DOI:
10.1039/C7RA07249J
(Paper)
RSC Adv., 2017,
7, 42856-42865
Evaluation of aptamer specificity with or without primers using clinical samples for C-reactive protein by magnetic-assisted rapid aptamer selection†
Received
30th June 2017
, Accepted 17th August 2017
First published on 5th September 2017
Abstract
Aptamers with primer binding sites are necessary for the SELEX (Systematic Evolution of Ligands by EXponential enrichment) process. Such primer sequences cause non-specific binding by their nature and raise the incidence of false positives. Thus, the use of aptamers without primer or with minimum primer, to shorten the length of selected aptamers, has become an interesting research topic. Recently, a primer-free aptamer selection protocol based on the magnetic-assisted rapid aptamer selection (MARAS) technology has been reported to generate primer-free aptamers with high affinity and specificity. Adversely, the yield of a suitable aptamer was low and the advantage of MARAS was deteriorated. In this study, multiple negative selection runs were used in the MARAS procedure to remove aptamers with non-specific binding to unwanted biomolecules. Smart aptamers with predetermined affinity for C-reactive protein were isolated by window-MARAS. The specificity of aptamers was validated by using blind serum samples and compared with those using monoclonal antibody-based nephelometry analysis. The relative specificity of aptamers with or without primers was evaluated, revealing that the specificity of the aptamer with primer is similar to that of the aptamer without primer. By using a randomized library with primers, the simplicity and rapidity of MARAS in generating aptamers are preserved, and by implementing multiple negative selection, MARAS could efficiently select aptamers with high binding affinity and specificity for clinical applications.
Introduction
In the 1990s, an in vitro selection method, the systematic evolution of ligands by exponential enrichment (SELEX), was developed to generate aptamers.1–3 Aptamers are short sequences of single-stranded DNA or RNA with the ability to bind to targets. The SELEX process is started with a random oligonucleotide library comprising 1014 to 1015 random DNA or RNA sequences that is incubated with target molecules of interest. After incubation, the unbound oligonucleotides are removed, and the target-bound oligonucleotides are eluted from targets. The eluted oligonucleotides are then amplified by polymerase chain reaction (PCR), and the single strand oligonucleotides are purified from the PCR product to act as a library for the next selection cycle. These processes are repeatedly executed for 5 to 15 selection cycles, until there is no further detectable enrichment of the functional nucleic acids.
The entire SELEX process is tedious and less effective. Several studies attempted to improve the selective efficiency, using capillary electrophoresis SELEX (CE-SELEX), FluMag-SELEX, Capture-SELEX, and one-round aptamer selection MonoLEX.4–7 However, these protocols require expensive resources and high technological training, and the selected aptamers suffer low affinity properties. A newly developed protocol, magnetic-assisted rapid aptamer selection (MARAS), can efficiently generate high-affinity aptamers without any repeat cycle.8,9 According to this straightforward protocol, the generated aptamer with a predetermined dissociation constant (Kd) is feasible, and the value of Kd is highly dependent on the external applied magnetic field (e.g. frequency and amplitude of magnetic field).
The random library used in either SELEX or MARAS contains oligonucleotides with a randomized central region and two fixed primer regions at both ends. The fixed primer regions are necessary for amplifying target-bound sequences by PCR. However, these extra sequences may cause non-specific binding or interfere with target-aptamer binding. Therefore, several attempts have been made to generate aptamers without primer or with minimum primers, which are discussed in a review article,10 including primer-annealing and primer-switching genomics SELEX,11 primer-free 2′-O-methyl random RNA fishing SELEX,12 primer-less random RNA library selection (tailored-SELEX),13 dual random RNA library selection (dual SELEX protocol),14 minimal-primer random DNA library selection15,16 and MARAS-based primer-free protocol.17 However, all of the primer-free or minimal-primer approaches mentioned above except the last one17 included several complicated molecular cloning steps for every selection cycle, which were time and resource intensive. Even the last one, which utilizes the efficiency of MARAS, still requires a complicated single-strand ligation process which ligates primers to the primer-free aptamer for PCR amplification prior to molecular cloning. These molecular cloning steps suffer a low success rate in generating a suitable aptamer. Therefore, the development of an efficient method to generate high-affinity and specificity aptamers is necessary in the future.
In this work, C-reactive protein (CRP), a common indicator of inflammation, heart attack, stroke and cardiovascular disease (CVD), was used as an experimental target, and a negative selection process with multiple negative selection rounds was added into the MARAS protocol.9 Smart aptamers18,19 with a predetermined dissociation constant were selected by using window-MARAS from a randomized oligonucleotide library containing primers at both the 5′ and 3′ termini. The binding affinity (Kd) and specificity of a selected aptamer were analyzed. The CRP concentration results analyzed by aptamer-based real-time quantitative PCR (q-PCR) and aptamer-based ELISA methods were compared with that by monoclonal antibody-based nephelometry analysis using blind clinical samples. The correlation result of the selected aptamer with primers was compared with that of the primer-free aptamer given in ref. 17 to investigate whether the MARAS protocol with a multiple negative selection step using randomized oligonucleotide libraries containing primers can still yield highly specific aptamers as those of the primer-free counterpart without loss of simplicity and rapidity of the protocol.
Results and discussions
Characteristics of isolated aptamers
Six aptamers were successfully selected by the window-MARAS procedure depicted in Fig. 1 and were used for preliminary reverse validation. The procedures for preliminary reverse validation and forward single-strand aptamer synthesis are described in the Experimental section and ESI,† respectively. The results of the preliminary reverse validation were shown in ESI Fig. S1.† Based on preliminary reverse validation result, CRP-aptamer-1 and CRP-aptamer-2 were selected for sequencing, and results are shown in Table 1. Their corresponding secondary structures and free energies ΔG were predicted by Mfold server20 and are shown in ESI Fig. S2.† The secondary structure tends to form one bigger and one smaller loop for CRP-aptamer-1, and one medium loop and a stem with several small loops for CRP-aptamer-2, with the participation of the primers at both 5′ and 3′ termini. The predicted free energies of CRP-aptamer-1 and CRP-aptamer-2 are −2.78 and −4.47 kcal mol−1, respectively. The low free energies of the selected aptamers ensured that aptamers could form an energetically stable complex by their nature.
