
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of 1,2-disubstituted benzimidazoles using an aza-Wittig-equivalent process†
Yuan Chen,
Fanghui Xu and
Zhihua Sun *
*
College of Chemistry and Chemical Engineering, Shanghai University of Engineering Science, 333 Longteng Road, Shanghai 201620, China. E-mail: sungaris@gmail.com
First published on 15th September 2017
Abstract
A synthetic approach for 1,2-disubstituted benzimidazoles has been successfully designed based on effective C–N bond construction, which demonstrated mild reaction conditions and excellent yields. The method involves treating derivatives of o-phenylenediamine with tert-butanesulfoxide and NBS under acidic conditions, which undergoes an aza-Wittig-equivalent process to afford the desired products. Using this method, a series of benzimidazoles containing multiple functional groups with varying electronic effects have been successfully constructed.
Introduction
In recent years, molecules based on 1,2-disubstituted benzimidazole have attracted extensive attention due to their wide applications in new drugs, including antihypertensive drugs, GABAA receptor agonists and the hepatitis C virus (HCV) NS5B polymerase inhibitors.1–3 We have witnessed great progress in the development of benzimidazole derivatives in recent decades. One of the key improvements is the effective synthesis of these functional benzimidazole compounds. The reported methods for the synthesis of 1,2-disubstituted benzimidazoles mostly include condensation of anilines with aldehydes under relatively harsh conditions (Scheme 1a),4 direct oxidative coupling of amines to imines (Scheme 1b)5 and transition metal-catalyzed intramolecular cyclization (Scheme 1c).6 However, the reported methods mostly employ harsh and strict conditions or expensive catalysts. Considering that harsh conditions could possibly cause decomposition of substrates or products, it is significant to explore milder conditions for the synthesis of 1,2-disubstituted benzimidazoles.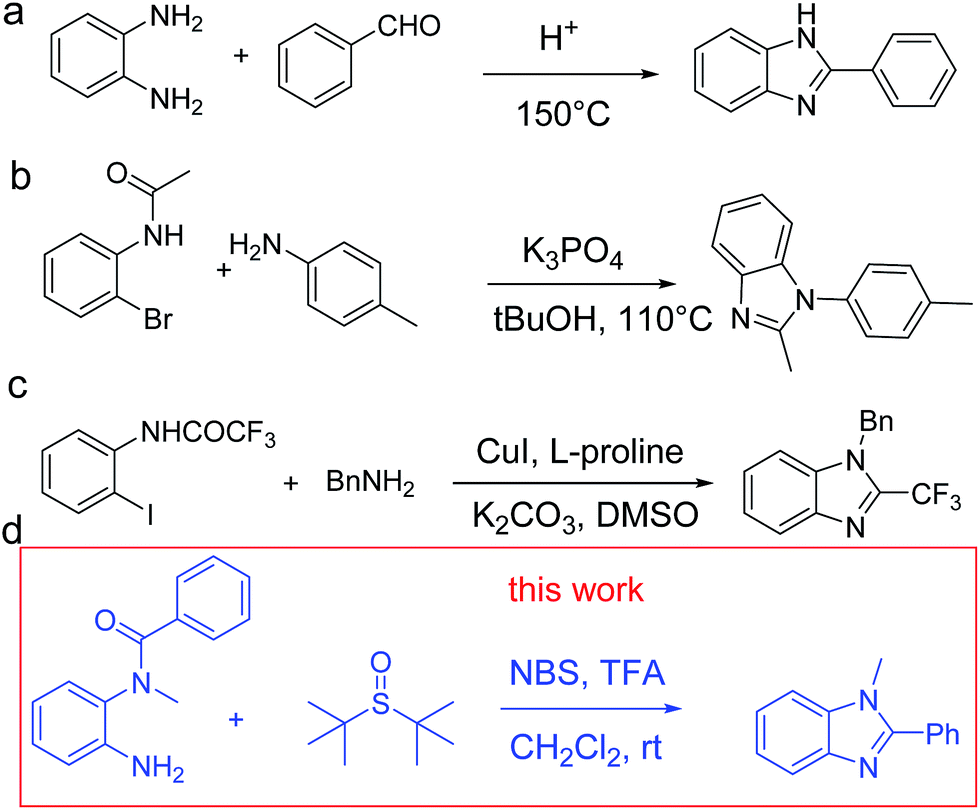 | ||
| Scheme 1 The synthetic methods of 1,2-disubstituted benzimidazoles. | ||
The synthesis of benzimidazoles involves the construction of C–N bond, an important class of transformation in organic synthesis.7,8 Of all C–N bond forming reactions, Schiff base represents an attractive and robust approach to the synthesis of nitrogen-containing compounds.9,10 However, in some cases, the activity of Schiff base reactions is not high enough. In this regard, assistant groups are introduced to enhance the activity.
