
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Variable self-assembly and in situ host–guest reaction of beta-cyclodextrin-modified graphene oxide composite Langmuir films with azobenzene compounds†
Yagui Gaoab,
Tifeng Jiao *ab,
Kai Mab,
Ruirui Xingbc,
Lexin Zhangb,
Jingxin Zhou*b and
Qiuming Penga
*ab,
Kai Mab,
Ruirui Xingbc,
Lexin Zhangb,
Jingxin Zhou*b and
Qiuming Penga
aState Key Laboratory of Metastable Materials Science and Technology, Yanshan University, Qinhuangdao 066004, P. R. China. E-mail: tfjiao@ysu.edu.cn
bHebei Key Laboratory of Applied Chemistry, School of Environmental and Chemical Engineering, Yanshan University, Qinhuangdao 066004, P. R. China. E-mail: zhoujingxin@ysu.edu.cn
cState Key Laboratory of Biochemical Engineering, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, P. R. China
First published on 22nd August 2017
Abstract
The preparation of graphene oxide (GO)-based composite Langmuir films play important roles in the application of self-assembly nanomaterials. In this work, beta-cyclodextrin-modified graphene oxide (GO–CD) and the azobenzene-modified PAA derivative (abbreviated as PAA-Azo) have been designed and synthesized. The interfacial assemblies and host–guest interactions of GO–CD material with used two azobenzene compounds (N-Azo and PAA-Azo) in the obtained Langmuir and Langmuir–Blodgett (LB) films were investigated. All the spreading films from different subphases were transferred onto solid substrates and their nanoscale morphologies as well as self-assembled characteristics were investigated by some methods including atomic force microscopy, UV-vis spectra, Raman spectra, etc. The obtained results indicated that there existed host–guest interaction between beta-CD segments with azobenzene groups. Besides, in the obtained GO–CD/N-Azo films, there also appeared J-aggregate of N-Azo molecules on GO sheet surface. However, there were only host–guest interactions without any aggregates of azobenzene segment in the GO–CD/PAA-Azo composite films. Hence, the presently obtained GO–CD/azobenzene composite Langmuir films provide new clues to develop GO-based self-assembled films as well as nanomaterials towards nano-device and sensor applications.
1. Introduction
Graphene oxide (GO)-based composites have been shown to be particularly attractive over recent years, owing to their special electronic, mechanical, and thermal properties.1–3 In recent years, more attention has been devoted to reasonable design and targeted synthesis of various functionalized GO-based nanocomposites, demonstrating better processability and adaptation for nanomaterial applications.4–6 The functionalized modification of GO materials with organic groups are needed for the purposes of good dispersion and variable applications.7–9 For example, porphyrin, dopamine, isocyanate derivatives, long-chain alkylamine or tetrathiafulvalene, have been used to decorate GO to obtain a well dispersed state in solvents for designed properties.10–14 Compared with the above used small molecules, some polymers, such as poly(2-(dimethylamino)ethyl methacrylate) and poly(vinyl alcohol), can be also employed to modify GO via different chemical reactions to enhance the properties of GO-based composites in obvious domains.15,16 However, some problems still appears, such as incomplete adsorption of organic molecules, aggregation between GO sheets, and undesired side effects. On the other hand, host–guest chemistry of cyclodextrin has drawn important development in recent years owing to easy modification, high selectivity and yields.17–20 For example, Li and coworkers have achieved numerous works about design and application of beta-CD functionalized carbon materials in guest enrichment or sensors.21–28 It is reported that the a host–guest reaction has been displayed to functionalize GO with cyclodextrin-based [2]rotaxane array for the development of GO-based photoswitches and cyclodextrin functionalized graphene–gold nanoparticle hybrids for electrochemical thrombin aptasensor.29,30 Therefore, it seems an interesting challenge to directly prepare GO-based composites with special host–guest reaction in a self-assembled systems.On the other hand, Langmuir and Langmuir–Blodgett (LB) techniques can be regarded as effective techniques to synthesize interfacial thin films.31–38 Since the initial reports from Huang's group about GO self-assembly process in Langmuir films,39,40 several studies show the preparation of GO monolayer or GO-based composite films via LB assembly method.41–47 Up to now, the inclusion complex formation of azobenzene-containing compounds and beta-cyclodextrin (beta-CD)/alpha-cyclodextrin at the air/water interface was investigated and characterized.48–52 It can be interesting that a combination of GO-based composites self-assembly films involved in host–guest interactions via beta-CD and azobenzene segments by Langmuir technique should be particularly advantageous due to excellent molecular recognition, moderate self-assembled nanostructures, and designed self-assembly models. Thus, aqueous solutions of two soluble azobenzene-containing compounds (abbreviated as N-Azo and PAA-Azo) as subphase solutions have been designed and chosen for the preparation of composite films.
In this article, we report the preparation of stable GO–CD composite Langmuir films by interfacial host–guest interactions with azobenzene compounds in subphase solutions in a facile and effective manner. We discover that self-assembly and host–guest interactions between beta-CD groups modified GO sheets and soluble azobenzene-containing small molecules or polymeric molecules are responsible for formation of present composite films. Also, the as-prepared Langmuir films can be easily transferred onto solid substrates via Langmuir–Blodgett (LB) assembly technique and characterized by morphological and spectral methods. Thus, the present GO-based composite Langmuir films will provide significant potential applications in nano-device and biosensor design.
2. Materials and methods
2.1 Materials
The experimental used materials, beta-cyclodextrin (98%, abbreviated as beta-CD), ethylenediamine (99.5%), chloroacetic acid, 4-aminoazobenzene (abbreviated as N-Azo), and poly(acrylic acid) (abbreviated as PAA, M.W. ∼ 450![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) were purchased from Aladdin Reagent (Shanghai, China), Alfa Aesar (Tianjin, China) Chemicals as well as TCI Shanghai Chemicals, and used without further purification. Graphite powder (325 mesh, 99%) was obtained from Alfa Aesar Chemicals (Shanghai, China). N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC-HCl) and N-hydroxysuccinimide (NHS) were obtained from Sigma-Aldrich without any purification. Other reagents were purchased from Beijing Chemicals and used without any purification. All used water was purified in a double-stage Millipore Milli-Q Plus purification system. Firstly, graphene oxide (GO) was synthesized from graphite powder by a modified Hummers method.53 Then, the carboxyl-modified GO (GO-COOH) was synthesized according to the reported literature,54,55 and lyophilized at a temperature of −50 °C. Amine-modified beta-CD derivative (abbreviated as NH2-beta-CD) were synthesized according to the literature reports,56,57 and the molecular structure (Fig. S1†) and purity was confirmed by 1H NMR and 13C NMR (Fig. S2 and S3†) as well as MS (Fig. S4†). Beta-CD functionalization on GO-COOH was finished following analogous dehydration reaction procedure described in the literatures,58,59 and modified in similar procedure. By filtration and dialysis process, the resulting beta-CD-modified GO solid (GO–CD) was obtained by freeze-drying. The azobenzene-modified PAA derivative (abbreviated as PAA-Azo) was prepared as described in the following dehydration reaction. The 100 mL aqueous PAA solution (10 mg mL−1) was mixed with NHS (3.4 g) and EDC-HCl (5.7 g) into a round-flask under ice-bath. After stirring vigorously for 2 h, N-Azo (137 mg, ca. 5 mol% of acrylic acid unit) was added to the mixture and left stirring overnight under ice-bath and next 3 days at room temperature. After that the solid was dispersed with water and dialyzed in water for 4–5 days with dialysis tubing (MWCO 12400). After dialysis, PAA-Azo powder was obtained by freeze-drying. The characteristic molecular structure was confirmed by XRD and IR spectra (Fig. S9 and S10†) as well as elemental analysis (yield 78%, found: C, 48.21; H, 6.15; N, 6.52; calculated PAA, (C3H4O2)n, C, 50.00; H, 5.59; O, 44.40).
000) were purchased from Aladdin Reagent (Shanghai, China), Alfa Aesar (Tianjin, China) Chemicals as well as TCI Shanghai Chemicals, and used without further purification. Graphite powder (325 mesh, 99%) was obtained from Alfa Aesar Chemicals (Shanghai, China). N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC-HCl) and N-hydroxysuccinimide (NHS) were obtained from Sigma-Aldrich without any purification. Other reagents were purchased from Beijing Chemicals and used without any purification. All used water was purified in a double-stage Millipore Milli-Q Plus purification system. Firstly, graphene oxide (GO) was synthesized from graphite powder by a modified Hummers method.53 Then, the carboxyl-modified GO (GO-COOH) was synthesized according to the reported literature,54,55 and lyophilized at a temperature of −50 °C. Amine-modified beta-CD derivative (abbreviated as NH2-beta-CD) were synthesized according to the literature reports,56,57 and the molecular structure (Fig. S1†) and purity was confirmed by 1H NMR and 13C NMR (Fig. S2 and S3†) as well as MS (Fig. S4†). Beta-CD functionalization on GO-COOH was finished following analogous dehydration reaction procedure described in the literatures,58,59 and modified in similar procedure. By filtration and dialysis process, the resulting beta-CD-modified GO solid (GO–CD) was obtained by freeze-drying. The azobenzene-modified PAA derivative (abbreviated as PAA-Azo) was prepared as described in the following dehydration reaction. The 100 mL aqueous PAA solution (10 mg mL−1) was mixed with NHS (3.4 g) and EDC-HCl (5.7 g) into a round-flask under ice-bath. After stirring vigorously for 2 h, N-Azo (137 mg, ca. 5 mol% of acrylic acid unit) was added to the mixture and left stirring overnight under ice-bath and next 3 days at room temperature. After that the solid was dispersed with water and dialyzed in water for 4–5 days with dialysis tubing (MWCO 12400). After dialysis, PAA-Azo powder was obtained by freeze-drying. The characteristic molecular structure was confirmed by XRD and IR spectra (Fig. S9 and S10†) as well as elemental analysis (yield 78%, found: C, 48.21; H, 6.15; N, 6.52; calculated PAA, (C3H4O2)n, C, 50.00; H, 5.59; O, 44.40).