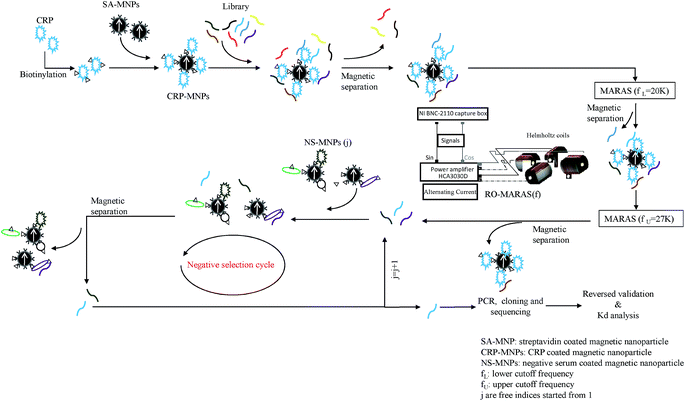 |
| | Fig. 1 Schematic illustration of the selection process for smart CRP aptamers with predetermined affinity. | |
Table 1 Random sequences (N20) of the selected aptamers
| Aptamer ID |
Oligo sequence |
| CRP-aptamer-1 |
TGTTATTTGTATGTGTTGTT |
| CRP-aptamer-2 |
CTGCATCACCAAGCCTCGCA |
Reverse validation of selected aptamers
The binding specificities of the CRP-aptamer-1 and CRP-aptamer-2 aptamer were analyzed by targeting the positive control (P1) and negative controls (N1, N2 and N3). The result is shown in Fig. 2. The CRP-aptamer-1 (Fig. 2a) and -2 aptamers (Fig. 2b) were bound to positive control MNPs, and the binding levels for negative control MNPs were low, near the noise level. This result was consistent with previous findings17 and demonstrated that MARAS provides a competitive mechanism to dissociate non-specific and low-affinity binding. Only high-specific and high-affinity binding between selected aptamers and targets could not be interrupted after applying the MARAS mechanism in the aptamer selection process.
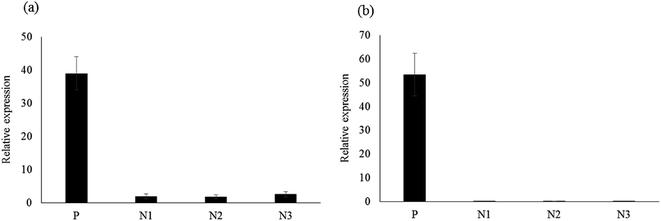 |
| | Fig. 2 Results of the reverse validation of selected aptamers with positive (P1: CRP) and negative controls (N1, N2, and N3). (a) CRP-aptamer-1 and (b) CRP-aptamer-2. | |
Determination of dissociation constants of selected aptamers
The apparent dissociation constants (Kd) of CRP-aptamer-1 and -2 were determined by q-PCR and fitted to the results with a nonlinear regression.8,9,17,21–23 The detailed procedures are described in the Experimental section. The fitting curve is shown in Fig. 3, and the Kd of selected CRP-aptamer-1 and -2 are 35.34 ± 1.67 nM and 33.34 ± 1.15 nM, respectively. The binding affinities of CRP-aptamer-1 and -2 slightly deteriorated when compared to those of the previous study with Kd values of 24.59 ± 1.15 to 28.80 ± 2.05 nM.9 The higher values of Kd in this work are attributed to the size effect