Our group recently successfully established an effective C–S functionalization strategy, and various useful Wittig-like processes have been developed based on these investigations. For example, the synthesis of 3-substituted aryl4,5 isothiazoles through an all-heteroatom Wittig-equivalent process offers a novel approach for isothiazoles.11 Based on this discovery, we were interested in investigating the possibility to construct C–N bond in order to synthesize substituted benzimidazoles with the help of an aza-Wittig-equivalent process.12 In this respect, the reaction presents extremely mild conditions compared with the reported methods. In this work, we use derivatives of o-phenylenediamine with the help of tert-butanesulfoxide and NBS in acid conditions to synthesize 1,2-disubstituted benzimidazoles, which demonstrates a novel synthetic route for functional benzimidazoles.
Results and discussion
The reaction details are outlined in Table 1. Our research initially began with the reaction of o-phenylenediamine derivatives 1 using tert-butanesulfoxide and NBS with the catalysis of TFA in CH2Cl2. After 1 h, the conversion of the starting material (1) was up to 95% and the product 1,2-disubstituted benzimidazole (2) was obtained in a 84% yield (entry 1). By prolonging the reaction time to 4 h, the conversion would be close to 100%, and the yield could slightly increase. To optimize the reaction conditions, we firstly screened the solvents (entry 1–5). The results demonstrated that CH2Cl2 is the best solvent. As shown in Table 1, other polar or non-polar solvents are not optimal for this reaction. When THF, toluene and DMF were introduced as the reaction media, the yields dramatically decreased, reagent ratio (1/addition/acid = 1/2/1.5).
|
||||
|---|---|---|---|---|
| Entry | Acid | Solvent | N-Halogen succinamide | Yield 5a (%) |
| a The reaction was treated at −20 °C.b The reaction was treated at 0 °C.c The reaction was treated at 40 °C. | ||||
| 1 | TFA | CH2Cl2 | NBS | 84 |
| 2 | TFA | THF | NBS | 52 |
| 3 | TFA | Tol | NBS | 34 |
| 4 | TFA | DMF | NBS | 60 |
| 5 | TFA | DMSO | NBS | 25 |
| 6 | PTSA | CH2Cl2 | NBS | 64 |
| 7 | AcOH | CH2Cl2 | NBS | 43 |
| 8 | PhCOOH | CH2Cl2 | NBS | 48 |
| 9 | CCl3COOH | CH2Cl2 | NBS | 72 |
| 10 | TFA | CH2Cl2 | NCS | 66 |
| 11 | TFA | CH2Cl2 | NIS | 75 |
| 12a | TFA | CH2Cl2 | NBS | 36 |
| 13b | TFA | CH2Cl2 | NBS | 50 |
| 14c | TFA | CH2Cl2 | NBS | 68 |
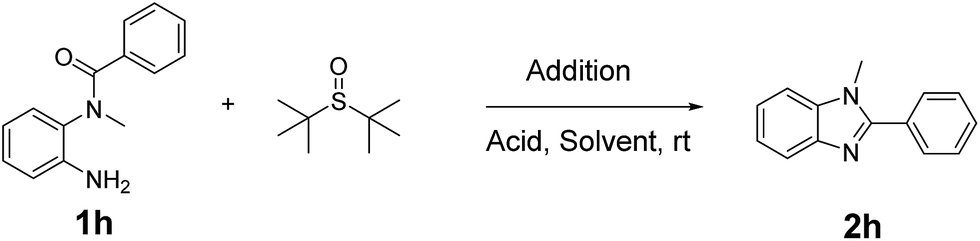
To further improve the reaction efficiency, we examined the influences of the acids in this reaction (entry 6–9). The results indicated that TFA with the appropriate acidity shows the best catalysis, while stronger acid or weaker acid would decrease the yields. It is possible that weaker acids are not suitable to form the sulfinyl halide, while stronger acids would decompose the intermediates. Furthermore, the N-halogen succinamide in this reaction could be NBS, NCS or NIS (entry 10–11), which helps to convert tert-butanesulfoxide to sulfinyl halogen. The results indicated that NBS is the best reagent while NCS and NIS could also play a similar role with the slightly decreased yields. Moreover, when the reaction was carried out at −20 °C, 0 °C or 40 °C, respectively, all yields would decrease compared with that at 25 °C (entry 12–14).
