clinical use of lapachol.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Program of National Foundation of Natural Sciences of China (Grant No. 81573683 and 81173121), the Key Program of Beijing Natural Science Foundation and Beijing Municipal Commission of Education (KM201110025024) and Beijing Laboratory for Biomedical Detection Technology and Instrument (PXM2014-014226-000021) for their financial supporting.Notes and references
- S. Vasanth, D. S. Jayakaran, V. P. Raj and V. Srinivasan, J. Indian Chem. Soc., 2002, 79, 765–767 CAS.
- H. Hussain, K. Krohn, V. U. Ahmad and G. A. Miana, Arkivoc., 2007,(2), 145–171 CAS.
- T. B. Machado, A. V. Pinto, M. C. Pinto and I. C. Leal, Int. J. Antimicrob. Agents, 2003, 21, 279–284 CrossRef CAS PubMed.
- I. M. F. Cavalcanti, J. G. Pontes-Neto, P. O. Kocerginsky, A. M. Bezerra-Neto, J. L. C. Lima, M. A. V. Maciel, R. P. Neves, M. F. Pimentel and N. S. Santos-Magalhaes, Journal of Global Antimicrobial Resistance, 2015, 3, 103–108 CrossRef CAS PubMed.
- V. A. Isidorov, R. Bagan, L. Szczepaniak and L. Swiecicka, Open Chem., 2015, 13, 125–137 CAS.
- E. Perez-Sacau, A. Estevez-Braun, A. G. Ravelo and Y. D. Gutierrez, Chem. Biodiversity, 2005, 2, 264–274 Search PubMed.
- C. Salas, R. A. Tapia, K. Ciudad and V. Armstrong, Bioorg. Med. Chem., 2008, 16, 668–674 CrossRef CAS PubMed.
- M. R. Suo and S. Y. Yan, Chem. Biodiversity, 2016, 13, 1611–1616 CAS.
- F. Epifano, S. Genovese, S. Fiorito, V. Mathieu and R. Kiss, Phytochem. Rev., 2014, 13, 37–49 CrossRef.
- R. A. Oliveira, J. Correia-Oliveira, L. J. Tang and R. C. Garcia, Int. Immunopharmacol., 2012, 14, 54–65 CrossRef CAS PubMed.
- F. D. Da Silva and V. F. Ferreira, Curr. Org. Synth., 2016, 13, 334–371 CrossRef.
- H. L. Xu, Q. Y. Chen, H. Wang, P. X. Xu, R. Yuan and M. Xue, J. Exp. Clin. Cancer Res., 2016, 35, 178–187 CrossRef PubMed.
- K. O. Eyong, H. S. Foyet, C. Eyong, L. S. Sidjui and M. C. Yimdjo, Med. Chem. Res., 2012, 22, 2902–2911 CrossRef.
- B. Lu, H. Ying, J. F. Yao, X. R. Li and M. Xue, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 944, 128–135 CrossRef PubMed.
- Y. Y. Kou, Y. Liu, M. Xue, Y. X. Xu and H. Liu, Int. J. Pharm., 2008, 353, 88–94 CrossRef CAS PubMed.
- X. L. Qin, X. Chen, G. P. Zhong, X. M. Fan, Y. Wang, X. P. Xue, Y. Wang and M. Huang, Phytomedicine, 2014, 21, 766–772 CrossRef CAS PubMed.
- C. H. Wang, C. S. Wu, J. L. Zhang and Y. Jin, Phytomedicine, 2015, 22, 487–497 CrossRef CAS PubMed.
- H. J. Liu, C. Yan, C. M. Li and L. Lin, J. Pharm. Biomed. Anal., 2013, 74, 56–61 CrossRef CAS PubMed.
- Y. Y. Chen, J. M. Guo, Y. F. Qian, S. Guo, C. H. Ma and J. A. Duan, Phytomedicine, 2013, 21, 82–89 CrossRef CAS PubMed.
- C. Y. Gao, X. R. Li, Y. H. Li, L. J. Wang and M. Xue, J. Pharm. Pharmacol., 2010, 62, 360–367 CrossRef CAS PubMed.
- B. K. Matuszewski, M. L. Constanzer and C. M. Chavez-Eng, Anal. Chem., 2003, 75, 3019–3030 CrossRef CAS PubMed.
- E. M. Leslie, R. G. Deeley and S. P. C Cole, Toxicol. Appl. Pharmacol., 2005, 204, 216–237 CrossRef CAS PubMed.
- Y. Y. Yang, L. Bai, X. R. Li, P. X. Xu and M. Xue, Toxicol. in Vitro, 2014, 28, 388–396 CrossRef CAS PubMed.
- W. W. Gong, P. X. Xu, S. S. Guo, X. R. Li, Z. L. Jin, Y. M. Zhao, M. Fan and M. Xue, RSC Adv., 2017, 7, 25414–25421 RSC.
- Z. Wang and B. L. Qin, Anal. Methods, 2016, 8, 8443–8451 RSC.
- Y. S. He, W. Sun, B. Y. Zhang, L. H. Xu, J. Yang, W. Gao, L. W. Qi and X. D. Wen, Anal. Methods, 2016, 8, 785–789 RSC.
- R. Nirogi, D. R. Ajjala, V. Kandikere, R. Aleti, H. R. Pantangi, S. R. Srikakolapu, V. Benade and H. Vurimindi, J. Pharm. Biomed. Anal., 2013, 82, 160–167 CrossRef PubMed.
- K. L. Hu, Y. B. Shi, W. M. Jiang, J. Y. Han, S. X. Huang and X. G. Jiang, Int. J. Pharm., 2011, 415(1/2), 273–283 CrossRef CAS PubMed.
Footnote |
| † Lu Bai and Ying Han was co-first authors. |
| This journal is © The Royal Society of Chemistry 2017 |