of aptamers (50 nt vs. 60 nt). Moreover, the Kds of the aptamers generated in this work satisfy the requirement specified prior to the selection process (Kd ≥ 23.58 ± 0.82 nM). The results demonstrate that the window-MARAS protocol can be used to generate smart aptamers with a predetermined Kd value without any repeat cycle.
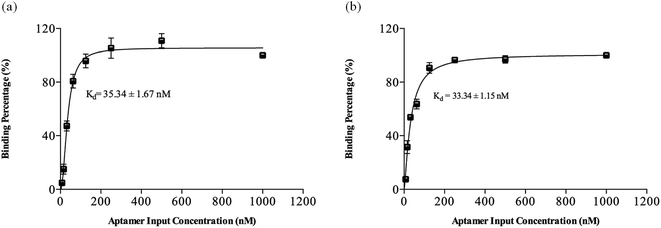 |
| | Fig. 3 The results of dissociation constant (Kd) of selected CRP aptamers. (a) CRP-aptamer-1, Kd: 35.34 ± 1.67 nM and (b) CRP-aptamer-2, Kd: 33.34 ± 1.15 nM. | |
Establishment of standard calibration curves using aptamer as a capture probe by q-PCR and ELISA methods
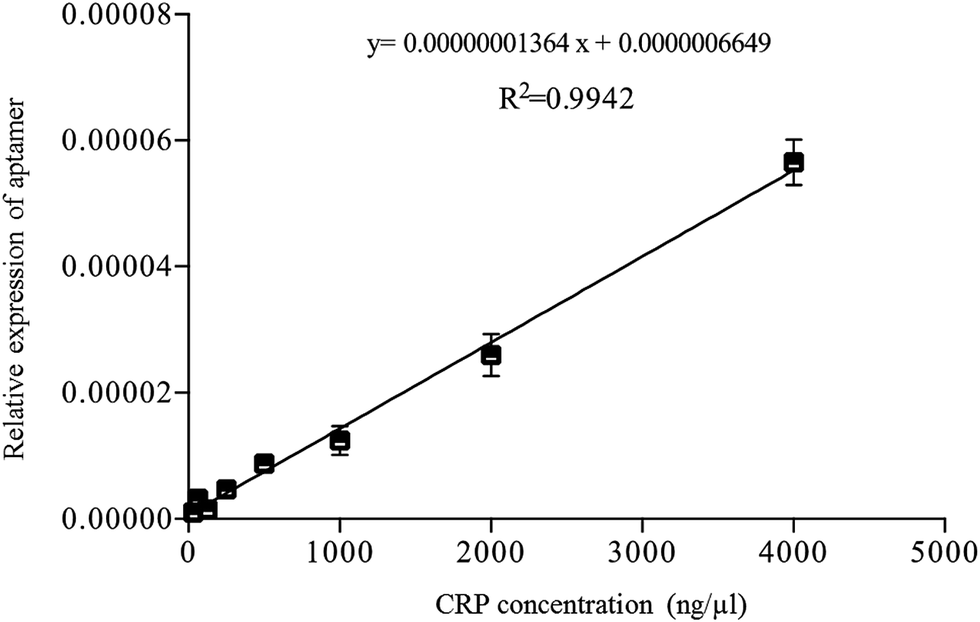
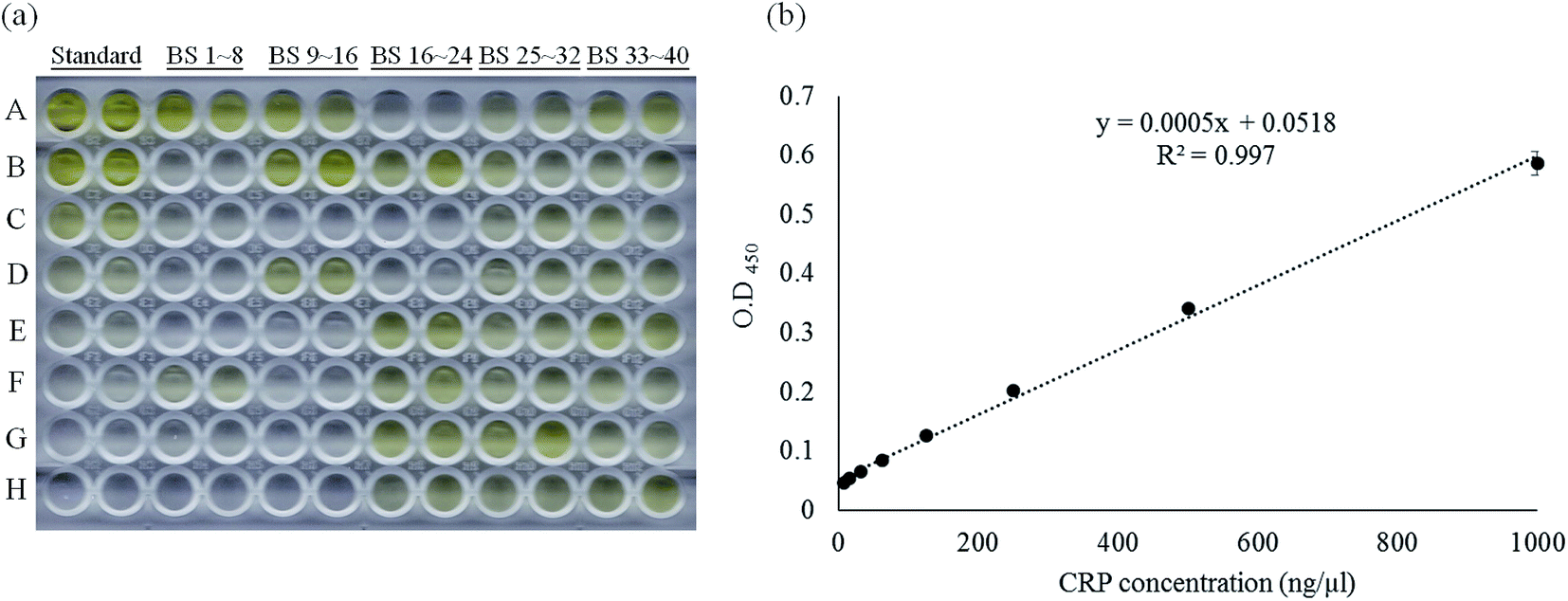
According to the reverse validation result, CRP-aptamer-2 has better specificity than CRP-aptamer-1 (Fig. 2). CRP-aptamer-2 was therefore selected to establish a standard calibration curve for both q-PCR and ELISA analyses and for the subsequent blind clinical test. The detailed aptamer-based q-PCR method has been described in the experimental section. The standard calibration curve for q-PCR analysis was linearly fitted in a CRP concentration series from 0 to 4000 ng μl−1, as shown in Fig. 4. In addition, ELISA is a common method for detecting target protein concentration in clinical diagnosis. Therefore, an aptamer-based ELISA protocol was developed in the study, and the detailed processes are described in the Experimental section. The original image of the ELISA plate is presented in Fig. 5a. Each measurement was performed in duplicate. Columns 1 and 2 of the microplate show the wells used to establish the standard calibration curve. The standard calibration curve of ELISA has been linearly fitted in the CRP concentration series from 0 to 1000 ng μl−1 as shown in Fig. 5b. Both standard calibration curves were used to calculate the concentration of the target in the blind clinical samples as mentioned above.
