
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Noncytotoxic polycaprolactone-polyethyleneglycol-ε-poly(L-lysine) triblock copolymer synthesized and self-assembled as an antibacterial drug carrier†
Chuncai Zhou *,
Xinyu Zhou and
Xiaokai Su
*,
Xinyu Zhou and
Xiaokai Su
School of Materials Science and Engineering, Tongji University, 4800 Caoan Road, Shanghai, 201804, China. E-mail: cczhou@tongji.edu.cn
First published on 15th August 2017
Abstract
A novel biodegradable and noncytotoxic polycaprolactone-polyethyleneglycol-ε-poly(L-lysine) (PCL-PEG-EPL) triblock copolymer was synthesized via the combination of ring-opening polymerization and facile coupling reactions. The triblock copolymer can self-assemble into vesicles in aqueous solution, and PCL forms the vesicle membrane, whereas the PEG and EPL blocks constitute the vesicle coronas. Atomic force microscopy (AFM) and transmission electron microscopy (TEM) had proved its vesicular structure. The excellent antibacterial ability of the ε-poly(L-lysine) makes the vesicles have good antibacterial activities towards both E. coli (Gram negative) and S. aureus (Gram positive), and the minimal inhibitory concentrations (MICs) were both 62.5 μg mL−1. Moreover, the special bacterial-killing mechanism of antibacterial peptides may avoid producing antibiotic-resistant bacteria. Besides, the cell viability assays showed that the triblock copolymer was noncytotoxic up to a tested concentration of 1000 μg mL−1. Therefore, the copolymer vesicles have high selectivity (1000/62.5). The DOX drug loading and release profile suggested that the triblock copolymer vesicles have drug releasing ability. At the same time, in vitro enzymatic biodegradation experiments proved that the triblock copolymer vesicles could be degraded in the existence of lipid enzyme. All these results indicate that these multifunctional triblock copolymer vesicles with much lower cytotoxicity, showing antibacterial activities (without antibiotic resistance), can be a promising substitute for antibiotics. In addition, it can be used as an “armed” delivery template for anticancer drugs due to its drug delivery capacity.
Introduction
More and more attention has been paid to the design and synthesis of amphiphilic block copolymers in the last few decades.1–3 Amphiphilic block copolymers have been widely reported to form particulate systems of various morphologies such as spherical or micelles, and vesicles.4–6 They are well established as building blocks for the preparation of vesicular or micellar drug carriers and can decrease the toxicity of the drugs and prolong drug release time.7–10 Recently, more and more attention has been paid to synthesize biodegradable and biocompatible amphiphilic block copolymers to prepare vesicles including antibacterial vesicles.11–15 Most of the cancer patients have lower resistance to bacterial infections during chemotherapy, which can be fatal to them.16,17 The antibacterial vesicles can not only be used as a drug carrier, but they are also able to resist bacterial infections.18 However, most of the reported antibacterial vesicles are silver nanoparticle vesicles19–21 or quaternized copolymer vesicles,4,22,23 which are not suitable for application in the human body due to their toxicity.24,25 Thus, it is urgent to develop highly effective and biocompatible antibacterial vesicles.Antimicrobial peptides (AMPs) have recently garnered significant attention as an emerging source of potential antibiotics, due to the swift emergence of multidrug-resistant bacteria.26–29 Most of the antimicrobial peptides are cationic (positive) antibacterial peptides, which are composed of hydrophobic amino acids (phenylalanine, alanine, leucine, etc.) and hydrophilic amino acids (lysine, arginine, etc.). Compared with traditional antibiotics, antimicrobial peptides have different antibacterial mechanism. It has been shown that antibacterial peptides can be absorbed onto the negative bacterial surface by electrostatic interaction, and then the hydrophobic amino acid residues insert into the lipid bilayer and induce formation of pore on the bacterial membrane.30–33 This type of antibacterial mechanism makes it difficult for bacteria to have drug resistance to antibacterial peptides.34,35 In addition, the antimicrobial peptides have broad spectrum and high antibacterial activity.34,36 They have potential application as a new generation of antibiotics to solve the drug-resistance problem.
Poly(ε-caprolactone) (PCL) is one of the most extensively studied biodegradable chains in various copolymers for drug carriers.8,37,38 PCL is usually used as the hydrophobic component of the amphiphilic block copolymers.39 Its wide applicability and excellent properties (controlled degradability, miscibility with other polymers, biocompatibility, etc.)38,40,41 make PCL a very useful polymer. At the same time, cationic antibacterial peptides can be selected as the hydrophilic chains to combine with PCL to synthesize amphiphilic block copolymers, which can self-assemble into PCL-polypeptide antibacterial vesicles. These types of vesicles have potential application in medical research. Our group has published a class of antibacterial vesicles prepared by PCL and poly(Phe12-stat-Lys15). However, its biocompatibility is not good enough due to the high toxicity of the polypeptide (poly(Phe12-stat-Lys15)).14
ε-Poly(L-lysine) (EPL) is a type of antibacterial peptide produced by filamentous fungus, which was accidentally found by Shima and Sakai.42 It consists of 25–35 lysine residues with linkages between the α-carboxyl and ε-amino groups.42,43 EPL has broad spectrum and efficient antibacterial ability, which is similar to that of common antibacterial peptides.43–46 However, there are no hydrophobic amino acid residues in EPL, which is different from common antibacterial peptides. The antibacterial mechanism is unclear to date. Scientists propose that antibacterial mechanism of EPL is similar to that of natural AMPs mentioned above. Due to the cationic property of EPL, it can be absorbed onto the bacterial surface via electrostatic interaction. Then, the hydrophobic groups (long carbon chains) insert into the lipid bilayer, destroying the integrity of the membrane. This leads to the stripping of the outer membrane and abnormal distribution of cytoplasm, finally causing the death of bacteria.47,48 EPL is widely used as food additive for its high antibacterial activity and nontoxicity to human cells.44,49 However, the EPL is difficult to dissolve in organic solvents, and therefore modification of EPL with hydrophobic polymer is seldom reported.
Herein, we synthesized a biodegradable and noncytotoxic polycaprolactone-polyethyleneglycol-ε-poly(L-lysine) triblock copolymer, and biodegradable and noncytotoxic antibacterial vesicles were also prepared by the triblock copolymer, as shown in Scheme 1. The hydrophobic PCL forms the membrane, and the EPL and PEG form the corona stabilizing the nanostructure in aqueous dispersion. The antibacterial ability was evaluated by MICs, and in vitro drug release was examined to assess their potential as a drug carrier. Cell Counting Kit-8 (CCK-8) results suggested that the triblock copolymer vesicles were biocompatible to human cells and at the same time, they were biodegradable. All results suggested that the triblock copolymer vesicles have potential application in cancer chemotherapy as an antibacterial drug carrier.