2.2 Procedure of LB films preparation
The interfacial preparation and LB films transfer were finished via KSV-NIMA Mini-trough LB system. The trough was carefully cleaned with chloroform and ethanol, and then filled with DI pure water, aqueous N-Azo solution (50 mg L−1), or PAA-Azo solution (25 mg L−1), respectively. After sonication 20 min, GO–CD dispersed solution (0.500 mg mL−1) with different spread solvents was dropwise spread onto the water surface using a glass syringe. Surface pressure was monitored using a tensiometer attached to a Wilhelmy plate. The film was compressed by barriers at a speed of 10 cm2 min−1 at room temperature. The prepared GO–CD monolayer was transferred to solid substrates at suitable pressure points by vertically pulling method with speed of 2 mm min−1. Fresh cleaved mica, glass plates, quartz, and CaF2 were used as the substrates to transfer monolayer or multilayer for next morphological and spectral characterizations. Quartz and glass plates were treated with 1:1:5 NH4OH:H2O2:H2O (by volume) and washed repeatedly with deionized water before use.
2.3 Characterization
The morphology of composite Langmuir films were characterized using a field-emission scanning electron microscope (SEM) (S-4800II, Hitachi, Japan) as well as a transmission electron microscope (TEM) (HT7700, Hitachi High-Technologies Corporation, Japan). Atomic force microscopy (AFM) pictures were measured with a Nanoscope model Multimode 8 Scanning Probe Microscope (Veeco Instrument, USA) with silicon cantilever probes using the tapping mode. UV-vis spectra were recorded with a Shimadzu UV-2550 system (Shimadzu Corporation, Japan). FT-IR spectra were obtained using a Fourier infrared spectroscopy (Thermo Nicolet Corporation) by the conventional KBr disk tablet method or composite films on CaF2 plates. Raman spectroscopy was measured by a Horiba Jobin Yvon Xplora PLUS confocal Raman microscope equipped with a motorized sample stage. The intensity of a Raman peak was extracted from the maximum value after baseline subtraction over corresponding spectral range. X-ray photoelectron spectroscopy (XPS) was measured by the Thermo Scientific ESCALab 250Xi using 200 W monochromated Al Ká radiation. Both survey scans and individual high-resolution scans for N(1s), O(1s) and C(1s) peaks were recorded. X-ray diffraction study was carried out by using an X-ray diffractometer (SMART LAB, Rigaku) equipped with a conventional Cu Kα X-ray radiation (λ = 1.54 Å) source and a Bragg diffraction setup. Circular dichroism (CD) spectra of the transferred LB films were recorded on a JASCO J-810 CD spectrometer. In the process of measuring CD spectra, the multilayer film was placed perpendicular to the light path and rotated within the film plane to avoid polarization-dependent reflections and eliminate the possible angle dependence of the CD signals.60–65 Elemental analysis was carried out with a Flash EA Carlo-Erba-1106 Thermo-Quest. 1H NMR and 13C NMR spectra were obtained on an ARX400 (Bruker, Fällanden, Switzerland) NMR spectrometer in D2O or d-DMSO with TMS as an internal standard. MALDI-TOF mass spectra (MS) were determined with BIFLEXIII in methanol solution. A UV lamp with wavelength of 365 nm (LUYOR-3405, 20 mW cm−2, LUYOR Corporation) was utilized to irradiate the obtained solutions and LB films to induce the photoisomerization reactions.3. Results and discussion
The obtained beta-CD-functionalized GO–CD material can act as building blocks to prepare Langmuir films via interfacial host–guest interactions with azobenzene-containing molecules by standard Langmuir self-assembly technique. For this, the prepared GO precursor, the intermediates GO-COOH and NH2-beta-CD, and the targeted building block GO–CD were synthesized and characterized by many methods. Firstly, the SEM and TEM images of the obtained GO (Fig. S5a and b†) show well-dispersed ultrathin GO sheet with micrometer scale size. In addition, after modification with beta-CD groups, the SEM images of as-prepared GO–CD powder (Fig. S5c and d†) demonstrate aggregated state of GO sheet due to possible hydrogen bonding between modified CD segments. And XRD curves of the as-prepared materials show the disappearance of Bragg peaks at 2θ value of 11.3° owing to the (001) diffraction peak and presence of new broad peak at 2θ value of 19.8° for GO–CD (Fig. S6†). Moreover, the FT-IR spectra of the as-prepared GO–CD powder and original materials were measured and shown in Fig. S7.† It can be easily observed that the characteristic peak of GO sheet appeared at 3424 cm−1 can be attributed to the –OH vibration stretching, while other obvious bands demonstrated carboxyl group at 1724 cm−1, epoxy group at 1226 cm−1, and alkoxy group at 1050 cm−1, respectively.66–70 In addition, vibrational peaks of NH2-beta-CD at 2927 cm−1 can be assigned to C–H vibration stretching, with other peaks at 946 and 756 cm−1 for pyranose ring in CD components. Simultaneously, characteristic peaks corresponding to amide–carbonyl (NH–CO) stretching vibration at 1650 cm−1, C–N stretching vibration at 1384 cm−1 suggest that the GO-COOH material was modified with NH2-beta-CD via amide bond. Thus, the obtained FT-IR data indicate successful modification of beta-CD groups in GO sheet and possible 3D structural composite formation due to chemical functionalization.59 The elemental analysis data (Table 1) of as-prepared GO-modified materials demonstrate obvious increment of hydrogen composition and appearance of nitrogen component with value of 3.91 wt% for obtained GO–CD material. The comparison of XPS data and elemental analysis data show similar calculated N/C ratios, which is almost in agreement with the above obtained results. Thus, we characterized the interfacial phase behaviors and the transferred GO–CD composite LB films by means of morphological and spectral methods.| Sample | C [wt%] | H [wt%] | N [wt%] |
|---|---|---|---|
| GO | 44.30 ± 0.02 | 2.01 ± 0.08 | — |
| GO-COOH | 44.70 ± 0.06 | 2.10 ± 0.07 | — |
| GO–CD | 42.84 ± 0.06 | 4.79 ± 0.05 | 3.91 ± 0.01 |
For the interfacial self-assembly, firstly, we investigated the surface pressure–area isotherms of as-obtained Langmuir films at room temperature. The optimization of used different spreading solvents and volumes for present GO–CD Langmuir film on pure water surface was demonstrated in Fig. S11.† It indicates that the optimized spread condition (methanol/chloroform 3:2 (v/v), 300 μL) seem suitable for material dispersion on surface and used in next interfacial characterization process. It can be easily observed from Fig. 1 that the isotherm of GO–CD spread on pure water surface with optimized solvent ratio and spread volume show slow increment of pressure at trough area of 125 cm2 until reaching 27 mN m−1 at compression end of 21 cm2. For the GO–CD film on N-Azo subphase, the isotherm show obvious increment of surface pressure from initial compression step and the final pressure reached above 25 mN m−1. In addition, for the GO–CD/PAA-Azo composite Langmuir film, a little increment appear for the initial pressure point at 136 cm2 and final pressure of 28 mN m−1 compared the film from pure water surface. The obvious change in all isotherms suggested the occurrence of interfacial interaction between beta-CD groups in GO–CD building blocks and N-Azo/PAA-Azo molecules in subphase solutions and formation of composite Langmuir films.