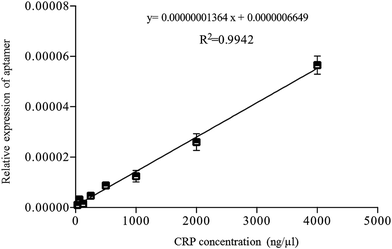 |
| | Fig. 4 Standard calibration curve of aptamer-based q-PCR method fitted by linear regression using CRP-aptamer-2 as a capture probe. | |
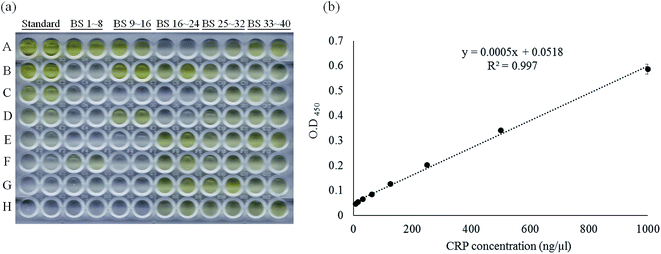 |
| | Fig. 5 Standard calibration curve of aptamer-based ELISA method fitted by linear regression using CRP-aptamer-2 as a capture probe. (a) The photo image of the aptamer-based ELISA measurement and (b) the standard calibration curve of the aptamer-based ELISA method. “Standard” indicates the pure CRP protein dilution series coated on the wells, “BS” indicates blind serum samples. | |
Using the selected aptamer as a capture probe in detecting CRP in blind clinical samples
For the specificity analysis using the CRP-aptamer-2 as a detection probe, the CRP-aptamer-2 was incubated with serum-MNPs of forty blind serums, and the amount of bound aptamer was analyzed by q-PCR. As shown in Fig. 6a, the filled circles represent the CRP concentration in blind serum samples where CRP-aptamer-2 was used as the detection probe. The results of CRP concentration in blind clinical serums were compared with those using monoclonal antibody-based nephelometry methods. Fig. 6a illustrates the correlation between aptamer-based q-PCR and monoclonal antibody-based nephelometry analyses in all blind clinical samples with Spearman's rho = 0.974, P < 0.001. Furthermore, a Bland–Altman plot24 was constructed to evaluate the level of agreement between the monoclonal antibody-based nephelometry method and the aptamer-based q-PCR method. Bland–Altman analysis revealed a bias of −9.604 (ng μl−1) with 95% limits of measurement within the range −45.61 to 26.40 (ng μl−1) (Fig. 6b). Results indicate that the two analysis methods, aptamer-based q-PCR and monoclonal antibody-based nephelometry, were highly consistent in detecting CRP concentration in blind serum samples. The concentration of the CRP in these forty blind clinical serums was also analyzed using ELISA method. The ELISA measurements in duplicate for the forty blind clinical samples are shown in Fig. 5a, from columns 3 through 12 of the 96-well microplate. The relationship between the CRP concentrations measured using aptamer-based ELISA analysis (vertical axis) and the monoclonal antibody-based nephelometry methods (horizontal axis) was analyzed by Spearman's rank correlation coefficient, and as shown in Fig. 6c. The Spearman's rho value of correlation between the aptamer-based ELISA and monoclonal antibody-based nephelometry method was 0.986, with p < 0.001. Furthermore, data from the Bland–Altman plot showed a high level of agreement between the two analysis methods, with a bias of 1.788 ng μl−1. The upper and lower limits at 95% were 17.52 ng μl−1 LOA and −13.94 ng μl−1 LOA, respectively (Fig. 6d). The data suggest a high level of agreement between the aptamer-based ELISA and the monoclonal antibody-based nephelometry method. Furthermore, the comparison result of q-PCR analysis (Fig. 6a) with that of ELISA analysis (Fig. 6c) using CRP-aptamer-2 as the capture probe indicates that both methods yield similar results (Spearman's rho value: 0.974 vs. 0.986) as compared with the monoclonal antibody-based nephelometry method, and that the specificity of the aptamer-based analysis, including q-PCR and ELISA, is guaranteed. It is worth mentioning that the data points were much more scattered for q-PCR than for ELISA method due to the inhomogeneity of the blind-MNP reagents used in q-PCR analysis.
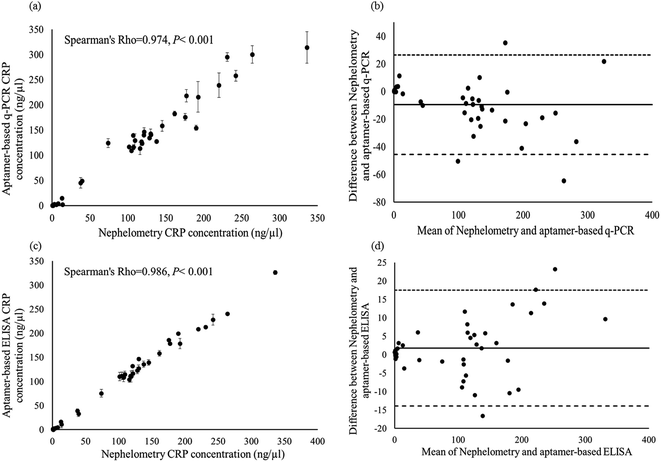 |
| | Fig. 6 Comparison of the CRP concentration measured using aptamer-based q-PCR method and aptamer-based ELISA method vs. monoclonal antibody-based nephelometry method for blind serum samples. (a) q-PCR vs. nephelometry and (c) ELISA vs. nephelometry; (b and d) Bland–Altman plot analyses of the CRP concentrations measured by monoclonal antibody-based nephelometry method vs. aptamer-based q-PCR and ELISA method, respectively. | |
Assessment of specificity of detection using aptamers with and without primer as a capture probe
Finally, to assess the specificity of the aptamers with or without primers as a capture probe, the correlation result shown in Fig. 6 was compared with those shown in Fig. 4 of ref. 17. The result is listed in Table 2, in which the aptamer selection condition and the results of the detection specificity for aptamer-based (with and without primer) vs. monoclonal antibody-based nephelometry analysis are included. In Table 2, the correlation results between aptamer-based q-PCR, with and without primer, vs. monoclonal antibody-based nephelometry were similar (Spearman's rho value: 0.974 vs. 0.985, respectively). It must be mentioned that in ref. 17, there were three mechanisms used to enhance the specificity of the selected aptamer, including a multiple negative selection to remove non-target bound aptamer during selection, a primer-free library to reduce the size of the selected aptamer to avoid the interference from nonbinding sequences, and usage of externally applied magnetic field to further suppress the nonspecific binding during the detection phase. Conversely, in the current work, only a multiple negative selection step was used to enhance the specificity of detection. Moreover, there is no need for nonspecific suppression by magnetic field during the detection phase using the selected aptamers as the capture probe and magnetic particles become unnecessary during the detection phase, as demonstrated in the current work, i.e., aptamer-based ELISA. Therefore, it can be concluded that by using libraries with primers, the advantages, rapidity and simplicity, of MARAS procedure can be fulfilled to generate aptamers with high affinity, and the specificity of the selected aptamer can be enhanced by multiple negative selection alone.