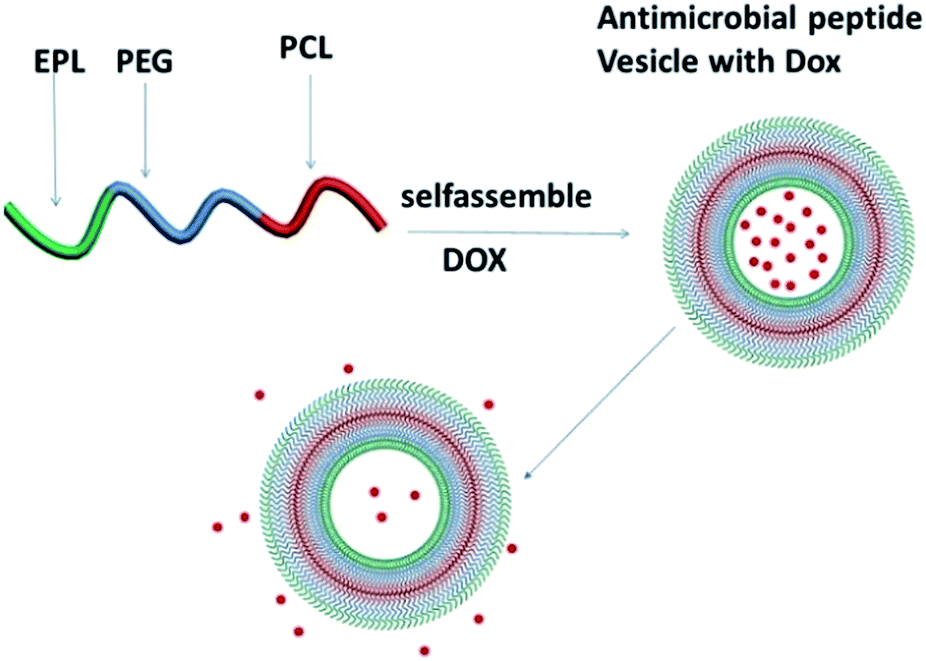 | ||
| Scheme 1 Illustration of PCL35-b-PEG45-b-EPL23 triblock copolymer vesicles as an “armed” drug carrier. | ||
Experimental
Materials
ε-Caprolactone (CL) was purchased from Sigma-Aldrich. Stannous 2-ethylhexanoate (Sn(Oct)2), dibutyltion dilaurate, tetrahydrofuran (THF), benzyl alcohol, toluene, dichloromethane (DCM), hexamethylene diisocyanate (HDI), polyethylene glycol (PEG, Mn = 2000), hexane, methanol and lipase were obtained from Aladdin. ε-Poly(L-lysine) (EPL, Mn = 3000) was purchased from Zhengzhou Bainafo Bioengineering Co., Ltd. Doxorubicin hydrochloride (DOX·HCl) was purchased from Xingcheng Chempharm Co., Ltd. Gram-negative bacteria E. coli (ATCC35218) and Gram-positive bacteria S. aureus (ATCC29213) were purchased from Nanjing Bianzhen Biological Technology Co. Ltd. L02 liver cells were purchased from Cell Bank, Shanghai Institute of Biochemistry and Cell Biology, Chinese Academic of Science. Water in ε-caprolactone was removed azeotropically with anhydrous toluene before using. Other chemicals were used directly as they were purchased.Synthesis of PCL35-OH
First, ε-caprolactone (20.000 g, 175.439 mmol) was dissolved in 100.0 mL of toluene in a round-bottom flask, and treated in an oil bath at 140 °C for 5 h to remove water. Then, 417.0 μL of the initiator benzyl alcohol and a drop of Sn(Oct)2 were added into the solution and reacted at 110 °C for 48 h under argon atmosphere. The reaction was terminated by cooling to room temperature. After removing the solvent using rotary evaporation, the crude product was dissolved in 20 mL of DCM and precipitated in excess methanol and filtered. Such purification steps were repeated three times. Eventually, the final product was dried in a vacuum oven at 40 °C for 2 days. Yield: 70%.Synthesis of PCL35-NCO
PCL35-OH (2.000 g) was dissolved in 10 mL DCM in argon atmosphere. Then, 0.842 g HDI and two drops of dibutyltion dilaurate (as catalyst) were added into the solution. After reaction at 0 °C for 4 h, the mixture was precipitated in excess hexane to remove excess HDI, and the supernatant was discarded after standing for a few minutes. Then, the precipitate was dissolved in 10 mL of DCM and recrystallized in excess hexane again. Such purification steps were repeated three times. Finally, the product dissolved in 30 mL of DCM was ready for the next reaction.Synthesis of PCL35-b-PEG45-OH
Herein, 20.000 g PEG in 40 mL of DCM was added into the PCL35-NCO solution, two drops of dibutyltion dilaurate were added to the mixture and reacted at room temperature for 24 h, and then the DCM was removed by rotary evaporation. The crude product was dialyzed against deionized water in a dialysis tube with 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MWCO (Molecular Weight Cutoff) for 24 h (deionized water was changed every 2 h). After that, the product was obtained by freeze-drying. Yield: 83%.
000 MWCO (Molecular Weight Cutoff) for 24 h (deionized water was changed every 2 h). After that, the product was obtained by freeze-drying. Yield: 83%.
Synthesis of PCL35-b-PEG45-NCO
PCL35-b-PEG45-OH (2.000 g) was dissolved in 10 mL of DCM in a flask in an anhydrous condition. Then, 0.561 g HDI and two drops of dibutyltion dilaurate were added into the solution and stirred at 0 °C for 4 h. After that, the crude product was precipitated in excess hexane to remove excess HDI, and the supernatant was discarded after standing for a few minutes. Then, the sticky solid was dissolved in 10 mL of DCM and precipitated in excess hexane again. The process was repeated three times. Finally, the product (1.000 g) dissolved in 50 mL of THF was ready for the next reaction.Synthesis of PCL35-b-PEG45-b-EPL23
Excess EPL (10.000 g) was dissolved in 100 mL of deionized water, and 50 mL of THF was added. Then, PCL35-b-PEG45-NCO solution was dropped into the prepared EPL turbid solution. The mixture reacted at room temperature for 24 h. After that, the solution was dialyzed against deionized water in a dialysis tube with 14000 MWCO for 4 days to remove excess EPL (deionized water was changed every 2 h). The product was dried by freeze-drying. Yield: 80%.
Self-assembly of PCL35-b-PEG45-b-EPL23
Herein, 5.000 mg PCL35-b-PEG45-b-EPL23 was dissolved in 3.0 mL of mixture of THF/H2O (1:1, v/v). Then, deionized water was added into the solution at a rate of 0.6 mL min−1 for 10 min using a gastight syringe with violent stirring. After stirring for another 12 h, the solution was dialyzed against deionized water using a dialysis tube (14000 MWCO) for 24 h to remove the THF (deionized water was changed every 2 h).
Preparation of DOX-loaded copolymer vesicles
Herein, 15.000 mg PCL35-b-PEG45-b-EPL23 and 3.500 mg DOX·HCl were dissolved in 5.0 mL of mixture of THF/H2O (1:1, v/v). Then, 10.0 mL of deionized water was added into the solution and stirred continuously for over 10 min. The drug-loaded copolymer vesicles were obtained by dialysis against 1000 mL of tris buffer (0.01 M, pH 7.4) at 25 °C, for removing the unloaded drug and THF (dialysis medium renewed each 0.5 h during the first 3 h).