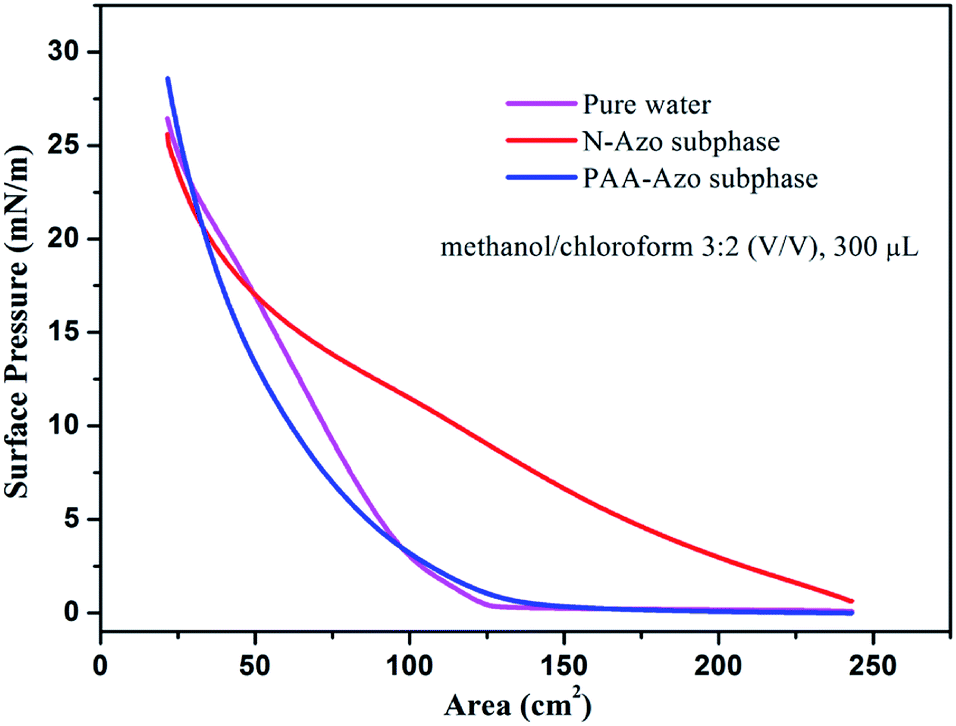 | ||
| Fig. 1 Surface pressure–area isotherms of Langmuir films of as-prepared GO–CD solution (methanol/chloroform v/v 3:2, 300 μL, 0.5 mg mL−1) spread on pure water surface, N-Azo, and PAA-Azo subphases at room temperature. | ||
The nanoscale morphologies of monolayer GO–CD Langmuir films were investigated by AFM (Fig. 2). The morphological difference is obviously observed. The overlapped GO sheet is representing the GO–CD Langmuir films with the height distribution of 1.1 ± 0.2 nm (Fig. 2a), while dispersed packed films with little wrinkles dominate rather than dispersed GO sheets in the case of composite films from N-Azo subphase with the height distribution of 1.6 ± 0.3 nm (Fig. 2b). As for the morphology of GO–CD/PAA composite film, messy reticular nanoscale aggregates appear in Fig. 2c. In addition, SEM and TEM images of GO–CD composite LB films from N-Azo and PAA-Azo subphase were also characterized. The SEM images in Fig. 3b and d clearly demonstrate distinct dispersed or cross-linked aggregate states after interaction with different azobenzene-containing molecules. The TEM images in Fig. 3e and f also verify the formation of large aggregation in the obtained films. The difference of morphology and height between the GO–CD Langmuir films from different subphase solutions can be mainly owing to the introduction of N-Azo or PAA-Azo molecules in the composite Langmuir film after host–guest interaction.
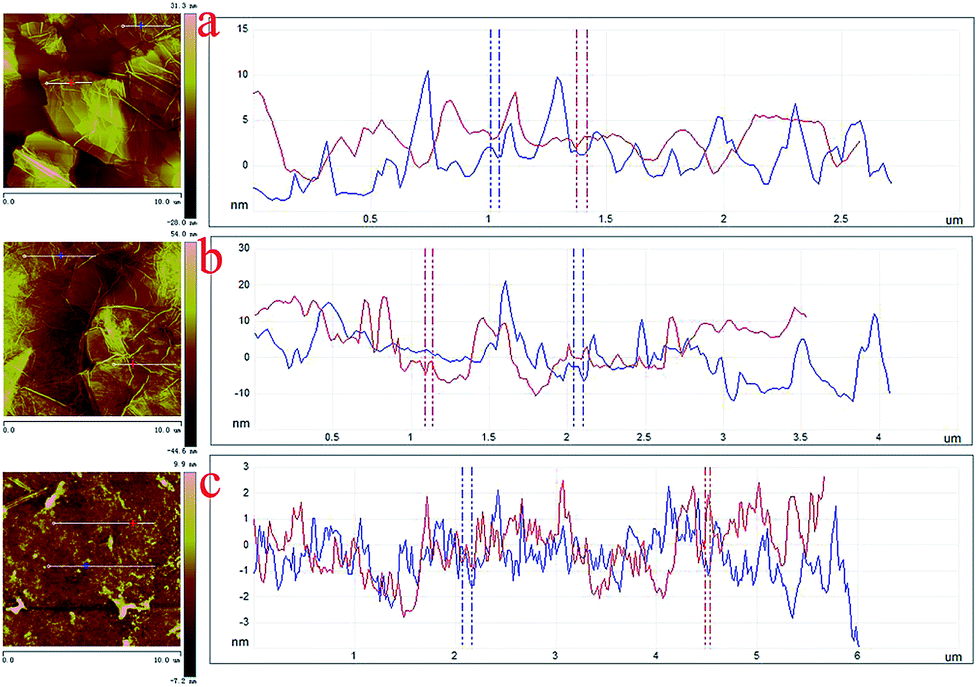 | ||
| Fig. 2 AFM images with section analysis of monolayer Langmuir films of GO–CD spread on different subphases transferred at 15 mN m−1: (a), pure water surface; (b), N-Azo subphase; (c), PAA-Azo subphase. | ||
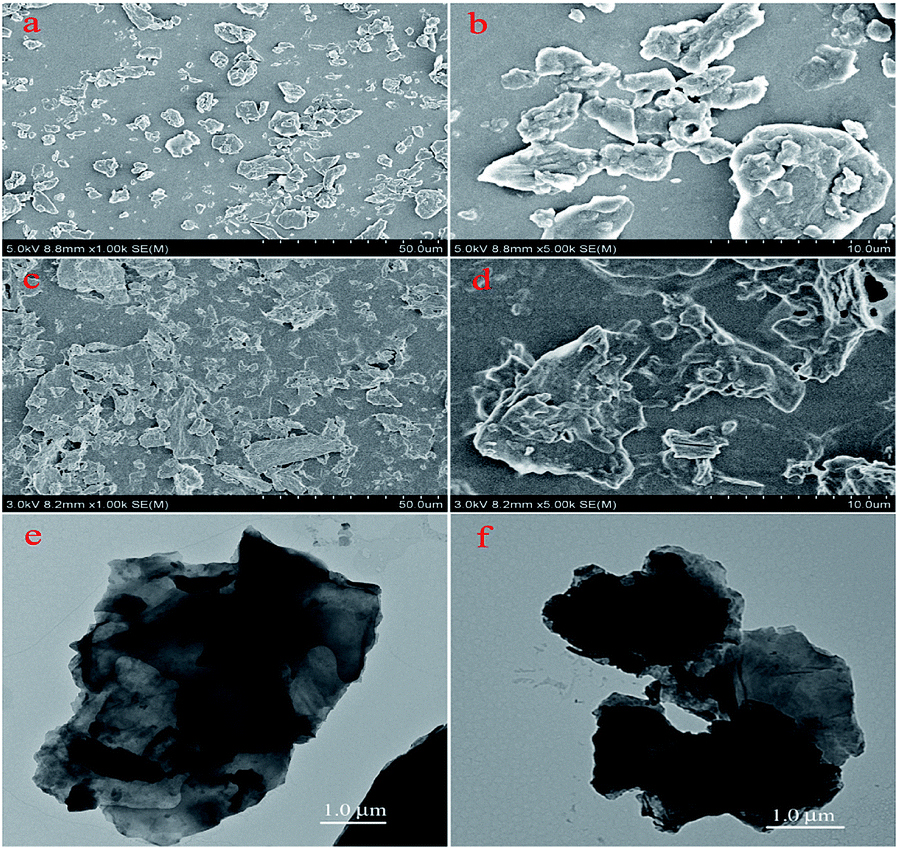 | ||
| Fig. 3 SEM of 10-layered LB films and TEM images of monolayer LB films of GO–CD from N-Azo subphase (a, b and e) and PAA-Azo subphase (c, d and f) transferred at 15 mN m−1. | ||
Azobenzene compounds have characteristic UV-vis absorption. We utilized the UV-vis spectra to investigate the obtained azobenzene derivatives and the multilayer LB films deposited on solid substrates. Fig. 4 shows the UV-vis spectra of the multilayer GO–CD/N-Azo and GO–CD/PAA-Azo composite LB films. At the same time, as shown in Fig. S8,† the GO aqueous dispersed solution displays a peak at 226 nm which is due to the π–π* transition of aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and a shoulder at 290–300 nm which corresponds to the n–π* transition of the CO bonds.71–73 Thus, the UV-vis spectrum of the GO–CD solution showed strong absorption peak at 282 nm, which could be reasonably ascribed to the charge transfer band of GO sheet with beta-CD segment. In addition, the obtained UV-vis spectrum of aqueous N-Azo solution showed two strong absorption peaks at 230 and 376 nm and a weak one at 442 nm. The former band can be ascribed to the localized absorption of the aromatic rings, while the peak at 376 nm and the weak peak at 442 nm can be assigned to the π–π* transition of trans isomers and n–π* transition of the cis-isomers, respectively.74 When the GO–CD film was deposited from N-Azo subphase, a great change in the absorption spectra was observed. The charge transfer band at 282 nm disappeared completely, which suggested the decrement of charge transfer between GO sheet with beta-CD segment as well as possible production of host–guest interaction between beta-CD segment and azobenzene part. Simultaneously, the red shift of the maximum absorption of azo group (from 376 nm to 393 nm) indicated the possible formation of J-aggregate of conjugated azobenzene segment in the composite films compared with monomers in the N-Azo solution. In addition, for the GO–CD/PAA-Azo composite films shown in Fig. 4b, the characteristic band appears at the position of 344 nm with the same value of PAA-Azo solution. This suggested most of azobenzene segment kept monomers state in the GO–CD/PAA-Azo composite films. In addition, the UV-vis spectra of aqueous mixed solutions of GO/GO–CD with azobenzene compounds were also measured and shown in Fig. S12 and S13.† No shift appeared for the mixed system of GO and N-Azo, indicating the adsorption process of N-Azo on GO surface in monodispersed state. However, for the mixed methanol solutions of GO–CD with N-Azo, the characteristic peak shifted from 376 to 384 nm, suggesting formation of weak J-aggregate. While for the case of mixed solution of GO–CD with PAA-Azo, no obvious shift was observed.