According to our previous works, as shown in Scheme 2, a possible mechanism is based on the aza-Wittig-equivalent process. In the beginning, tert-butanesulfoxide reacted with NBS to afford tert-butanesulfinic bromine (3). Subsequently, tert-butanesulfinic bromine reacted with the substrate (1) to form tert-butanesulfinamide (4). Then the sulfinyl amide attacks the carbonyl group to form the intermediate (5). In the end, by intramolecular rearrangements and charge transfer, accompanied by the elimination of tert-butanesulfinic acid, 1,2-disubstituted benzimidazole (2) can be formed. Consistent with this mechanism, the intermediate (4) could be detected by NMR and HRMS (see ESI†). From the mechanism, we can clearly see that one of the key steps is the formation and stability of intermediate (5). When the substrate is less reactive to form 5 or 5 is more inclined to decomposition rather than rearrangements, the product (2) could not be successfully synthesized. It is worth to note that by treating intermediate (4) in the optimized condition, the yield of benzimidazole could be effectively enhanced.
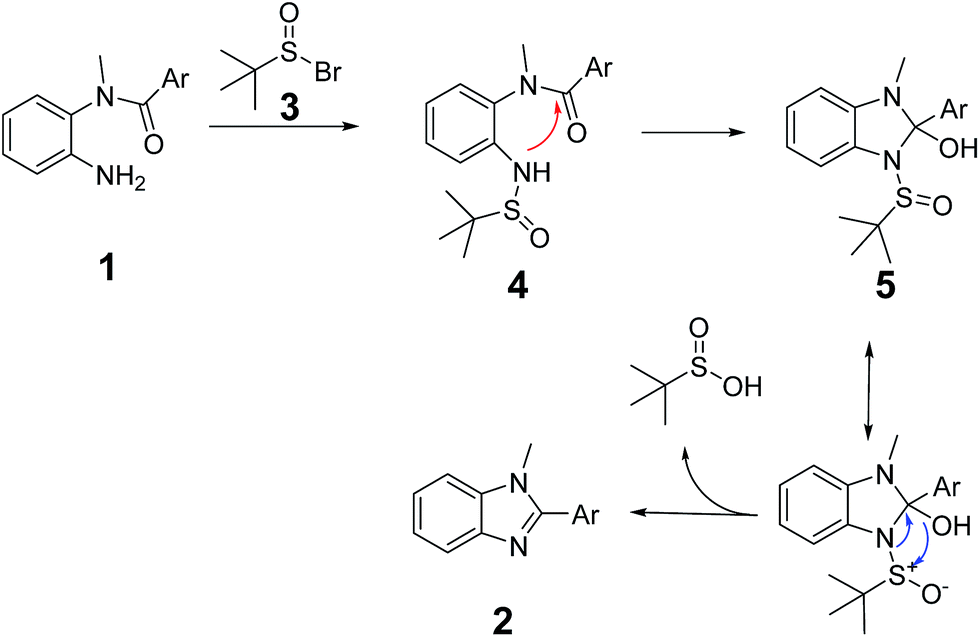 | ||
| Scheme 2 The possible mechanism of forming benzimidazole. | ||
With the optimized reaction conditions in hand, a collection of starting materials (1) bearing various functional or electronic effecting groups were subjected to the NBS/TFA/CH2Cl2 protocol to produce 2. As shown in Scheme 3, as expected, when R1 and R2 are alky or alkoxy groups (2a–2d), the reactions proceed smoothly and the yields are satisfying (73–80%). It is worthy to note that for 2a, we obtained the dibromo substituted product, which is possibly related to the high activity of 1a.