To determine the drug loading content (DLC) and drug loading efficiency (DLE) of the polymer vesicles, a series of DOX/tris buffer solutions with different concentrations were measured by fluorescence spectroscopy, and then a calibration curve representing dependence of DOX fluorescence intensity on concentration was obtained to help quantify the DOX concentration in the polymer vesicles. The DLC and DLE of the polymer vesicles were calculated by the following equations:
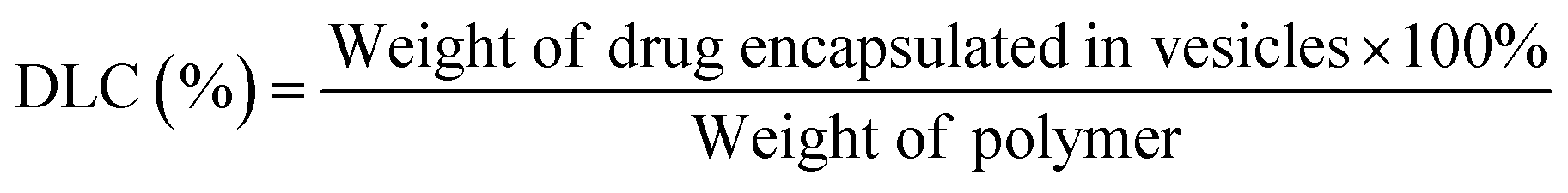
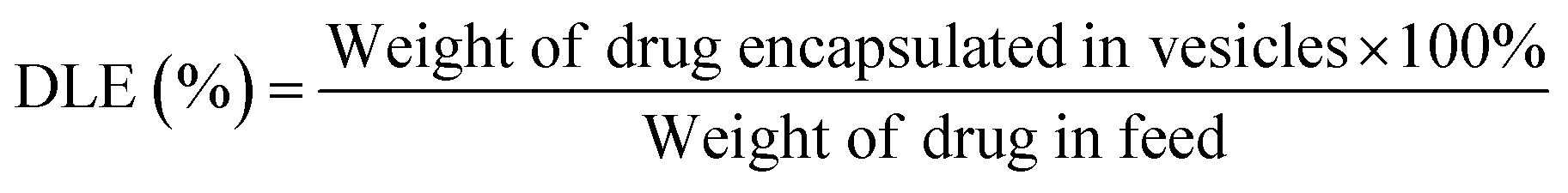
Antibacterial test
The antibacterial assay was conducted based on the optical density methods.50 E. coli and S. aureus cells were cultured to a mid-log phase at 37 °C in Luria-Bertani (LB) broth medium. Before addition of PCL35-b-PEG45-b-EPL23 vesicle solution, the concentration of bacteria solution was diluted to 104–105 colony forming unit (CFU) mL−1. Moreover, a series of vesicle solutions with different concentrations (from 1000 to 8 μg mL−1) were prepared by semi-dilution and mixed with the bacteria solution in each Petri dish. Each dish was incubated at 37 °C for 24 h and its optical density was measured at 600 nm every 2 h. LB broth with bacteria cells alone was used as control. The tests were repeated at least three times.Cytotoxicity test
The cytotoxicity of antibacterial peptides was determined via Cell Counting Kit-8 (CCK-8) assay according to literature.51,52 The cytotoxicity to normal liver cells (L02) was evaluated. L02 cells were cultivated at 37 °C with equal density (4000 cells per well) in each well, which was filled with 100.0 μL of Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) in a humidified 5% CO2-containing atmosphere. After 24 h of incubation of L02 cells, 20.0 μL of polymer vesicles solutions with varied concentrations (250, 500, 1000 and 2000 μg mL−1) was incubated with cells for another 24, 48 and 72 h. The untreated L02 cells served as controls. Finally, CCK-8 dye was added to each well, and after incubation for 1 h at 37 °C, the absorbance was eliminated using a microplate reader in the method of dual wavelength spectrophotometry at 450 nm and 630 nm. Each treatment was repeated five times. Finally, the relative cell viability (%) was calculated by comparing the absorbance at 450 nm with control wells with cell culture medium alone.In vitro drug release test
The drug release experiment was designed based on the protocols in literature.39,53 Briefly, the drug release process was carried out by dividing drug-loaded copolymer vesicles into three dialysis tubes (14000 MWCO), each containing 3.0 mL solution. Each dialysis tube was dialyzed against 80 mL tris buffer (0.01 M, pH 7.4) in a 100 mL of beaker at 37 °C. The release media in the beakers were measured with fluorescence spectroscopy at different time intervals. Three parallel experiments were conducted simultaneously, for minimizing experimental error. Finally, the cumulative release curve of DOX was obtained based on the calibration curve (DOX fluorescence intensity vs. concentration).
In vitro enzymatic degradation test
Due to the presence of the biodegradable PCL block, the triblock copolymer vesicles in this study were capable of being degraded as well. To evaluate the degradation degree of the polymer vesicles, in vitro degradation test was carried out. The aqueous lipase solution (0.034 mL) and 1.66 mL of vesicles solution (0.05 mg mL−1) were mixed together. Then, the mixture was put into an incubator at 37 °C. The count rate reflecting the degradation degree was detected at regular intervals by dynamic light scatting (DLS).Characterization
Results and discussion
Synthesis of PCL35-b-PEG45-b-EPL23 triblock copolymer
The PCL35-b-PEG45-b-EPL23 triblock copolymer was synthesized in five steps, and the synthetic route is shown in Scheme 2: first, the PCL35-OH was prepared by ring opening polymerization of caprolactone with benzyl alcohol as the initiator, and a single hydroxyl group (OH) existed on the end of PCL. Then, PCL35-OH was modified by HDI to form the PCL35-NCO with excess HDI and the crude product was purified by precipitating in hexane for three times. After that, excess PEG was added to react with PCL35-NCO in THF to form the PCL35-b-PEG45 diblock copolymer, and then precipitated in methanol for three times. PCL35-b-PEG45-NCO was prepared by the reaction between PCL35-b-PEG45 diblock copolymer and excess HDI, then precipitated in large amounts of hexane to remove excess HDI for three times. Finally, PCL35-b-PEG45-NCO reacted with excess EPL in the mixture of THF/H2O (1:1, v/v) at room temperature and was purified by dialysis in deionized water to obtain PCL35-b-PEG45-b-EPL23 triblock copolymers. Their chemical structure was characterized by 1H-NMR (Fig. 1 and S1–S4†) and GPC (Fig. S5†). All of these results proved that the triblock copolymer was successfully synthesized.