C bonds and a shoulder at 290–300 nm which corresponds to the n–π* transition of the CO bonds.71–73 Thus, the UV-vis spectrum of the GO–CD solution showed strong absorption peak at 282 nm, which could be reasonably ascribed to the charge transfer band of GO sheet with beta-CD segment. In addition, the obtained UV-vis spectrum of aqueous N-Azo solution showed two strong absorption peaks at 230 and 376 nm and a weak one at 442 nm. The former band can be ascribed to the localized absorption of the aromatic rings, while the peak at 376 nm and the weak peak at 442 nm can be assigned to the π–π* transition of trans isomers and n–π* transition of the cis-isomers, respectively.74 When the GO–CD film was deposited from N-Azo subphase, a great change in the absorption spectra was observed. The charge transfer band at 282 nm disappeared completely, which suggested the decrement of charge transfer between GO sheet with beta-CD segment as well as possible production of host–guest interaction between beta-CD segment and azobenzene part. Simultaneously, the red shift of the maximum absorption of azo group (from 376 nm to 393 nm) indicated the possible formation of J-aggregate of conjugated azobenzene segment in the composite films compared with monomers in the N-Azo solution. In addition, for the GO–CD/PAA-Azo composite films shown in Fig. 4b, the characteristic band appears at the position of 344 nm with the same value of PAA-Azo solution. This suggested most of azobenzene segment kept monomers state in the GO–CD/PAA-Azo composite films. In addition, the UV-vis spectra of aqueous mixed solutions of GO/GO–CD with azobenzene compounds were also measured and shown in Fig. S12 and S13.† No shift appeared for the mixed system of GO and N-Azo, indicating the adsorption process of N-Azo on GO surface in monodispersed state. However, for the mixed methanol solutions of GO–CD with N-Azo, the characteristic peak shifted from 376 to 384 nm, suggesting formation of weak J-aggregate. While for the case of mixed solution of GO–CD with PAA-Azo, no obvious shift was observed.
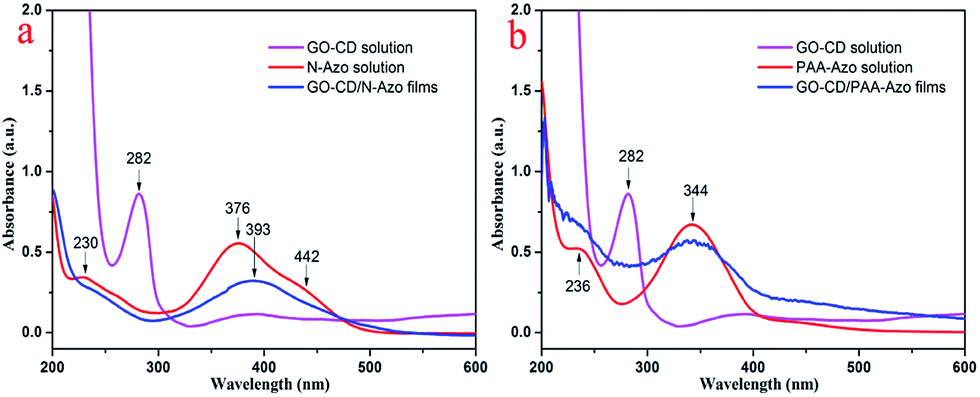 | ||
| Fig. 4 UV-vis spectra of GO–CD solution and multilayer LB films of GO–CD/N-Azo and GO–CD/PAA-Azo transferred at 15 mN m−1. | ||
In order to further confirm the interaction between GO–CD building blocks and azobenzene-containing molecules in the subphases, the FT-IR spectra of the obtained LB films were also measured. Obvious changes were observed for the IR spectra of prepared composite LB films after interacting with N-Azo and PAA-Azo molecules (Fig. 5a). In comparison with the multilayer LB films of GO–CD from water surface, the spectra of composite films show obvious intensity for bands at 3239 cm−1 (N–H stretching and hydrogen bonding), 2923 and 2849 cm−1 (C–H modes of methylene moieties), 1718 cm−1 (CO stretching), and 1636 cm−1 (amide CO stretching). In addition, the bands at 1454 and 1414 cm−1 can be assigned to the azobenzene groups, while the additional peaks at 1154, 1082 and 1031 cm−1 can be assigned to the vibrations from beta-CD skeletons. At the same time, in comparison with the IR spectra of N-Azo and as-synthesized PAA-Azo material in Fig. S10,† the obvious changes for the LB films indicated the successful composition of two azobenzene-containing molecules with GO–CD materials in the organized self-assembly films.
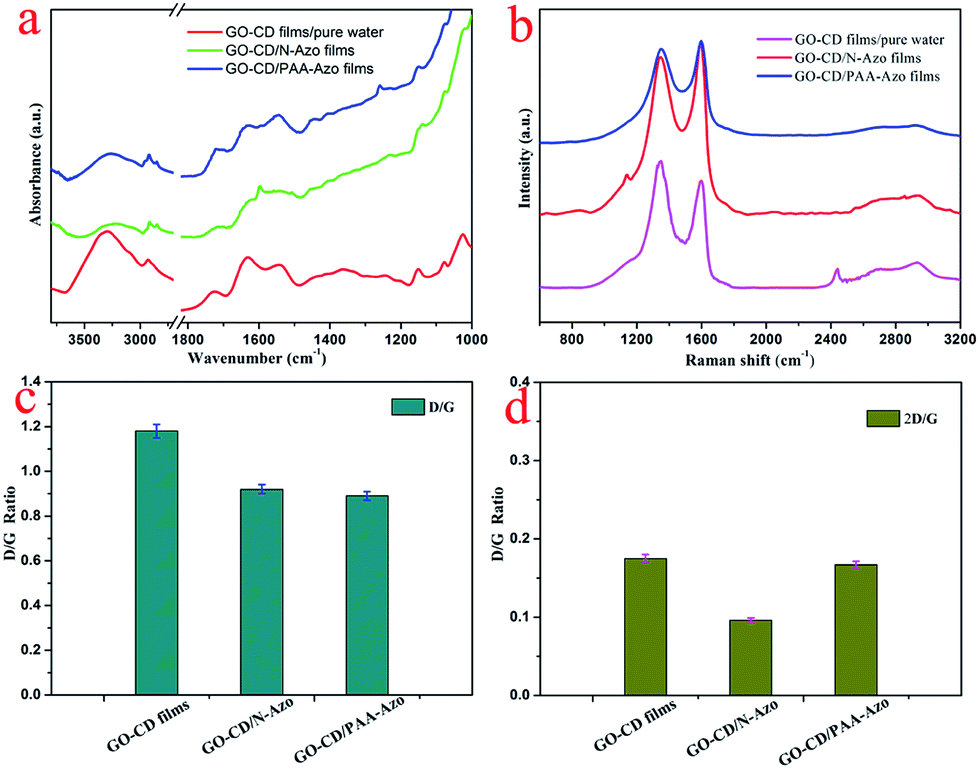 | ||
| Fig. 5 IR spectra (a) and Raman spectra (b) of the multilayer LB films of GO–CD from water surface, N-Azo and PAA-Azo subphase transferred at 15 mN m−1. Pictures (c and d) indicate the D/G ratio and 2D/G ratio analysis in Raman spectra. | ||
It is well established that Raman spectroscopy has been widely used to investigate GO-based composite materials. The measurements for present as-obtained composite LB films of GO–CD from N-Azo and PAA-Azo subphase were performed. Three characteristic bands of graphene sheets in Raman spectra appear in Fig. 5b. One band at 1597 cm−1 can be owing to the G band originating from the first-order scattering of the E2g phonons of the sp2-hybridized carbon atoms. Another band at 1351 cm−1 can be assigned to the D band, which comes from a breathing mode of κ-point phonons of A1g symmetry of the defects involved in the sp3-hybridized carbon bonds such as hydroxyl and/or epoxide bonds.75,76 In addition to the G and D bands, a broad peak at 2690 cm−1 (i.e., 2D band) was also observed. The intensity of 2D band is correlated to the stacking mode of graphene sheets. It was well-known that the D/G peak intensity ratio could be utilized as a measurement of the sp2 domain size of graphene sheets containing sp3 and sp2 bonds due to the origination of G and D bands.77,78 For present system in Fig. 5c, the D/G ratio shifted from 1.18 for GO–CD LB films from water surface to 0.92 and 0.89 for the obtained GO–CD/N-Azo and GO–CD/PAA-Azo composite LB films. The obvious change confirmed the successful reaction of N-Azo molecules and polymeric PAA-Azo molecules with azobenzene groups in the formation process of composite films. Furthermore, the 2D/G intensity ratios for the single and bilayer GO sheets were found in the range of 1.53–1.68 and 0.82–0.89, respectively.79,80 Previous studies have also reported that the 2D/G intensity ratios for single-, double-, triple-, and multi- (>4) layer graphene sheets are 1.6, 0.8, 0.30 and 0.07, respectively. Fig. 5d shows that the 2D/G ratios of the obtained LB films prepared in this study have values between 0.096 and 0.175, further suggesting the multilayer nature in present GO–CD LB films.