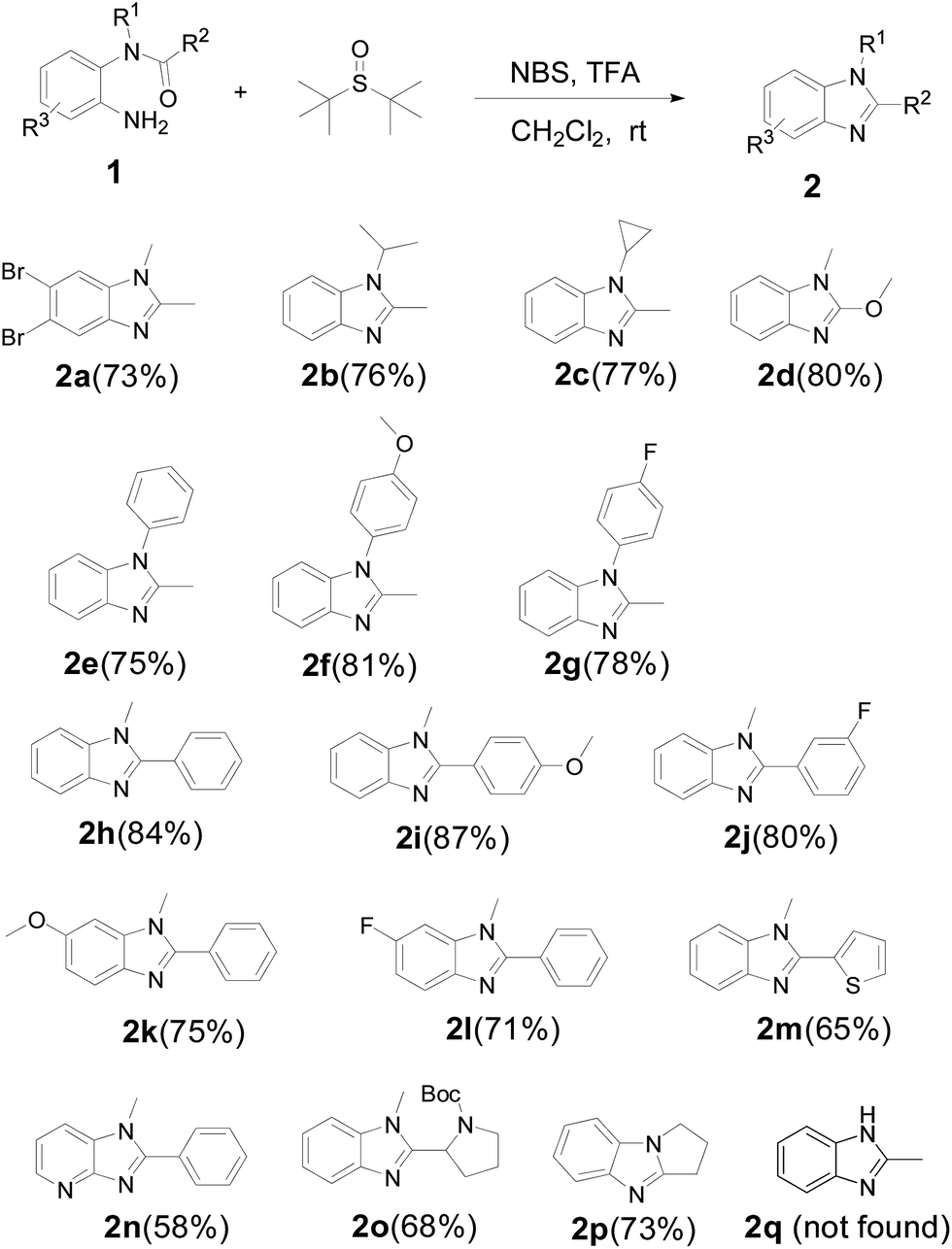 | ||
| Scheme 3 Additional examples of 1,2-disubstituted benzimidazoles synthesis. Reaction conditions: a mixture of 1 (0.5 mmol), NBS (1.0 mmol), TFA (0.75 mmol), and CH2Cl2 (5 mL) was stirred at room temperature for 2 h under a nitrogen atmosphere. | ||
Furthermore, substrates with substituent (R1), regardless of an electron-withdrawing or electron-donating group, could react smoothly to afford the desired product in high yields (75–81%) (2e–2g). This reaction also tolerated a range of electronic effect groups in the substituent (R2) (80–87%), which enabled potential applications of the final products in further functionalization (2h–2j). The substituents (R3) on aromatic ring present few influence with the reaction (2k, 2l), and the yields are excellent. On the other hand, apart from introducing benzene units to benzimidazoles, we have successfully synthesized heterocycle fused products (2m–2o). However, the yields are slightly lower than the benzo analogues. Interestingly, the fused ring product was also obtained in a good yield (2p), which affords a new route to synthesize novel benzimidazoles. In comparison, 2k, 2l and 2n could also be obtained without using the aza-Wittig process, but the yields were dramatically decreased (see ESI†), indicating that our new aza-Wittig protocol may be a very useful complement to the existing methods of benzimidazole synthesis. However, N-mono-substituted amide could not be transformed to corresponding benzimidazole (2q).
Experimental
Materials and instrumentation
All the reactions were performed in oven-dried reaction vessels under N2. Commercially available solvents and reagents were used without further purification unless otherwise mentioned. CH2Cl2, THF, toluene, DMF, DMSO were dried before using. Thin-layer chromatography (TLC) was carried out on aluminum sheets coated with silica gel 60 F254 (MERCK). 1H NMR spectra were obtained using a Bruker AM 400 spectrometer, and chemical shifts were reported relative to DMSO-d6 (δ = 2.52), MeOD (δ = 3.34) and CDCl3 (δ = 7.26) in ppm. 13C NMR spectra were recorded at 100 MHz (Bruker AM 400) and chemical shifts were reported relative to DMSO-d6 (δ = 40.4), MeOD (δ = 49.8) and CDCl3 (δ = 77.00) in ppm. The characterization data of 2b, 2c, 2d, 2e, 2f, 2g, 2h, 2i, 2j, 2k, 2m, 2n, 2p could be found in the previous literature.13General procedure for 2a–2o