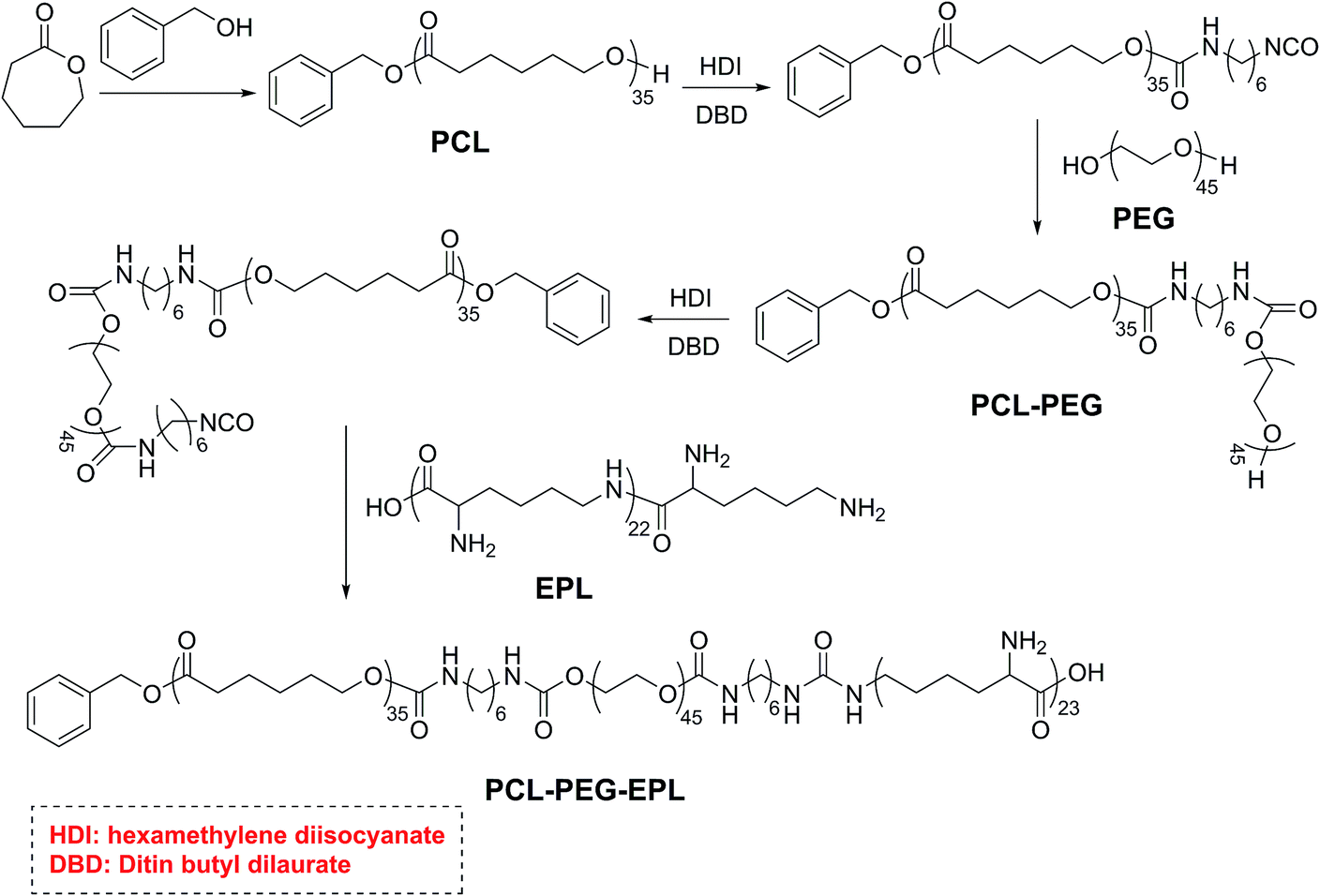 | ||
| Scheme 2 Synthetic strategy toward antibacterial PCL35-b-PEG45-b-EPL23 triblock copolymer. | ||
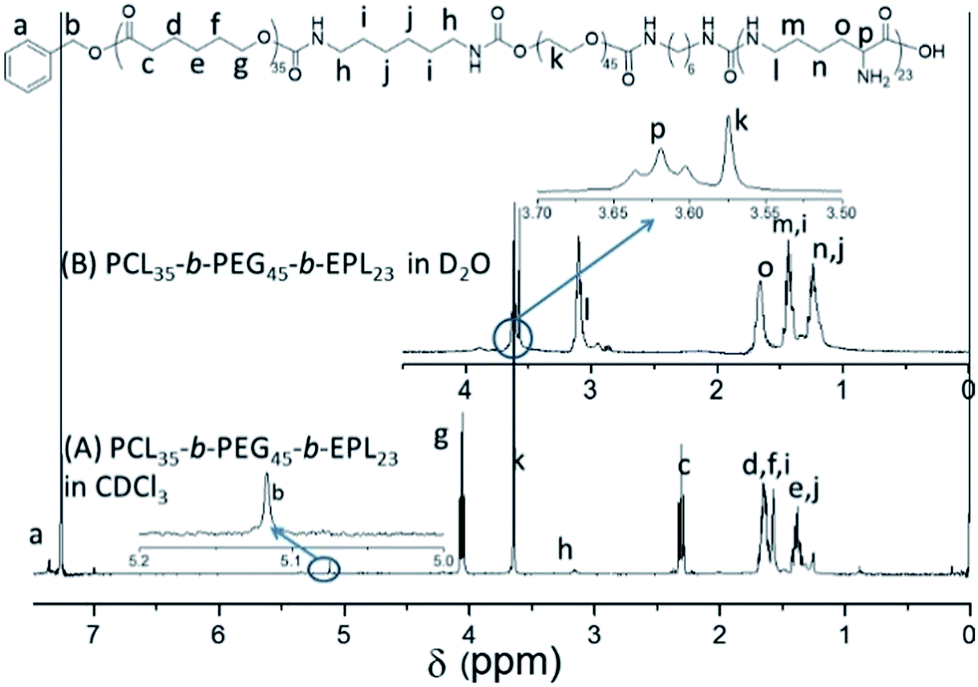 | ||
| Fig. 1 1H NMR spectra of PCL35-b-PEG45-b-EPL23 in D2O and CDCl3. | ||
Self-assembly of PCL35-b-PEG45-b-EPL23 triblock copolymer
The amphiphilic triblock copolymer can self-assemble into nanoparticles in THF/H2O (1:4, v/v), as shown in Scheme 1. THF can be removed by dialysis in deionized water for 2 days. The DLS results by intensity showed that the average diameter of the nanoparticles was about 373 nm. AFM study (Fig. 2) suggested a diameter of 260 nm, which was reasonably in agreement with the DLS results. At the same time, the ratio of diameter/height of the nanoparticle was about 16.3; the height profile of AFM study confirms the hollow structure of the triblock copolymers vesicles since their soft and deformable vesicle has collapsed on the silicon substrate.54 The vesicular structures were also observed by TEM (Fig. S6†).