To investigate the composition and chemical state of present prepared composite films, X-ray photoelectron spectroscopy (XPS) measurements were performed. The survey data for the obtained composite films show the characteristic C(1s), O(1s), and N(1s) peaks (Fig. 6). The deconvolution of C(1s) peaks from the multilayer LB films of GO–CD from pure water surface in Fig. 7a demonstrate the peaks centered at positions of 286.5, 288.4, and 289.9 eV, which can be assigned to the C–O, CO, and CO–O oxygen-containing bonds, respectively.81,82 For composite films GO–CD/N-Azo and GO–CD/PAA-Azo, the deconvolution of C(1s) peaks (Fig. 7d and g) display the peaks centered at positions of 284.8 and 285.4 eV attributed to C–C, CC & C–H as well as C–N bonds. This confirmed the successful hybrid of N-Azo and PAA-Azo molecules in composite films. In addition, the deconvolution of O(1s) peaks from the LB films from pure water surface in Fig. 7b demonstrate the peaks centered at positions of 532.5 and 533.8 eV, which can be assigned to the CO and C–O oxygen-containing bonds, respectively. However, the area ratio assigned to CO bond in O(1s) peak (Fig. 7h) from GO–CD/PAA-Azo composite films show the value of 72% in comparison with the value of 16% in GO–CD films from pure water surface, indicating the increment of carboxyl groups and addition of PAA-Azo molecules in the obtained composite LB films. Moreover, the deconvolution analysis of N(1s) peaks from the LB films of GO–CD from pure water surface in Fig. 7c demonstrate the single peak centered at positions of 401.6 eV assigned to the N+ species from beta-CD segments in GO–CD building blocks.83 After interacting with N-Azo or PAA-Azo, the deconvolution of N(1s) peaks (Fig. 7f and i) show also the new peak at 400.5 eV assigned to the C–N bond, which indicate the successful addition of azobenzene part in composite films.
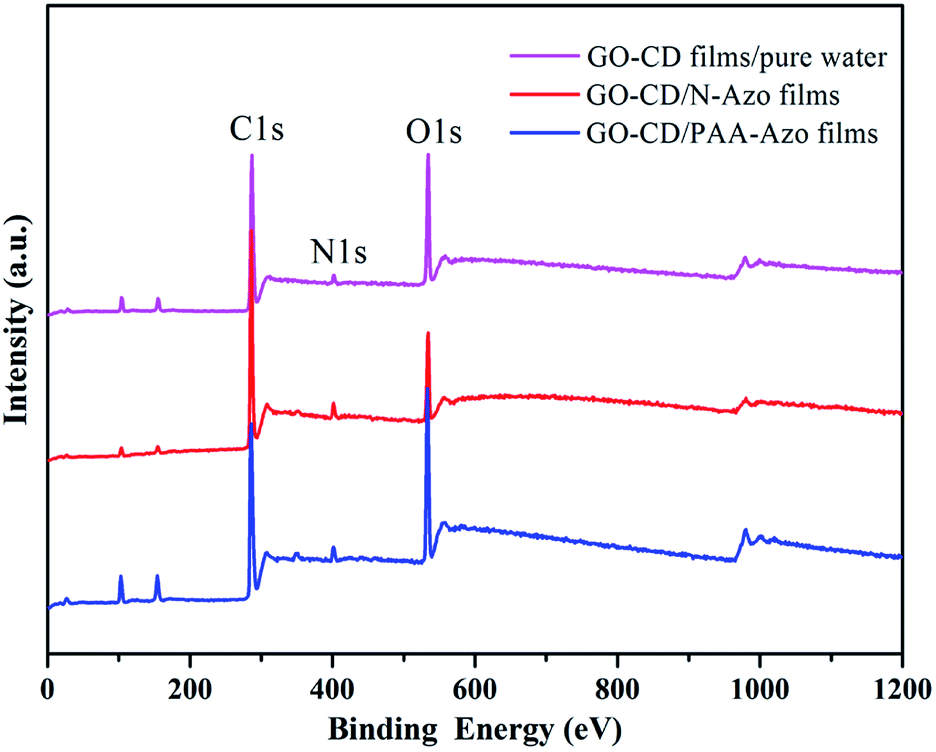 | ||
| Fig. 6 Survey XPS spectra of the multilayer LB films of GO–CD from different subphases transferred at 15 mN m−1. | ||
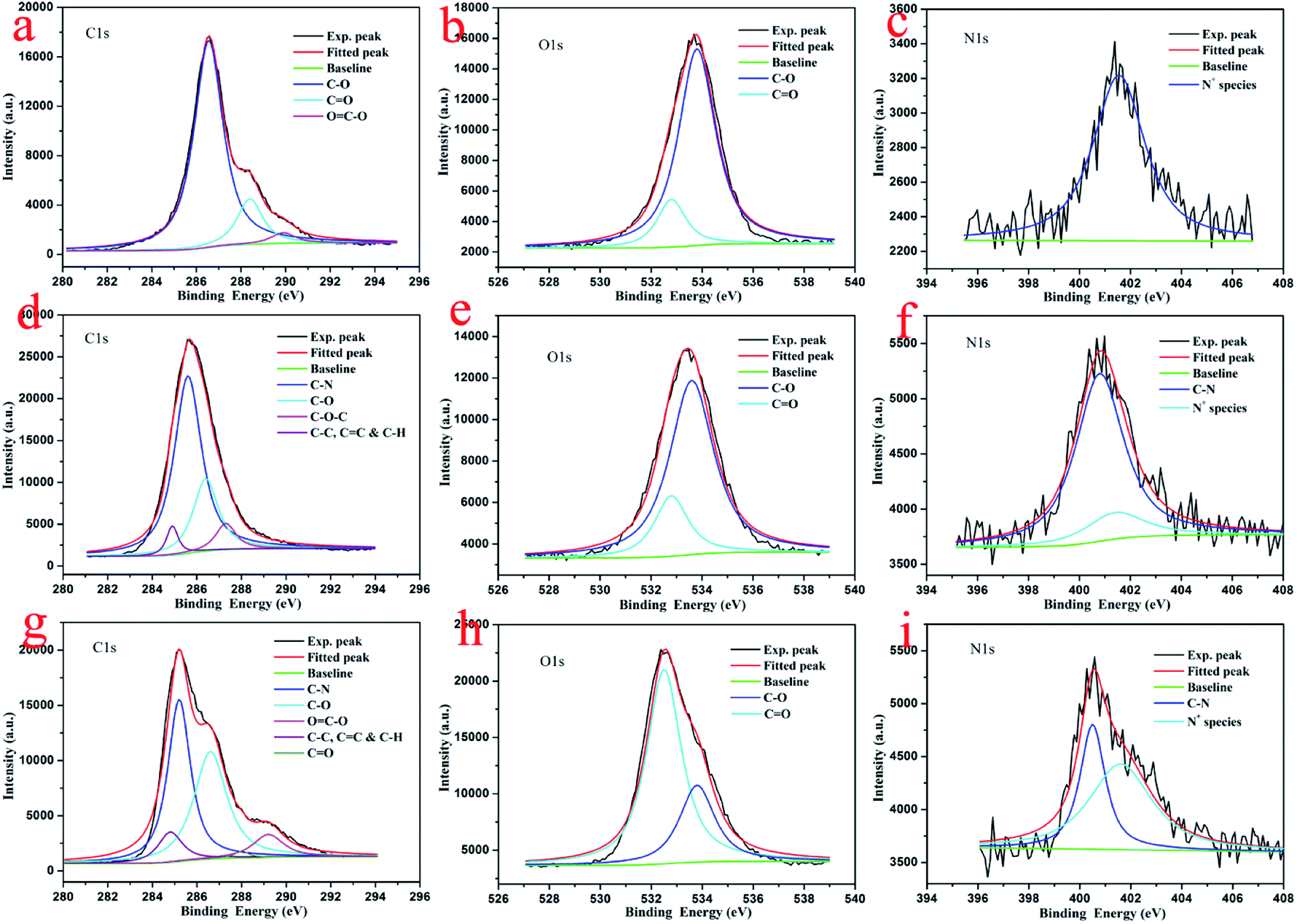 | ||
| Fig. 7 Deconvolution of XPS peaks of multilayer LB films of GO–CD from pure water surface (a–c), GO–CD/N-Azo (d–f), and GO–CD/PAA-Azo (g–i) transferred at 15 mN m−1. | ||
To investigate the photoresponsiveness of present azobenzene-containing compounds, the aqueous solutions and composite LB films were irradiated by a UV lamp with wavelength of 365 nm, and the change of the configuration of the azobenzene units was monitored by UV-vis spectroscopy. Fig. 8a and b shows the UV-vis absorption spectra of the obtained aqueous solutions of two azobenzene compounds upon UV irradiation in different time. As for the case of N-Azo solution, the π–π* transition band of trans isomers with λmax at 376 nm decreases slightly. After irradiation for 10 min, the UV-vis spectra do not change any more, indicating azobenzene groups reach to a trans–cis equilibrium state. For the PAA-Azo solution, with time increment, the π–π* transition band of trans isomers with λmax at 344 nm decreases remarkably and finally shift to 325 nm, at the same time, the n–π* transition band of cis isomers at around 438 nm increases slightly. After irradiation for 5 min, the UV-vis spectra do not change any more, indicating azobenzene groups reach to a cis saturated state. In addition, the obtained GO–CD/N-Azo and GO–CD/PAA-Azo composite LB films were investigated upon irradiation with UV irradiation in different time. As shown in Fig. 8c, the obtained GO–CD/N-Azo composite LB films showed slight decrement at 393 nm even upon UV irradiation for 30 min, suggesting possible large spacial hindrance for trans–cis photoisomerization due to the J-aggregates of azobenzene parts in LB films. Moreover, Fig. 8d demonstrates the obvious spectral change in GO–CD/PAA-Azo composite LB films with UV irradiation in different time. The obtained spectral changes suggested that there was no strong aggregation between azobenzene groups in GO–CD/PAA-Azo composite LB films. The present results were in well accordance with the above obtained results.