Table 2 Specificity comparison of aptamers, with and without primers, as the capture probe vs. monoclonal antibody-based nephelometry using blind clinical samples
| |
Primer-free aptamera |
Smart aptamer |
| The information is referenced from ref. 17. Process time is from the material preparation to completion of the cloning step. Spearman's rho is the correlation of the results using aptamer-based and monoclonal antibody-based methods in detecting forty blind clinical samples. For this work, the aptamer-based q-PCR and ELISA analyses were only performed without any non-specific suppression by magnetic fields. NSS indicates the non-specific suppression by externally applied magnetic fields adopted in ref. 17. |
| Starting library |
5′-N20-3′ |
5′-AGCAGCACAGAGGTC-N20-GCGTGCTACCGTGAA-3′ |
| MARAS condition |
27 kHz and 14 gauss |
20 kHz and 14 gauss (lower bound) |
| 27 kHz and 14 gauss (upper bound) |
| Negative selection |
3 rounds |
3 rounds |
| Special molecular technique |
Single strand ligation reverse transcription |
N/A |
| N20 sequence |
GTTGACGGGCGATTGGTCTT |
CTGCATCACCAAGCCTCGCA |
| Process timeb |
<1 week |
<2 days |
| Yield |
Low |
High |
| Technical difficulty |
Moderate |
Low |
| Waste cost |
Moderate |
Low |
| Kd value (nM) |
23.58 ± 0.82 |
33.34 ± 1.15 |
| Blind sample |
40 |
40 |
| Spearman's rhoc |
q-PCR |
0.985 (before NSSd) |
0.974 |
| 0.987 (after NSS) |
N/A |
| ELISA |
N/A |
0.986 |
Experimental
Oligonucleotide library and primers
A ssDNA 50 nt randomized oligonucleotide library (50N DNA library sequence: 5′-AGCAGCACAGAGGTC-N20-GCGTGCTACCGTGAA-3′) was used as a starting library. The sequences of oligonucleotides consist of a central region of 20 random oligonucleotides flanked by two fixed primer regions at both ends for PCR amplification. One set of primers (50N-LAB-S: 5′-AGCAGCACAGAGGTC-3′; 50N-LAB-AS: 5′-TTCACGGTAGCACGC-3′) was used to anneal the 5′ and 3′ degenerating regions of the oligonucleotides during PCR. A 5′ biotinylated primer (Biotin-50N-LAB-AS) was used to extract forward single-stranded oligonucleotides from the PCR product. All of the oligonucleotides were chemically synthesized at 1 mM with randomized sequences and purified by polyacrylamide gel electrophoresis, and were purchased from MDBio, unless specified otherwise.
CRP and serum protein-coated, bio-functionalized magnetic particles
CRP served as the positive target (P1) in this work (MYBIOSOURCE, San Diego USA). Three human serums served as negative samples (N1, N2, and N3), in which the CRP content was removed by latex particles covered by a hydrophilic shell covalently bonding with anti-CRP monoclonal antibodies (Siemens Health-care Diagnostics, Eschborn, Germany). Forty human serums, in which the CRP content was unremoved, served as blind samples. Magnetic nanoparticles (MNPs) were bio-functionalized by coating streptavidin on the outermost surface and were dispersed in PBS (pH = 7.4) to form a SA-MNP reagent (purchased from Magqu, Taipei, Taiwan). The average hydrodynamic diameter of the streptavidin-coated magnetic nanoparticles (SA-MNPs) in the reagent was 50 nm. The reagent had a concentration of SA-MNPs of 0.3 emu g−1. The biotinylation kit (EZ-Link Sulfo-NHS-Biotinylation Kit) was purchased from Pierce (Rockford, IL, USA). The CRP-MNP (P1), negative serum-MNP (N1: negative serum-MNP-1, N2: negative serum-MNP-2, and N3: negative serum-MNP-3), and blind serum-MNP reagents used in this work were the same as those used in ref. 17 in which the preparation of materials was described. As needed, the CRP-MNPs, negative serum-MNPs, or blind serum-MNPs were obtained from the corresponding reagent through magnetic separation. The collected CRP-MNPs, negative serum-MNPs, or blind serum-MNPs were washed three times with a binding buffer (BD buffer: 50 mM NaH2PO4, pH 8.0, 150 mM NaCl, 5 mM KCl, 2 mM MgCl2, and 0.05% (v/v) Tween-20) and collected with a magnetic stand. All clinical serum samples were stored at −20 °C for further analysis. This study was conducted according to the Declaration of Helsinki principles. Clinical serums were obtained from Chung Shan Medical University Hospital (Taichung, Taiwan) with approval from the medical ethics committee of Chung Shan Medical University Hospital (CS15071), and written informed consent was obtained from all participants. All experiments, procedures, and methods were carried out in accordance with the IRB approved guidelines and regulations.
Window-MARAS selection process to generate smart aptamers
The experimental setup of MARAS is identical to that of ref. 9 and 17. The detailed working principle is described in this section. In order to be able to compare the specificity of selected aptamers with and without primers, and to prevent the affinity from affecting the amount of captured target in the samples, the concept of smart aptamer was adopted. A smart aptamer is defined to have a predetermined dissociation constant (Kd) prior to the selection process.18,19 As reported in ref. 17, the primer-free aptamer with a dissociation constant of 23.58 ± 0.82 nM was generated by RO-MARAS using a rotating magnetic field with frequency of 27 kHz and strength of 14 gauss. To minimize the effect on the specificity evaluation, the potential aptamers with a dissociation constant equal or slightly higher than that of the primer-free counterpart are desired, i.e., Kd ≥ 23.58 ± 0.82 nM. The reason for the decision on the Kd value of the potential aptamer is that the capture amount of target in the clinical samples for the potential aptamer (larger Kd) would be less than that using primer-free counterpart (smaller Kd) as a capture probe. Then, the concentration of target in the clinical samples measured using the potential aptamer departs farther from that using monoclonal antibody-based nephelometry analysis compared with that using the primer-free counterpart, and the specificity evaluation of the current work becomes conservative. Unlike the method of capillary electrophoresis used in ref. 18 and 19 to partition the aptamers with different dissociation constants to obtain a smart aptamer, a window-MARAS procedure was adopted here. The window-MARAS employs a frequency range of the externally applied magnetic field, i.e., a lower bound and an upper bound of magnetic fields. Based on the frequency-dependent characteristic of the Kd value given in ref. 9 and the magnetic field condition used to obtain the primer-free aptamer in ref. 17, a lower bound and an upper bound of magnetic field with frequency of 20 kHz and 27 kHz, respectively, and a constant strength of 14 gauss were proposed to be used to select the smart aptamers having the desired dissociation constant mentioned above. The selection mechanism and physical interpretation of MARAS, which uses externally applied magnetic field coupled with oligonucleotide-target bound magnetic particles to produce the competitive mechanism for aptamer selection, are detailed in ref. 8 and 9 and briefly described here. When a rotating magnetic field is applied, the oligonucleotide-target-magnetic particle bound mixtures undergo rotational or angular oscillating motion due to the action of the magnetic field on the magnetic dipole of the magnetic particles. As the magnetic particle bound mixture in an aqueous solution moves, a dissipative force opposite to the direction of motion is developed. The magnetic driving force and the dissipative force produce a stretch force on all bonds of the bound mixture. The stretch force acting on the oligonucleotide-target bond depends on the magnetic field as well as the size of the outmost portion of the bound mixture, i.e., the size of the oligonucleotide. The smaller the oligonucleotide, the less stretch force is produced. Therefore, a smaller size of oligonucleotide used yields aptamers with lower affinity or larger dissociation constant toward the target. It must be noted that the size of oligonucleotides used in this work (50 nt) differed from that used in ref. 9 (60 nt) and ref. 17 (20 nt). However, the size of oligonucleotides used only slightly affects the affinity of the selected aptamers.9,17 Therefore, the designated frequency range (20 kHz < f < 27 kHz) used in this work is expected to yield the potential aptamers with the desired dissociation constant (Kd ≥ 23.58 ± 0.82 nM).