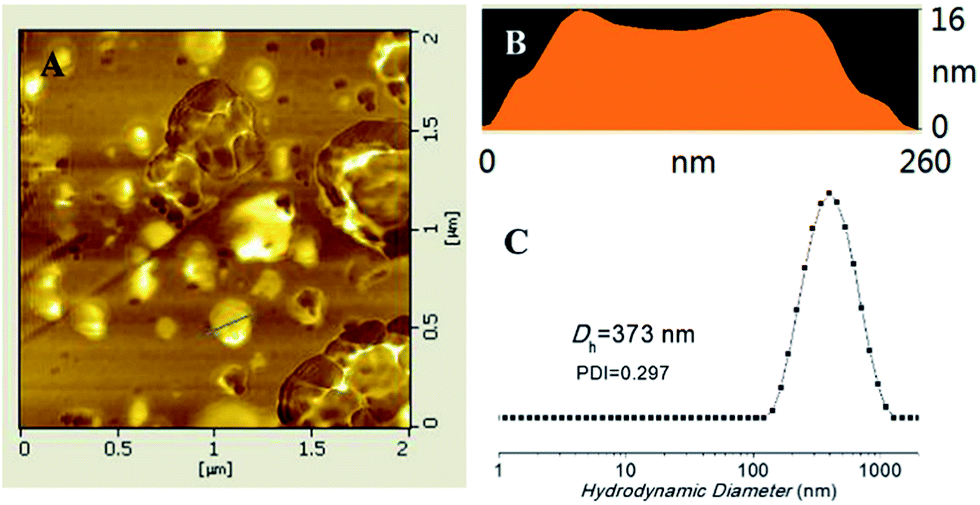 | ||
| Fig. 2 PCL35-b-PEG45-b-EPL23 vesicles: (A) height contrast of atomic force microscopy (AFM) images; (B) the corresponding height profile reveals an aspect ratio of 16, suggesting a capsular structure; (C) size distribution by DLS. | ||
Minimal inhibitory concentrations (MICs)
MIC is an important value for antibacterial materials, and the MICs of the PCL35-b-PEG45-b-EPL23 copolymers were measured using broth microdilution method. Briefly, PCL35-b-PEG45-b-EPL23 triblock copolymer vesicles solution was diluted with LB broth to prepare copolymer solutions with different concentrations in a 96-well plate. Then, the microorganism solution with an optical density reading of 0.7–0.8 at 600 nm was added to each well. The minimum inhibitory concentrations (MICs) are defined as the lowest peptide concentration that arrested bacterial growth after 18 h, representing the concentration where bacteria are lower than 99% growth compared with the control. Fig. 3 showed that the triblock copolymer vesicles had the MIC values of 62.5 μg mL−1 against both E. coli and S. aureus. The value was a little higher than some natural or synthetic antibacterial peptides, which is due to reduction of the effective antibacterial component (EPL) caused by incorporation of non-antibacterial polymer segments (PCL, PEG) into the triblock copolymer. Nevertheless, its antibacterial ability was still better than a lot of AMPs55–57 and cationic polymers.58,59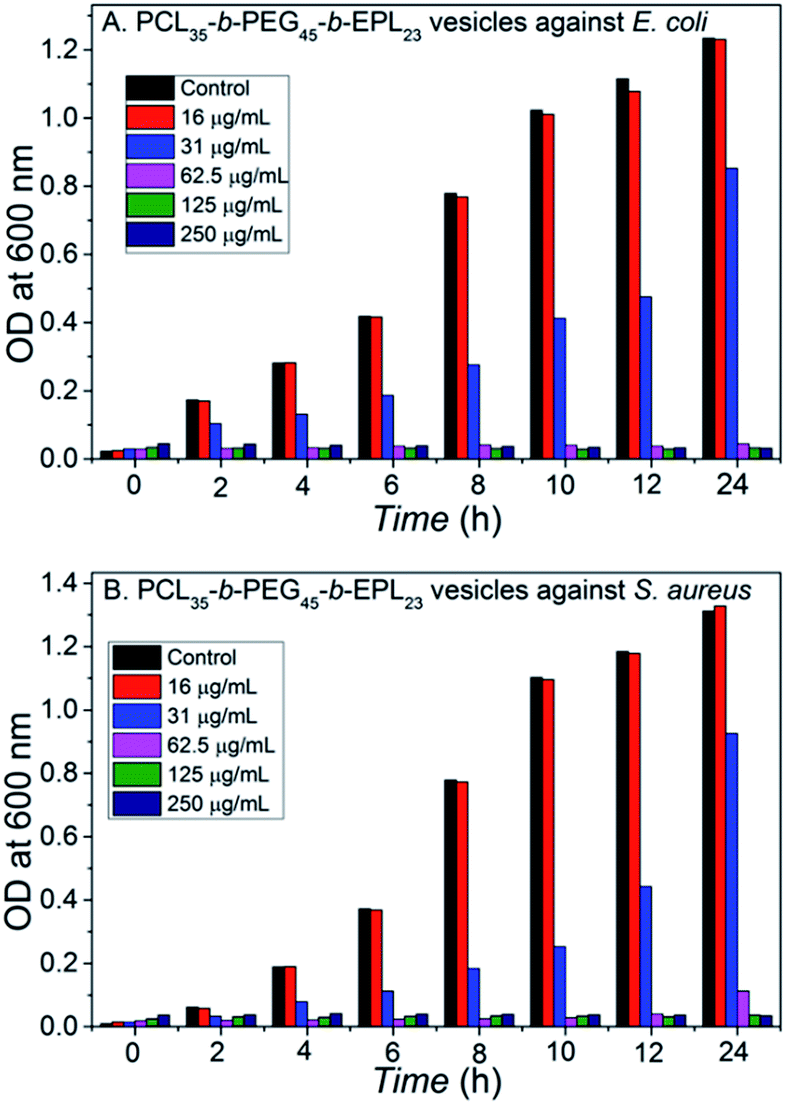 | ||
| Fig. 3 Dose-dependent growth inhibition: (A) E. coli treated with the vesicles; (B) S. aureus treated with the vesicles. | ||
Cytotoxicity study
The in vitro cytotoxicity tests were conducted by culturing PCL35-b-PEG45-b-EPL23 triblock copolymer vesicles with human L02 liver cells for 3 days. The concentration of copolymer vesicles varied from 250 to 2000 μg mL−1, respectively. The CCK-8 assay in Fig. 4 showed that the relative cell viability of the triblock copolymer vesicles remained over 75% up to a concentration of 1000 μg mL−1 after culturing