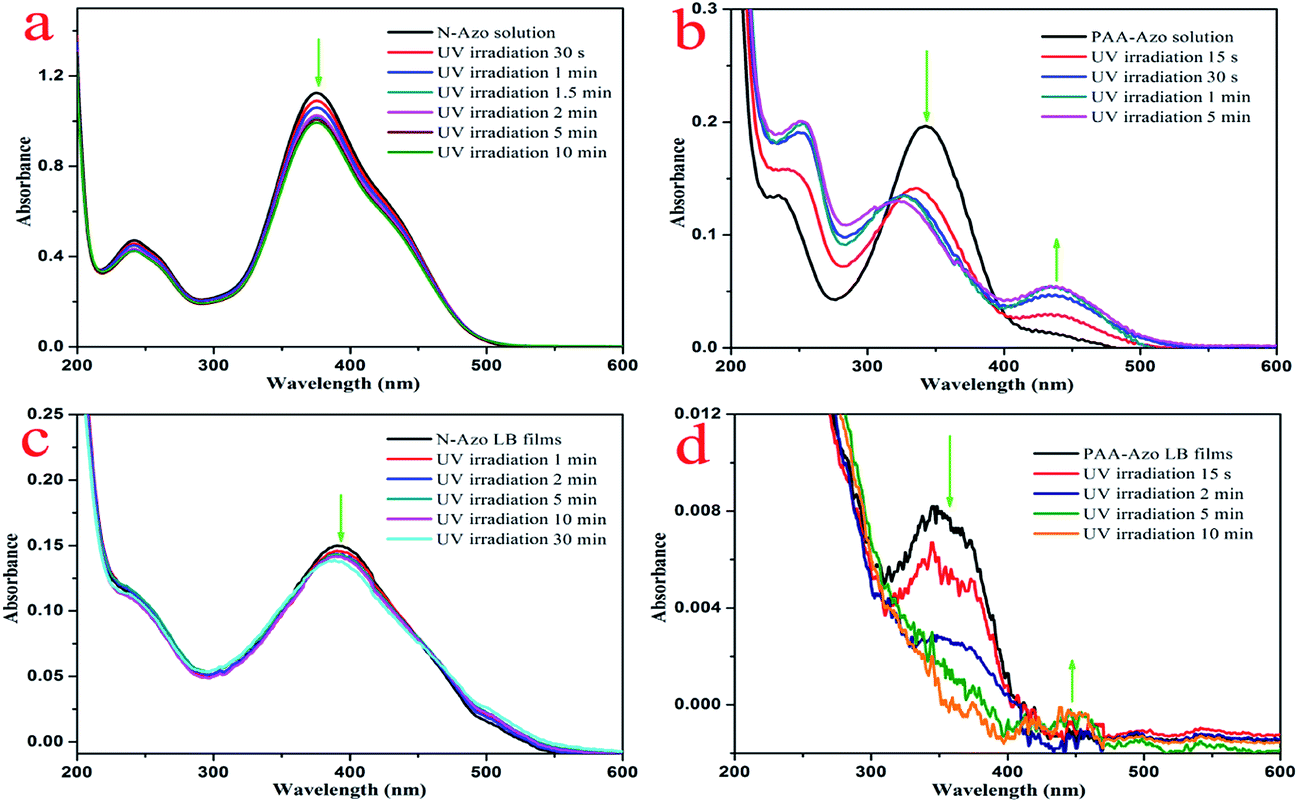 | ||
| Fig. 8 UV-vis spectral changes of N-Azo and PAA-Azo solutions (a and b) as well as two composite LB films (c and d) upon UV irradiation at 365 nm for different time. | ||
Considering the above results, the self-assembly modes and stacking change of all prepared composite Langmuir films at the air/water interface can be proposed, as schematically shown in Fig. 9, which was in well accordance with our initial proposal to design this work. For the GO–CD/N-Azo composite Langmuir film, because of the hydrophilic amine moieties and aromatic azobenzene segments in N-Azo molecules, in the self-assembly process on water surface, the adjacent N-Azo molecules can be adsorbed on GO sheet surface and pack organized to overlap each other to form the J-aggregate domains (Fig. 9a). At the same time, some N-Azo molecules can penetrate into the beta-CD cavity to form inclusion complex with dynamic equilibrium.84,85 This deduction was verified from the surface pressure–area isotherms, UV-vis spectra, and morphological characterization. As for GO–CD/PAA-Azo composite Langmuir film, the situation is greatly different. Azobenzene segments were linked with PAA long alkyl chains via amide bonds and well dispersed in the composite film. Some azobenzene part can form inclusion complex with beta-CD cavity, while others can be adsorbed on GO sheet surface or embedded in the PAA chain kinking with dispersed state (Fig. 9b). In this state, the limiting molecular areas in isotherm become similar to that from pure water surface and none of J-aggregate of azobenzene parts appeared. Thus GO sheet cross-linked morphologies with surface of messy thread were observed. In addition, it should be noted that the CD spectra of the transferred multilayer GO–CD/N-Azo and GO–CD/PAA-Azo composite LB films have been measured, as shown in Fig. S14.† No obvious CD signals could be observed. This result can be reasonably explained by the minor beta-CD component in the synthesized GO–CD building blocks and subsequent fewer formed beta-CD/azobenzene inclusion complex in the composite films. The follow-up research work can focus on the preparation of new GO–CD materials with nanoscale GO sheet (multiple carboxyl group sites) or poly-beta-CD segment (multiple CD cavities) to improve the inclusion effect in the proposed LB films.