In a 25 mL vial along with a stirring bar, to a mixture of tert-butanesulfoxide (1.0 mmol), TFA (0.75 mmol) and NBS (1.0 mmol) in CH2Cl2 (5 mL) under N2. A CH2Cl2 (5 mL) solution of 1 (0.5 mmol) was added to the mixture and the reaction was stirred at RT for additional 1 h. The reaction mixture was washed with water; dried over anhydrous Na2SO4 and evaporated in vacuo. The residue was purified by silica-gel column chromatography with CH2Cl2/PE as an eluent. Recrystallization from CH2Cl2/hexane gave the white solid 2.Synthesis of intermediate 4
In a 25 mL vial along with a stirring bar, to a mixture of tert-butanesulfoxide (1.0 mmol), TFA (0.75 mmol) and NBS (1.0 mmol) in CH2Cl2 (5 mL) under N2. A CH2Cl2 (5 mL) solution of 1h (0.5 mmol) was added to the mixture and the reaction was stirred at RT for additional 10 min. The reaction mixture was washed with water; dried over anhydrous Na2SO4 and evaporated in vacuo. The residue was purified by silica-gel column chromatography with CH2Cl2/PE as an eluent. Recrystallization from CH2Cl2/hexane gave the white solid 4.Conclusions
In summary, by treating the derivatives of o-phenylenediamine with the help of tert-butanesulfoxide and NBS in the acid condition, we have demonstrated an effectively intramolecular cyclization sequence to construct substituted benzimidazoles containing various alky or aromatic groups, which reveals a new approach for aza-Wittig-equivalent process. This synthetic method involves mild conditions instead of reported harsh conditions and the yields are excellent. Furthermore, a series of substituted benzimidazoles containing diverse electronic effect groups have been successfully obtained based on this method. Further work towards expanding the use of photoredox catalysis in the construction of heterocyclic products is underway.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors are grateful for financial support from the Science and Technology Commission of Shanghai Municipality (17ZR1412000).Notes and references