for 72 h, which confirmed that these copolymer vesicles maintained the metabolic activities of L02 liver cells at various vesicle concentrations (up to 1000 μg mL−1). The result is better than that of most of natural AMPs,60 synthetic AMPs61 and cationic antibacterial polymers,62 which is due to the fact that all three blocks (PCL, PEG, EPL) in this study are noncytotoxic and biocompatible.39–41,44 The cytotoxicity is very low (1000 μg mL−1), thus such triblock copolymers maintain high selectivity (1000/62.5). Therefore, the antibacterial copolymer vesicles may have potential application in nanomedicine and clinical fields due to their noncytotoxicity properties.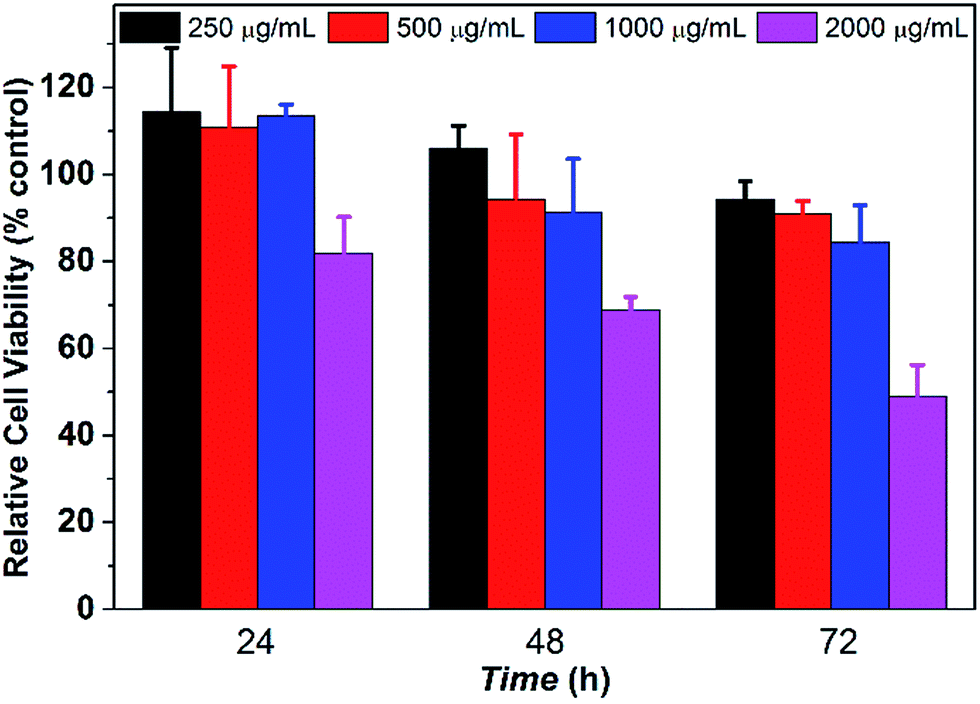 | ||
| Fig. 4 Cytotoxicity of PCL35-b-PEG45-b-EPL23 against L02 (CCK-8 assay, n = 5). | ||
Drug loading/release profiles of copolymer vesicles
The amphiphilic PCL35-b-PEG45-b-EPL23 copolymer can self-assemble into vesicles in aqueous solutions, which enables the triblock copolymers to be a potential drug carrier for clinical applications. Doxorubicin (DOX), an anticancer drug, was applied to evaluate the drug-loading and release ability of the triblock copolymer vesicles. The drug loading content (DLC) and drug loading efficiency (DLE) were calculated as follows: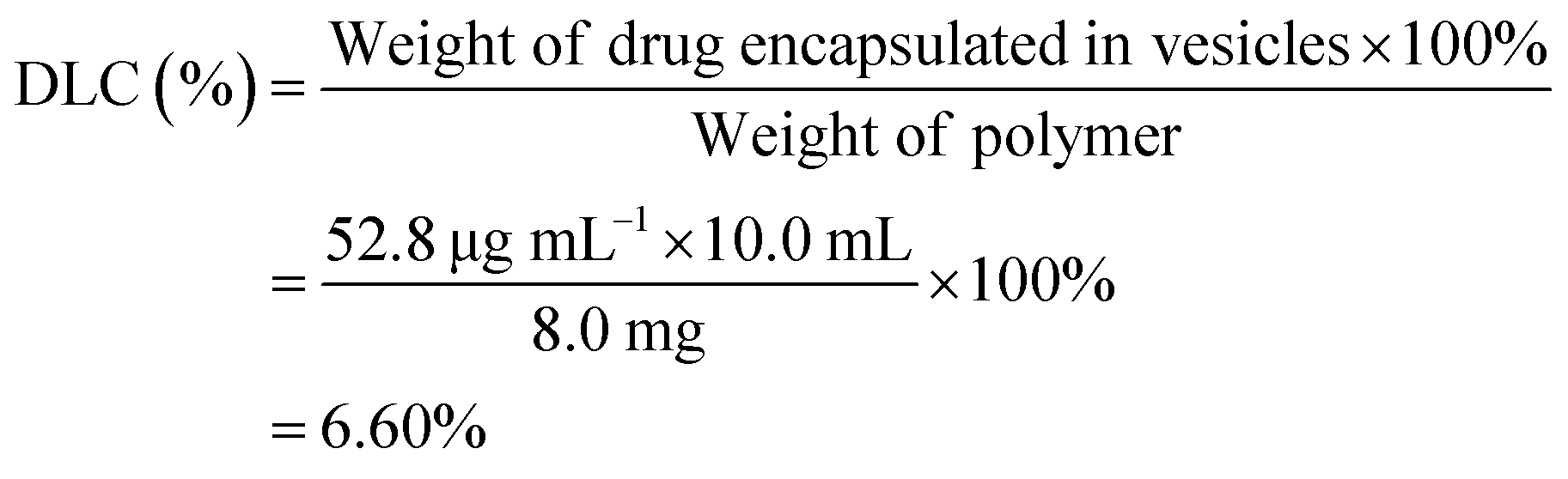
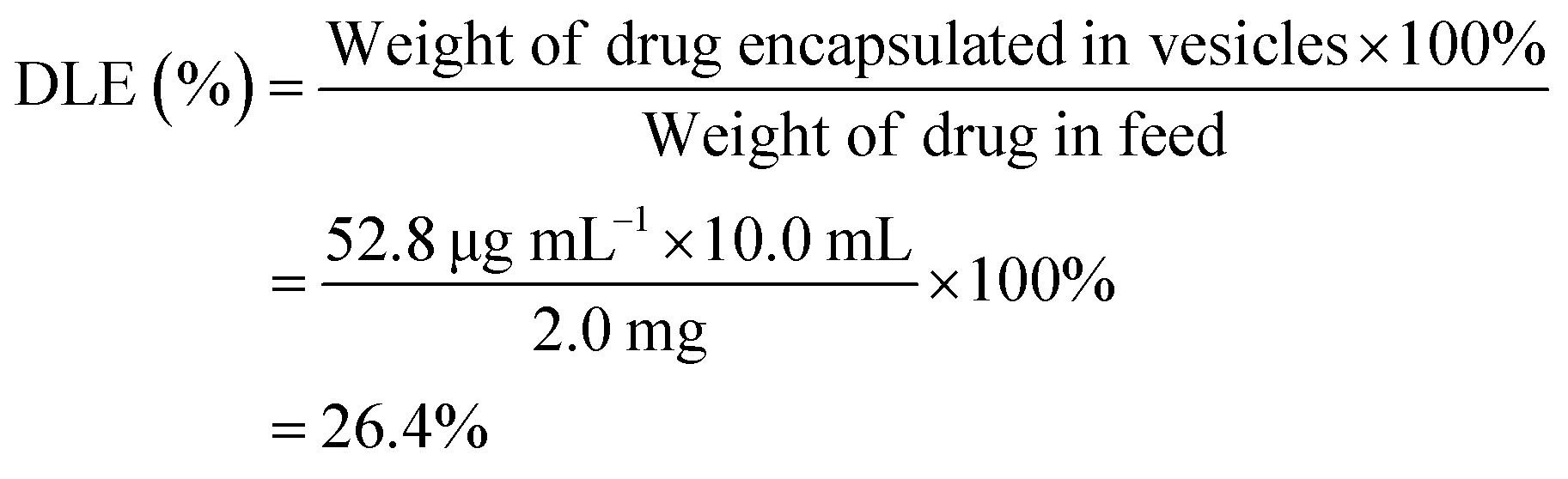
As a control of the triblock copolymer vesicles, the free DOX (0.050 mg mL−1) solution without vesicles was prepared, and the release profile was obtained in a similar method. The release rate of the DOX-loaded copolymer vesicles was slower than the release rate of the free DOX (in Fig. 5). These results showed that the vesicles have drug release ability and can prolong the release cycle to a certain extent. Compared with usual amphiphilic copolymers and their vesicles, our synthetic triblock copolymers can self-assemble into vesicles with antibacterial ability and drug-release ability, which enables the vesicles an “armed” drug carrier. These types of vesicles can be applied in chemotherapy for cancer patients.