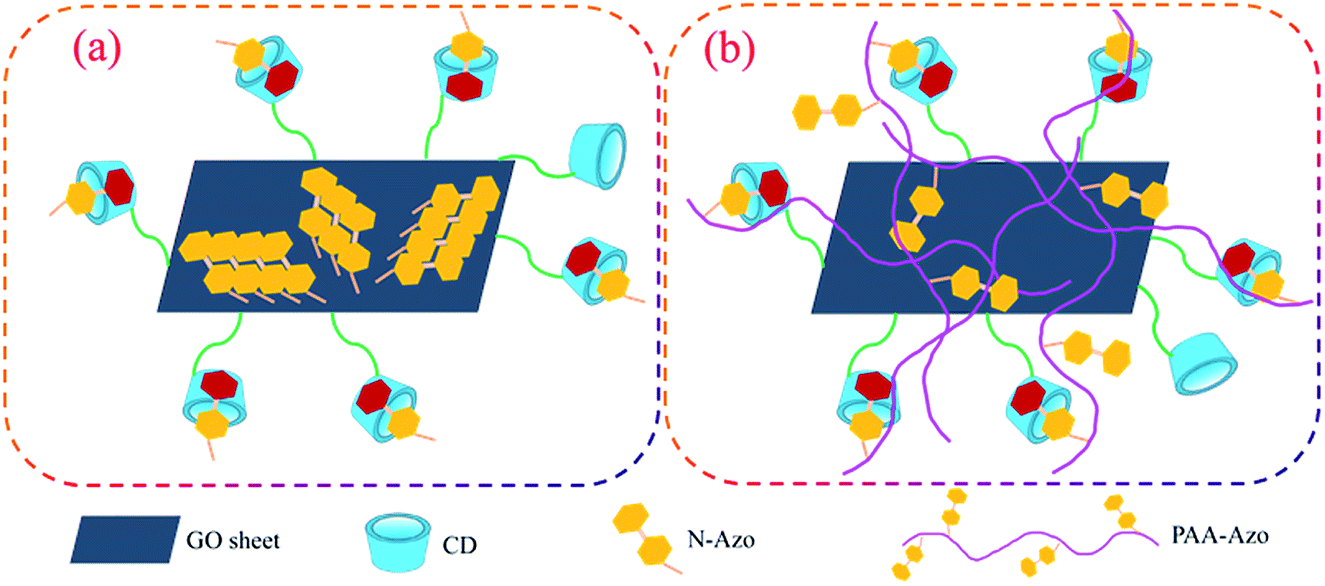 | ||
| Fig. 9 Schematic illustration of reasonable self-assembly models in GO–CD/N-Azo (a) and GO–CD/PAA-Azo (b) composite Langmuir films. | ||
4. Conclusions
In summary, we have presented the design and characterization of beta-cyclodextrin-modified graphene oxide composite Langmuir films with some azobenzene compounds. The synthesized beta-cyclodextrin-modified graphene oxide GO–CD acts as either a host agent or a building block/amphiphile to form spreading films on different subphases. Depending on the different molecular structures of N-Azo and PAA-Azo molecules, variable self-assembly processes in the obtained composite films were achieved. When the building block GO–CD formed composite Langmuir films on N-Azo subphase, both host–guest interaction between beta-CD with azobenzene and J-aggregate of N-Azo molecules on GO sheet surface appeared. When GO–CD was spread on the subphase containing PAA-Azo, cross-linked nanostructures with messy thread and inclusion complex in composite films were obtained without any aggregates of azobenzene segment. Owing to the specific properties and self-assembled nanostructures in the obtained composite films, the present research work can demonstrate great potentials for the preparation of self-assembled nano-device films and nanosensors materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21473153), Support Program for the Top Young Talents of Hebei Province, China Postdoctoral Science Foundation (No. 2015M580214), and Scientific and Technological Research and Development Program of Qinhuangdao City (No. 201701B004 and 201502A006).Notes and references
- R. Narayan, J. E. Kim, J. Y. Kim, K. E. Lee and S. O. Kim, Adv. Mater., 2016, 28, 3045–3068 CrossRef CAS PubMed.
- Y. Y. Zhang, S. S. Gong, Q. Zhang, P. Ming, S. J. Wan, J. S. Peng, L. Jiang and Q. F. Cheng, Chem. Soc. Rev., 2016, 45, 2378–2395 RSC.
- F. Li, X. Jiang, J. J. Zhao and S. B. Zhang, Nano Energy, 2015, 16, 488–515 CrossRef CAS.
- M. Daud, M. S. Kamal, F. Shehzad and M. A. Al-Harthi, Carbon, 2016, 104, 241–252 CrossRef CAS.
- G. H. Jeong, S. Baek, S. Lee and S. W. Kim, Chem.–Asian. J., 2016, 11, 949–964 CrossRef CAS PubMed.
- T. Jiao, Y. Liu, Y. Wu, Q. Zhang, X. Yan, F. Gao, A. Bauer, J. Liu, T. Zeng and B. Li, Sci. Rep., 2015, 5, 12451 CrossRef CAS PubMed.
- J. Texter, Curr. Opin. Colloid Interface Sci., 2015, 20, 454–464 CrossRef CAS.
- S. Gambhir, R. Jalili, D. L. Officer and G. G. Wallace, NPG Asia Mater., 2015, 7, e186 CrossRef CAS.
- R. Xing, T. Jiao, Y. Liu, K. Ma, Q. Zou, G. Ma and X. Yan, Polymers, 2016, 8, 181 CrossRef.
- S. Niyogi, E. Bekyarova, M. E. Itkis, J. L. Mcwilliams, M. A. Hamon and R. C. Haddon, J. Am. Chem. Soc., 2006, 128, 7720–7721 CrossRef CAS PubMed.
- S. Stankovich, R. D. Piner, S. B. T. Nguyen and R. S. Ruoff, Carbon, 2006, 44, 3342–3347 CrossRef CAS.
- Z. B. Liu, Y. F. Xu, X. Y. Zhang, X. L. Zhang, Y. S. Chen and J. G. Tian, J. Phys. Chem. B, 2009, 113, 9681–9686 CrossRef CAS PubMed.
- I. Kaminska, A. Barras, Y. Coffinier, W. Lisowski, J. Niedziolka-Jonsson, P. Woisel, J. Lyskawa, M. Opallo, A. Siriwardena, R. Boukherroub and S. Szunerits, ACS Appl. Mater. Interfaces, 2012, 4, 5386–5393 CAS.
- I. Kaminska, M. R. Das, Y. Coffinier, J. Niedziolka-Jonsson, J. Sobczak, P. Woisel, J. Lyskawa, M. Opallo, R. Boukherroub and S. Szunerits, ACS Appl. Mater. Interfaces, 2012, 4, 1016–1020 CAS.
- Y. F. Yang, J. Wang, J. Zhang, J. C. Liu, X. L. Yang and H. Y. Zhao, Langmuir, 2009, 25, 11808–11814 CrossRef CAS PubMed.
- H. J. Salavagione, M. A. Gómez and G. Martínez, Macromolecules, 2009, 42, 6331–6334 CrossRef CAS.
- A. F. Hirschbiel, B. V. K. J. Schmidt, P. Krolla-Sidenstein, J. P. Blinco and C. Bamer-Kowollik, Macromolecules, 2015, 48, 4410–4420 CrossRef CAS.
- L. Peng, H. J. Zhang, A. C. Feng, M. Huo, Z. L. Wang, J. Hu, W. P. Gao and J. Y. Yuan, Polym. Chem., 2015, 6, 3652–3659 RSC.
- S. B. Xie, J. Zhang, Y. L. Yuan, Y. Q. Chai and R. Yuan, Chem. Commun., 2015, 51, 3387–3390 RSC.
- Y. L. Wu, R. F. Shi, Y. L. Wu, J. M. Holcroft, Z. C. Liu, M. Frasconi, M. R. Wasielewski, H. Li and J. F. Stoddart, J. Am. Chem. Soc., 2015, 137, 4111–4118 CrossRef CAS PubMed.
- H. Zhao, L. Yang, Y. Li, X. Ran, H. Ye, G. Zhao, Y. Zhang, F. Liu and C. P. Li, Biosens. Bioelectron., 2017, 89, 361–369 CrossRef CAS PubMed.
- L. Yang, H. Zhao, Y. Li, Y. Zhang, H. Ye, G. Zhao, X. Ran, F. Liu and C. P. Li, Biosens. Bioelectron., 2017, 87, 737–744 CrossRef CAS PubMed.
- L. Yang, S. Fan, G. Deng, Y. Li, X. Ran, H. Zhao and C. P. Li, Biosens. Bioelectron., 2015, 68, 617–625 CrossRef CAS PubMed.
- L. Yang, H. Zhao, C. P. Li, S. Fan and B. Li, Biosens. Bioelectron., 2015, 64, 126–130 CrossRef CAS PubMed.
- L. Yang, H. Zhao, Y. Li and C. P. Li, Sens. Actuators, B, 2015, 207, 1–8 CrossRef CAS.
- X. Ran, L. Yang, J. Zhang, G. Deng, Y. Li, X. Xie, H. Zhao and C. P. Li, Anal. Chim. Acta, 2015, 892, 85–94 CrossRef CAS PubMed.
- L. Yang, H. Zhao, S. Fan, G. Zhao, X. Ran and C. P. Li, RSC Adv., 2015, 5, 64146–64155 RSC.
- X. Ran, L. Yang, G. Zhao, H. Ye, Y. Zhang, S. Fan, X. Xie, H. Zhao and C. P. Li, RSC Adv., 2015, 5, 60775–60785 RSC.
- H. Yan, L. L. Zhu, X. Li, A. Kwok, X. Y. Pan and Y. L. Zhao, Asian J. Org. Chem., 2012, 1, 314–318 CrossRef CAS.
- Q. Xue, Z. G. Liu, Y. J. Guo and S. J. Guo, Biosens. Bioelectron., 2015, 68, 429–436 CrossRef CAS PubMed.
- A. Abbadessa, M. M. Blokzijl, V. H. M. Mouser, P. Marica, J. Malda, W. E. Hennink and T. Vermonden, Carbohydr. Polym., 2016, 149, 163–174 CrossRef CAS PubMed.