To proceed with this work, 5 μM randomized oligonucleotide library was used as the starting library and diluted to 10 μl by BD buffer in a micro-tube. The library was heated to 95 °C for 5 minutes and then quickly cooled at 4 °C to form secondary structures, and then stayed at room temperature for 30 minutes. A positive selection was performed by adding CRP-MNPs, obtained from 5 μl of CRP-MNP reagent (P1) by magnetic separation, into the microtube and incubating with the library for 30 minutes at room temperature. The unbound oligonucleotides were removed with a magnetic stand, and the bound mixture was washed twice with 1 ml of BD buffer. 100 μl of BD buffer was added to re-disperse the bound mixture in the microtube. The bound mixture solution was placed inside the MARAS platform and subjected to a rotating magnetic field with a lower bound frequency of 20 kHz (fL) and strength of 14 gauss for 10 minutes. In order to prevent agglomeration, the bound mixture was stirred by pipetting every 2.5 minutes. Magnetic separation was performed to remove supernatant, and the bound mixture was retained. After washing three times with 200 μl BD buffer, the retained bound aptamer–CRP-MNP complexes were re-suspended into 100 μl BD buffer and placed inside the MARAS setup. The complex solution was then subjected to another rotating magnetic field with an upper bound frequency of 27 kHz (fU) with strength of 14 gauss and stirred by pipetting every 2.5 minutes, for 10 minutes. A portion of bound oligonucleotides in the mixtures was detached from the CRP-MNPs due to the stretch force acting on the bond between aptamer and target as the mixtures were driven by the upper bound magnetic field. Magnetic separation was performed to collect the supernatant, which contained detached aptamers, and the remaining mixtures were discarded. Under this selection procedure, the obtained aptamers bound with target-magnetic particles under the lower bound and detached under the upper bound rotating magnetic field. Subsequently, a negative selection procedure was performed by incubating the final supernatant collected from the positive selection with negative serum-MNPs, obtained from 5 μl of negative serum-MNP-1 reagent (N1) by magnetic separation, for 30 minutes at room temperature. After magnetic separation, aptamers bound with negative serum-MNPs were removed. The collected supernatant was then incubated with negative serum-MNP-2 (N2) and negative serum-MNP-3 (N3), obtained from 5 μl of the corresponding negative serum-MNP reagent by magnetic separation, and magnetically separated to remove the bound mixture, sequentially. Finally, the collected supernatant, containing the aptamers which cannot bind with all biomolecules in the negative serums, was purified with a DNA Extraction Miniprep System (Viogene, Taipei, Taiwan) and dispersed in ddH2O for the following cloning procedure.
Cloning and sequencing processes
Aptamers obtained from the window-MARAS were at low concentration; therefore, a PCR amplification was performed with primers (50N-LAB-S and 50N-LAB-AS). The PCR reaction, which contained 1.25 U of DNA polymerase (Invitrogen Life Technologies, Grand Island, NY, USA), 0.1 mM of dNTPs, 0.5 mM of MgSO4, and 0.5 nM primers, was performed under the following condition: 95 °C for 5 minutes; 35 cycles of 95 °C for 50 seconds, 57 °C for 50 seconds and 72 °C for 50 seconds; and elongation at 72 °C for 10 minutes. After PCR amplification, amplified DNA fragments were extracted and purified from PCR reaction using a DNA Extraction Miniprep System. The recovered PCR product was sub-cloned into yT&A-vector (Yeastern Biotech, Taipei, Taiwan) according to the manufacturer's instructions and transformed into DH5α competent cells. The plasmids of randomly picked-up colonies were purified using a High-Speed Plasmid Mini Kit (Geneaid, Taipei, Taiwan). Then, the plasmids were sequenced using an Applied Biosystems PRISM 3730 DNA automatic sequencer and a Big Dye terminator cycle sequencing kit (Applied Biosystems, Foster City, CA, USA). Mfold program was used to predict the secondary structures of the selected aptamers.20
Reverse validation of selected aptamers
To validate the binding specificity of the selected aptamer, 100 nM of the selected aptamer was heated in 20 μl of BD buffer to 95 °C for five minutes and cooled at 4 °C to form secondary structures. The aptamers were individually incubated with P1, N1, N2, and N3 MNPs, which were obtained from 5 μl of corresponding reagents through magnetic separation, for 30 minutes at room temperature. The supernatant was removed through magnetic separation, and the bound mixture was collected. The bound mixture was re-dispersed with 100 μl of ddH2O and then heated to 95 °C for 5 minutes to elute aptamers from the CRP-MNPs (P1) and negative serum-MNPs (N1, N2 and N3). The supernatant containing the eluted aptamer was collected using the magnetic stand. The amount of the aptamers in the collected supernatant was measured by q-PCR and is represented by the relative expression level resulted from q-PCR analysis. The reaction condition was as follows: 95 °C for 3 minutes; 40 cycles at 94 °C for 30 seconds, 60 °C for 30 seconds, and 72 °C for 30 seconds. The mixture for each q-PCR run was 10 μl, containing 5 μl of re-dispersed nucleic acids (from P1, N1, N2, or N3), 2.5 μl of SYBR Green PCR master mix (Applied Biosystems), and 0.5 nM of primers.