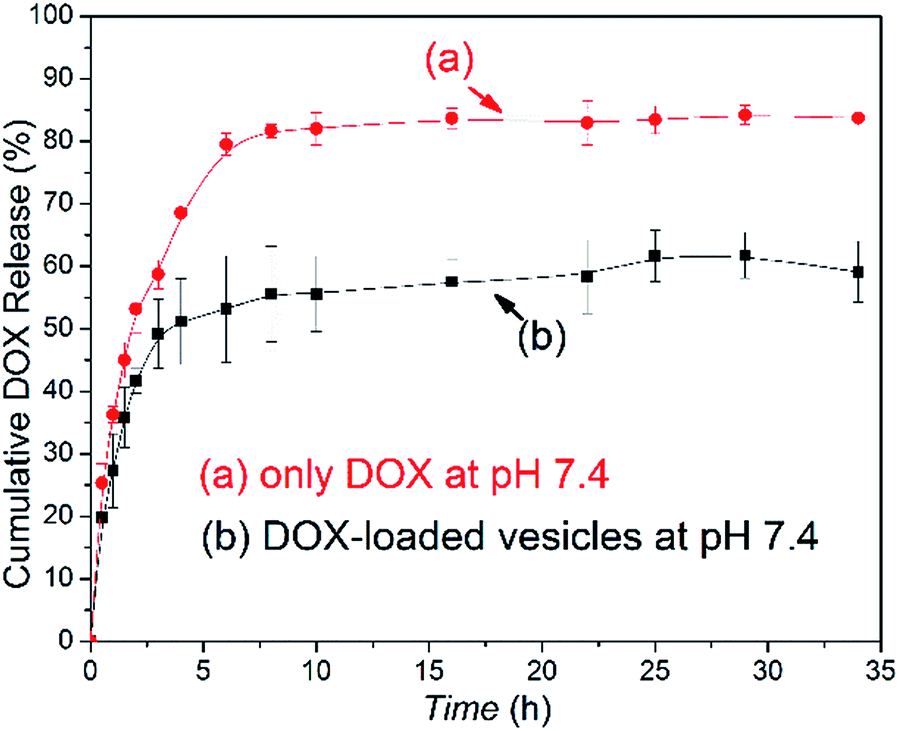 | ||
| Fig. 5 DOX release profiles of (a) free DOX in 0.01 M tris buffer and (b) DOX-loaded vesicles at pH 7.4 and 37 °C. | ||
In vitro enzymatic biodegradation
PCL is a type of enzymatically degradable material. The PCL forms the shell of the antibacterial copolymer vesicles, and upon the PCL being degraded, the vesicles are disintegrated simultaneously. To evaluate the biodegradation ability of the PCL35-b-PEG45-b-EPL23 triblock copolymer vesicles, in vitro enzymatic biodegradation experiments were carried out in the presence of lipase. As shown in Fig. 6, the degradation curve indicated that 80% of copolymer vesicles had been degraded in the presence of lipase. Compared with the system “PCL-b-poly(Phe12-stat-Lys15)” that our group had developed before,14 the degradation rate of the PCL35-b-PEG45-b-EPL23 triblock copolymer is much faster than that of the previous one which reached 80% degradation rate in 3 h.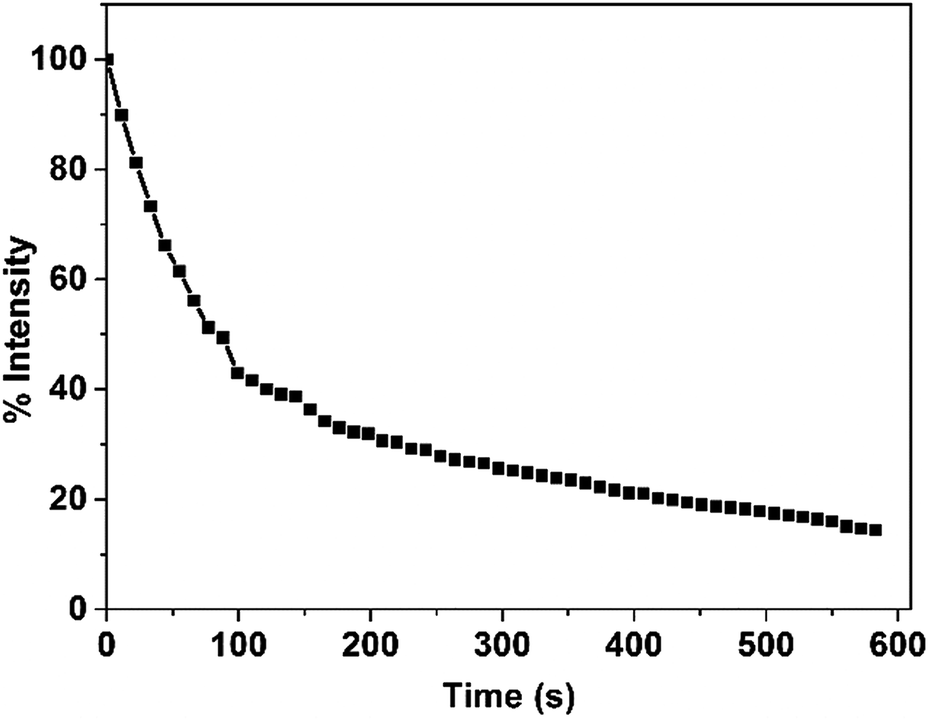 | ||
| Fig. 6 Count rate of PCL35-b-PEG45-b-EPL23 copolymer vesicles during lipase catalysed degradation. The concentration of the triblock copolymer vesicles and the lipase are 0.50 mg mL−1 and 0.03 mg mL−1, respectively. | ||
Conclusions
In brief, we have designed and synthesized a new triblock copolymer PCL35-b-PEG45-b-EPL23, which can form antibacterial vesicles as a multifunctional drug carrier. EPL affords the antibacterial peptide vesicle coronas, which are highly effective against Gram-negative and Gram-positive bacteria. Moreover, EPL does not induce drug-resistance in bacteria owing to its special pore-forming antibacterial mechanism, which is similar to that of common AMPs. The antibacterial copolymer vesicles can be degraded and dissociated after drug release due to biodegradable PCL chains. Most importantly, all the three block parts are biocompatible. Thus, the antibacterial vesicles have higher selectivity than most AMPs and cationic antibacterial copolymers. Overall, such multifunctional triblock copolymer vesicles with low cytotoxicity have potential application in clinical treatment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by NSFC 21274110.References
- A. Klaikherd, C. Nagamani and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 4830–4838 CrossRef CAS PubMed.