- T. Wang, N. Wu, H. Li, Q. L. Lu and Y. Jiang, J. Appl. Polym. Sci., 2016, 133, 43836 Search PubMed.
- H. Cao, X. Zhu and M. Liu, Angew. Chem., Int. Ed. Engl., 2013, 52, 4122–4126 CrossRef CAS PubMed.
- M. Deng, L. Zhang, Y. Jiang and M. Liu, Angew. Chem., Int. Ed. Engl., 2016, 55, 15062–15066 CrossRef CAS PubMed.
- P. Guo, L. Zhang and M. Liu, Adv. Mater., 2006, 18, 177–180 CrossRef CAS.
- X. Huang, C. Li, S. G. Jiang, X. S. Wang, B. W. Zhang and M. H. Liu, J. Am. Chem. Soc., 2004, 126, 1322–1323 CrossRef CAS PubMed.
- J. Jiang, Y. Meng, L. Zhang and M. Liu, J. Am. Chem. Soc., 2016, 138, 15629–15635 CrossRef CAS PubMed.
- C. Liu, D. Yang, Q. Jin, L. Zhang and M. Liu, Adv. Mater., 2016, 28, 1644–1649 CrossRef CAS PubMed.
- L. J. Cote, F. Kim and J. Huang, J. Am. Chem. Soc., 2009, 131, 1043–1049 CrossRef CAS PubMed.
- F. Kim, L. J. Cote and J. Huang, Adv. Mater., 2010, 22, 1954–1958 CrossRef CAS PubMed.
- K. L. Harrison, L. B. Biedermann and K. R. Zavadil, Langmuir, 2015, 31, 9825–9832 CrossRef CAS PubMed.
- M. M. Jaafar, G. P. M. K. Ciniciato, S. A. Ibrahim, S. M. Phang, K. Yunus, A. C. Fisher, M. Iwamoto and P. Vengadesh, Langmuir, 2015, 31, 10426–10434 CrossRef CAS PubMed.
- F. Han, S. M. Yang, W. X. Jing, Z. D. Jiang, H. Liu and L. Li, Appl. Surf. Sci., 2015, 345, 18–23 CrossRef CAS.
- T. H. Han, J. Nanosci. Nanotechnol., 2015, 15, 1191–1194 CrossRef CAS PubMed.
- J. D. Mangadlao, C. M. Santos, M. J. L. Felipe, A. C. C. de Leon, D. F. Rodrigues and R. C. Advincula, Chem. Commun., 2015, 51, 2886–2889 RSC.
- B. Gur, M. Sinoforoglu and K. Meral, RSC Adv., 2015, 5, 552–557 RSC.
- B. T. McGrail, J. D. Mangadlao, B. J. Rodier, J. Swisher, R. Advincula and E. Pentzer, Chem. Commun., 2016, 52, 288–291 RSC.
- J. Wen, Y. Jiang, Y. Yang and S. Li, J. Mater. Sci.: Mater. Electron., 2014, 25, 063–1071 Search PubMed.
- P. F. Duan, L. Qin and M. H. Liu, Langmuir, 2011, 27, 1326–1331 CrossRef CAS PubMed.
- S. J. Li, D. Taura, A. Hashidzume and A. Harada, Chem.–Asian J., 2010, 5, 2281–2289 CrossRef CAS PubMed.
- Y. G. Li and M. H. Liu, J. Colloid Interface Sci., 2007, 306, 386–390 CrossRef CAS PubMed.
- E. Rivera, M. D. P. Carreon-Castro, L. Rodriguez, G. Cedillo, S. Fomine and O. G. Morales-Saavedra, Dyes Pigm., 2007, 74, 396–403 CrossRef CAS.
- Y. Matsuzawa, S. Noguchi, H. Sakai, M. Abe and M. Matsumoto, Thin Solid Films, 2006, 510, 292–296 CrossRef CAS.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- S. J. Jeon, S. Y. Kwak, D. Yim, J. M. Ju and J. H. Kim, J. Am. Chem. Soc., 2014, 136, 10842–10845 CrossRef CAS PubMed.
- X. N. Luo, K. Ma, T. F. Jiao, R. R. Xing, L. X. Zhang, J. X. Zhou and B. B. Li, Nanoscale Res. Lett., 2017, 12, 99 CrossRef PubMed.
- C. Y. Quan, J. X. Chen, H. Y. Wang, C. Li, C. Chang, X. Z. Zhang and R. X. Zhuo, ACS Nano, 2010, 4, 4211–4219 CrossRef CAS PubMed.
- W. Tang and S. C. Ng, Nat. Protoc., 2008, 3, 691–697 CrossRef CAS PubMed.
- E. S. Orth, J. E. S. Fonsaca, S. H. Domingues, H. Mehl, M. M. Oliveira and A. J. G. Zarbin, Carbon, 2013, 61, 543–550 CrossRef CAS.
- G. Chen, S. Zhai, Y. Zhai, K. Zhang, Q. Yue, L. Wang, J. Zhao, H. Wang, J. Liu and J. Jia, Biosens. Bioelectron., 2011, 26, 3136–3141 CrossRef CAS PubMed.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed.
- Y. Liu, T. Wang, Y. Huan, Z. Li, G. He and M. Liu, Adv. Mater., 2013, 25, 5875–5879 CrossRef CAS PubMed.
- Z. Shen, T. Wang and M. Liu, Angew. Chem., Int. Ed. Engl., 2014, 53, 13424–13428 CrossRef CAS PubMed.
- J. Yuan and M. Liu, J. Am. Chem. Soc., 2003, 125, 5051–5056 CrossRef CAS PubMed.
- L. Zhang, Q. Lu and M. Liu, J. Phys. Chem. B, 2003, 107, 2565–2569 CrossRef CAS.
- Q. Bao, D. Zhang and P. Qi, J. Colloid Interface Sci., 2011, 360, 463–470 CrossRef CAS PubMed.
- R. Zhang, R. Xing, T. Jiao, K. Ma, C. Chen, G. Ma and X. Yan, ACS Appl. Mater. Interfaces, 2016, 8, 13262–13269 CAS.
- X. Zhao, K. Ma, T. Jiao, R. Xing, X. Ma, J. Hu, H. Huang, L. Zhang and X. Yan, Sci. Rep., 2017, 7, 44076 CrossRef PubMed.
- Y. Liu, K. Ma, T. Jiao, R. Xing, G. Shen and X. Yan, Sci. Rep., 2017, 7, 42978 CrossRef CAS PubMed.
- R. Xing, W. Wang, T. Jiao, K. Ma, Q. Zhang, W. Hong, H. Qiu, J. Zhou, L. Zhang and Q. Peng, ACS Sustainable Chem. Eng., 2017, 5, 4948–4956 CrossRef CAS.
- F. Xie, G. Ouyang, L. Qin and M. Liu, Chem.–Eur. J., 2016, 22, 18208–18214 CrossRef CAS PubMed.
- R. Xing, K. Liu, T. Jiao, N. Zhang, K. Ma, R. Zhang, Q. Zou, G. Ma and X. Yan, Adv. Mater., 2016, 28, 3669–3676 CrossRef CAS PubMed.
- S. Huo, P. Duan, T. Jiao, Q. Peng and M. Liu, Angew. Chem., Int. Ed., 2017 DOI:10.1002/anie.201706308.
- E. D. Grayfer, A. S. Nazarov, V. G. Makotchenko, S. J. Kim and V. E. Fedorov, J. Mater. Chem., 2011, 21, 3410–3414 RSC.
- T. F. Jiao, Y. J. Wang, F. Q. Gao, J. X. Zhou and F. M. Gao, Prog. Nat. Sci.: Mater. Int., 2012, 22, 64–70 CrossRef.
- A. C. Ferrari and J. Robertson, Phys. Rev. B, 2000, 61, 14095–14107 CrossRef CAS.
- L. M. Malard, M. A. Pimenta, G. Dresselhaus and M. S. Dresselhaus, Phys. Rep., 2009, 473, 51–87 CrossRef CAS.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J. H. Ahn, P. Kim, J. Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS PubMed.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prudhomme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36–41 CrossRef CAS PubMed.
- O. Akhavan, Carbon, 2015, 81, 158–166 CrossRef CAS.
- H. R. Thomas, A. J. Marsden, M. Walker, N. R. Wilson and J. P. Rourke, Angew. Chem., Int. Ed., 2014, 53, 7613–7618 CrossRef CAS PubMed.
- X. Chen, Y. C. Dai, Z. B. Zheng and K. Z. Wang, J. Colloid Interface Sci., 2013, 402, 107–113 CrossRef CAS PubMed.
- H. Guo, T. Jiao, Q. Zhang, W. Guo, Q. Peng and X. Yan, Nanoscale Res. Lett., 2015, 10, 272 CrossRef PubMed.
- Z. Z. Tong, R. Y. Wang, J. Huang, J. T. Xu and Z. Q. Fan, Polym. Chem., 2015, 6, 2214–2225 RSC.
- Q. Jin, G. Liu, X. Liu and J. Ji, Soft Matter, 2010, 6, 5589–5595 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07109d |
| This journal is © The Royal Society of Chemistry 2017 |