Analysis of the equilibrium dissociation constant of selected aptamer by q-PCR
The affinity of aptamers can be represented by the equilibrium dissociation constant (Kd). In this work, the Kd value of selected aptamers was determined by q-PCR.8,9,17,21–23 The process is described briefly as follows: a series of progressively diluted reacting aptamers reagents (1000 nM to 7.8125 nM) in 20 μl of BD buffer were heated to 95 °C for 5 minutes and cooled at 4 °C to form secondary structures. A partial series of diluted aptamers was retained for input control (input). Equal amounts of CRP-MNPs (magnetically separated from 5 μl CRP-MNP reagent) were added into each diluted aptamer tube and incubated for 30 minutes at room temperature. The supernatant was removed with a magnetic stand, and the bound mixture was collected. The bound aptamers were eluted from the MNPs by applying heat at 94 °C for 10 minutes in a final volume of 20 μl of ddH2O. The MNPs were removed through magnetic separation. Both the input controls and eluted aptamers were precipitated with 1 ml of 100% ice-cold ethanol and dissolved in test tubes filled with 100 μl of ddH2O, individually. The quantity of aptamers in each test tube, including the input control tube and eluted aptamer tubes, was calculated by q-PCR. q-PCR was performed using MicroAmp optical 96-well reaction plates, and the threshold cycle (ct) value was calculated automatically using the maximum correlation coefficient approach using StepOnePlus Real-Time PCR Systems software, version 2.0 (Applied Biosystems). The Kd of selected aptamers was measured by quantitating the concentration of the negative control and eluted nucleic acids resulted from q-PCR and fitting the results in a nonlinear regression. 1000 nM of input aptamer concentration was served as the maximum binding, and the Kd values of the selected aptamers were determined by fitting a saturation binding curve based on experimental data through a curve fitting program, Graphpad PRISM 6 (http://www.graphpad.com). Each q-PCR run was performed in duplicate for the determination of dissociation constant to reduce the experimental error, and the Kd value representing dissociation constant was expressed as a mean ± standard deviation from three independent experiments performed.
Development of standard calibration curve by q-PCR
Standard calibration curves were established by using a serial dilution of CRP-MNPs, obtained from 1 μl of CRP-MNP reagent by magnetic separation, in 10 μl BD buffer, of which corresponding CRP concentrations ranging from 4000 to 31.25 ng μl−1. The selected aptamer (1 μM), which has been heated and quenched to form secondary structures, was added and then incubated with CRP-MNPs for 30 minutes at room temperature. The bound mixture was collected and the supernatant was removed with a magnetic stand. The bound aptamers were eluted from the MNPs by heat at 94 °C for 10 minutes in a final volume of 100 μl of ddH2O, and then MNPs were removed through magnetic separation. q-PCR analyses were performed in duplicate, as described above, for each collected supernatant. The CRP concentration, represented by the relative expression level resulted from q-PCR analysis, was determined from the PCR cycle number at which fluorescence intensity reached a set cycle threshold value (ct). The standard calibration curve was linearly fitted from sixteen measured data points to obtain the linear equation and R2 value. The standard calibration curve was used to determine the measured CRP concentrations for the subsequent experiments.
Evaluation of the specificity of selected aptamers by aptamer-based q-PCR analysis
The blind-MNP reagents were prepared as mentioned above and were the same as those used in ref. 17. The concentrations of CRP in blind serum samples were measured by monoclonal antibody-based nephelometry analysis (Siemens Health-care Diagnostics, Eschborn, Germany). The detailed process of aptamer-based q-PCR analysis is described as follows. The selected aptamer (5 μM), CRP-aptamer-2 aptamer synthesized and purchased from MDBio, was heated to 95 °C for five minutes, and cooled at 4 °C to form secondary structures in 20 μl of BD buffer. The reagents containing the aptamers were individually incubated with blind serum-MNPs, obtained from 1 μl of blind serum-MNP reagent through magnetic separation, for 30 minutes at room temperature. The supernatant was removed with a magnetic stand, and the bound mixture was collected and re-dispersed in 100 μl of ddH2O. The bound mixture solutions were heated to 95 °C for 5 minutes to elute aptamers from the serum-MNPs. Magnetic separation was performed to remove MNPs and collect supernatant. CRP concentrations in blind serum samples were determined by q-PCR in duplicate, as previously described through the linear equation of the standard calibration curve. The results of the blind serum samples determined by aptamer-based q-PCR were compared to those determined using monoclonal antibody-based nephelometry analysis.
Verification of the aptamer binding by enzyme-linked immunosorbent assay
An aptamer-based enzyme-linked immunosorbent assay (aptamer-based ELISA) was performed, and the detailed experimental procedure is described as follows: biotinylated CRP-aptamer-2 aptamer was synthesized and purchased from MDBio. A series of progressively dilute pure CRP protein (1000 to 7.8125 ng μl−1 in 100 μl PBS) and forty blind serums (10 μl serum in final 100 μl PBS) were coated in duplicate on an ELISA microplate and stored at 4 °C overnight. All the wells on the ELISA microplate were blocked for two hours at 37 °C with 100 μl PBS with 10% (w/v) bovine serum albumin (BSA, Sigma-Aldrich, St. Louis, MO). After that, 200 μl washing buffer (PBS with 0.05%, Tween-20: PBS-T) was used to wash wells three times to eliminate the unbound mixtures. Biotin-labelled aptamers in BD buffer were heated and cooled to form secondary structures as described previously. For each well, 100 μl of 5 nM biotin-labelled aptamers was overdosed for all binding sites of CRP protein and incubated for 2 hours at room temperature. Wells on the plate were washed three times with 200 μl PBS-T and sequentially incubated with 100 μl streptavidin-HRP (Sigma-Aldrich, Missouri, USA) for 1 hour at room temperature. After washing the plate three times with 200 μl PBS-T, 100 μl 3,3′,5,5′-tetramethyl-benzidine (TMB, Sigma-Aldrich) substrate solution was added to develop colour and then incubated for 20 minutes at room temperature. 100 μl of 2 N HCl was added to complete the reaction, and the absorbance was measured at 405 nm by an EMax precision microplate reader (Molecular Devices, CA, USA). The standard calibration curve was established from the absorbance result of the CRP concentration series. The CRP concentrations of blind samples were determined by ELISA through the linear equation of the standard calibration curve.