-
(a) Y. Li, M. Kataoka, M. Tatsuta, K. Yasoshima, T. Yura, K. Urbahns, A. Kiba, N. Yamamoto, J. B. Gupta and K. Hashimoto, Bioorg. Med. Chem. Lett., 2005, 15, 805 CrossRef CAS PubMed
; (b) M. Sabat, J. C. Vanrens, M. J. Laufersweiler, T. A. Brugel, J. Maier, A. Golebiowski, B. De, V. Easwaran, L. C. Hsieh, R. L. Walter, M. J. Mekel, A. Evdokimov and M. J. Janusz, Bioorg. Med. Chem. Lett., 2006, 16, 5973 CrossRef CAS PubMed
-
(a) J. L. Falco, M. Pique, M. Gonzalez, I. Buira, E. Mendez, J. Terencio, C. Perez, M. Princep, A. Palomer and A. Guglietta, Eur. J. Med. Chem., 2006, 41, 985 CrossRef CAS PubMed
-
(a) R. B. Baudy, H. Fletcher III, J. P. Yardley, M. M. Zaleska, D. R. Bramlett, R. P. Tasse, D. M. Kowal, A. H. Katz, J. A. Moyer and M. Abou-Gharbia, J. Med. Chem., 2001, 44, 1516 CrossRef CAS PubMed
- D. N. Kommi, P. S. Jadhavar, D. Kumar and A. K. Chakraborti, Green Chem., 2013, 15, 798 RSC
-
(a) T. B. Nguyen, L. Ermolenko and A. Mourabit, Green Chem., 2013, 15, 2713 RSC
-
(a) J. E. R. Sadig, R. Foster, F. Wakenhut and M. C. Willis, J. Org. Chem., 2012, 77, 9473 CrossRef CAS PubMed
-
(a) M. A. Koch, A. Schuffenhauer, M. Scheck, S. Wetzel, M. Casaulta, A. Odermatt, P. Ertl and H. Waldmann, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17272 CrossRef CAS PubMed
-
(a) R. Hili and A. K. Yudin, Nat. Chem. Biol., 2006, 2, 284 CrossRef CAS PubMed
-
(a) F. Faridbod, M. R. Ganjali, R. Dinarvand, P. Norouzi and S. Riahi, Sensors, 2008, 8, 1645 CrossRef CAS PubMed
-
(a) L. Wang, W. K. Wong, L. Wu and Z. Y. Li, Chem. Lett., 2005, 34, 934 CrossRef CAS
-
(a) F. Xu, Y. Chen, E. Fan and Z. Sun, Org. Lett., 2016, 18, 2777 CrossRef CAS PubMed
-
(a) H. Huang, X. Ji, W. Wu, L. Huang and H. Jiang, J. Org. Chem., 2013, 78, 3774 CrossRef CAS PubMed
-
(a) R. Infante-Castillo and S. P. Hernandez-Rivera, Adv. Chem. Res., 2012, 11, 159 CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09010b |
| This journal is © The Royal Society of Chemistry 2017 |