Statistical analysis
To determine the correlation of measured results between two assaying methods, Spearman correlation coefficients and associated P values were calculated. Spearman's rho analysis was applied with SPSS software (Version 13.0 SPSS Inc., Chicago, IL), for which P < 0.05 was considered statistically significant. The Bland–Altman plot was used to compare two methods with EXCEL software (Office 2013). We calculated the mean difference between the monoclonal antibody-based nephelometry and two aptamer-based CRP concentration methods, including aptamer-based q-PCR and aptamer-based ELISA. The R–R interval measurements (bias) and the 95% limits of measurement (bias ± 1.96 SE) were analyzed to compare the two methods.
Conclusion
Smart aptamers, with a predetermined affinity and capable of specifically binding to CRP, have been successfully selected using the window-MARAS protocol. The assessment of specificity of aptamers with and without primer in detecting the target concentration in blind serum samples reveals that a multiple negative selection step alone is enough to achieve a satisfactory result. Without going through the primer-free procedure, using libraries with primers can preserve the rapidity and simplicity of MARAS. The result of the aptamer-based ELISA measurement demonstrates that the use of magnetic particles in detection is unnecessary and that the aptamer can be used to replace monoclonal antibody as a detecting probe in immunoassays. Two aptamer-based measurement approaches, including aptamer-based q-PCR and aptamer-based ELISA, have also been developed to unveil the great potential of the aptamer in possible clinical applications.
Author contributions statements
Huan-Hao Li, MS and Ji-Ching Lai, PhD wrote the main text and prepared all figures and tables. Chih-Yung Wen PhD reviewed the related articles. Chin-Yih Hong PhD and Ji-Ching Lai PhD created the initial idea and assembled and submitted this manuscript.
Conflicts of interest
The author(s) declare no competing financial interests.
Acknowledgements
The authors would like to thank the financial support for this research from the Research Grants Council, Hong Kong, under Contract No. GRF PolyU 152168/14E. The authors also wish to thank Mr Yu-Cheng Chang for his theoretical and technical support.
References
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CAS.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1992, 355, 850–852 CrossRef CAS PubMed.
- S. D. Mendonsa and M. T. Bowser, J. Am. Chem. Soc., 2004, 126, 20–21 CrossRef CAS PubMed.
- R. Stoltenburg, C. Reinemann and B. Strehlitz, Anal. Bioanal. Chem., 2005, 383, 83–91 CrossRef CAS PubMed.
- R. Stoltenburg, N. Nikolaus and B. Strehlitz, J. Anal. Methods Chem., 2012, 2012, 415697 Search PubMed.
- A. Nitsche, A. Kurth, A. Dunkhorst, O. Pänke, H. Sielaff, W. Junge, D. Muth, F. Scheller, W. Stöcklein, C. Dahmen, G. Pauli and A. Kage, BMC Biotechnol., 2007, 7, 48–60 CrossRef PubMed.
- J. C. Lai and C. Y. Hong, J. Mater. Chem. B, 2014, 2, 4114–4121 RSC.
- J. C. Lai and C. Y. Hong, ACS Comb. Sci., 2014, 16, 321–327 CAS.
- W. Pan and G. A. Clawson, Molecules, 2009, 14, 1353–1369 CrossRef CAS PubMed.
- J. D. Wen and D. M. Gray, Biochemistry, 2004, 43, 2622–2634 CrossRef CAS PubMed.
- C. Boiziau and J. J. Toulme, Antisense Nucleic Acid Drug Dev., 2011, 11, 379–385 CrossRef PubMed.
- A. Vater, F. Jarosch, K. Buchner and S. Klussmann, Nucleic Acids Res., 2003, 31, e130 CrossRef PubMed.
- F. Jarosch, K. Buchner and S. Klussmann, Nucleic Acids Res., 2006, 34, e8 CrossRef PubMed.
- W. Pan and G. A. Clawson, Methods Mol. Biol., 2010, 629, 369–385 Search PubMed.
- W. Pan and G. A. Clawson, BioTechniques, 2008, 44, 351–360 CrossRef CAS PubMed.
- S. M. Tsao, J. C. Lai, H. E. Horng, T. C. Liu and C. Y. Hong, Sci. Rep., 2017, 7, 45478 CrossRef CAS PubMed.
- A. P. Drabovich, M. Berezovski and S. N. Krylov, J. Am. Chem. Soc., 2005, 127, 11224–11225 CrossRef CAS PubMed.
- A. P. Drabovich, M. Berezovski, V. Okhonin and S. N. Krylov, Anal. Chem., 2006, 78, 3171–3178 CrossRef CAS PubMed.
- M. Zuker, Nucleic Acids Res., 2003, 31, 3406–3415 CrossRef CAS PubMed.
- D. F. Bibby, A. C. Gill, L. Kirby, C. F. Farquhar, M. E. Bruce and J. A. Garson, J. Virol. Methods, 2008, 151, 107–115 CrossRef CAS PubMed.
- L. A. Jones, L. E. Clancy, W. D. Rawlinson and P. A. White, Antimicrob. Agents Chemother., 2006, 50, 3019–3027 CrossRef CAS PubMed.
- L. Yang and A. D. Ellington, Anal. Biochem., 2008, 380, 164–173 CrossRef CAS PubMed.
- J. M. Bland and D. G. Altman, J. Biopharm. Stat., 2007, 17, 571–582 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07249j |
| ‡ Huan-Hao Li and Chih-Yung Wen contributed equally to this work, and both should be considered as first authors. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Chih-Yung Wen‡a,
Chin-Yih Hongb and
Ji-Ching Lai
a,
Chih-Yung Wen‡a,
Chin-Yih Hongb and
Ji-Ching Lai