- T. Shimizu, M. Masuda and H. Minamikawa, Chem. Rev., 2005, 105, 1401–1444 CrossRef CAS PubMed.
- S. Forster and M. Antonietti, Adv. Mater., 1998, 10, 195–217 CrossRef.
- A. Muñoz-Bonilla and M. Fernández-García, Eur. Polym. J., 2015, 65, 46–62 CrossRef.
- J. Wang, B. Li, X. Wang, F. Yang, H. Shen and D. Wu, Langmuir, 2016, 32, 13706–13715 CrossRef CAS PubMed.
- W.-N. He, B. Zhou, J.-T. Xu, B.-Y. Du and Z.-Q. Fan, Macromolecules, 2012, 45, 9768–9778 CrossRef CAS.
- A. Rösler, G. W. M. Vandermeulen and H.-A. Klok, Adv. Drug Delivery Rev., 2012, 64(suppl.), 270–279 CrossRef.
- M. Álvarez-Paino, A. Muñoz-Bonilla and M. Fernández-García, Nanomaterials, 2017, 7, 48 CrossRef PubMed.
- F. Wang, T. K. Bronich, A. V. Kabanov, R. D. Rauh and J. Roovers, Bioconjugate Chem., 2005, 16, 397–405 CrossRef CAS PubMed.
- B.-S. Kim, S. W. Park and P. T. Hammond, ACS Nano, 2008, 2, 386–392 CrossRef CAS PubMed.
- L. Zhang, Y. Feng, H. Tian, M. Zhao, M. Khan and J. Guo, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3213–3226 CrossRef CAS.
- C. Zhang, Y. Zhu, C. Zhou, W. Yuan and J. Du, Polym. Chem., 2013, 4, 255–259 RSC.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- M. Wang, C. Zhou, J. Chen, Y. Xiao and J. Du, Bioconjugate Chem., 2015, 26, 725–734 CrossRef CAS PubMed.
- F. Ye, Å. Barrefelt, H. Asem, M. Abedi-Valugerdi, I. El-Serafi, M. Saghafian, K. Abu-Salah, S. Alrokayan, M. Muhammed and M. Hassan, Biomaterials, 2014, 35, 3885–3894 CrossRef CAS PubMed.
- C.-C. Lai, L.-J. Teng, P.-R. Hsueh, A. Yuan, K.-C. Tsai, J.-L. Tang and H.-F. Tien, Clin. Infect. Dis., 2004, 38, 149–153 CrossRef CAS PubMed.
- A. M. Bressler, K. S. Kaye, J. J. LiPuma, B. D. Alexander, C. M. Moore, L. B. Reller and C. W. Woods, Infect. Contr. Hosp. Epidemiol., 2007, 28, 951–958 CrossRef PubMed.
- C. Zhou, M. Wang, K. Zou, J. Chen, Y. Zhu and J. Du, ACS Macro Lett., 2013, 2, 1021–1025 CrossRef CAS.
- Y. Deng, J. Li, J. Yu, J. Zhao and J. Tang, Mater. Sci. Eng., 2016, 60, 92–99 CrossRef CAS PubMed.
- S. Pal, Y. K. Tak and J. M. Song, Appl. Environ. Microbiol., 2007, 73, 1712–1720 CrossRef CAS PubMed.
- A. R. Shahverdi, A. Fakhimi, H. R. Shahverdi and S. Minaian, Nanomedicine, 2007, 3, 168–171 CrossRef CAS PubMed.
- N. Joondan, S. Jhaumeer Laulloo, P. Caumul, D. E. P. Marie, P. Roy and E. Hosten, Colloids Surf., A, 2016, 511, 120–134 CrossRef CAS.
- L. Timofeeva and N. Kleshcheva, Appl. Microbiol. Biotechnol., 2011, 89, 475–492 CrossRef CAS PubMed.
- M. Ahamed, M. S. AlSalhi and M. K. J. Siddiqui, Clin. Chim. Acta, 2010, 411, 1841–1848 CrossRef CAS PubMed.
- A. Muñoz-Bonilla and M. Fernández-García, Prog. Polym. Sci., 2012, 37, 281–339 CrossRef.
- A. K. Marr, W. J. Gooderham and R. E. W. Hancock, Curr. Opin. Pharmacol., 2006, 6, 468–472 CrossRef CAS PubMed.
- R. E. W. Hancock and H.-G. Sahl, Nat. Biotechnol., 2006, 24, 1551–1557 CrossRef CAS PubMed.
- R. E. W. Hancock and A. Patrzykat, Curr. Drug Targets: Infect. Disord., 2002, 2, 79–83 CrossRef CAS.
- H. Jenssen, P. Hamill and R. E. Hancock, Clin. Microbiol. Rev., 2006, 19, 491–511 CrossRef CAS PubMed.
- A. J. Karlsson, W. C. Pomerantz, B. Weisblum, S. H. Gellman and S. P. Palecek, J. Am. Chem. Soc., 2006, 128, 12630–12631 CrossRef CAS PubMed.
- K. A. Brogden, Nat. Rev. Microbiol., 2005, 3, 238–250 CrossRef CAS PubMed.
- M. Zasloff, Nature, 2002, 415, 389–395 CrossRef CAS PubMed.
- F. Siedenbiedel and J. C. Tiller, Polymers, 2012, 4, 46 CrossRef CAS.
- C. Chen, Y. Chen, C. Yang, P. Zeng, H. Xu, F. Pan and J. R. Lu, ACS Appl. Mater. Interfaces, 2015, 7, 17346–17355 CAS.
- S. J. Lam, E. H. H. Wong, N. M. O'Brien-Simpson, N. Pantarat, A. Blencowe, E. C. Reynolds and G. G. Qiao, ACS Appl. Mater. Interfaces, 2016, 8, 33446–33456 CAS.
- C. Chen, J. Hu, P. Zeng, Y. Chen, H. Xu and J. R. Lu, ACS Appl. Mater. Interfaces, 2014, 6, 16529–16536 CAS.
- J. S. Chawla and M. M. Amiji, Int. J. Pharm., 2002, 249, 127–138 CrossRef CAS PubMed.
- V. R. Sinha, K. Bansal, R. Kaushik, R. Kumria and A. Trehan, Int. J. Pharm., 2004, 278, 1–23 CrossRef CAS PubMed.
- Y. Zhang and R.-x. Zhuo, Biomaterials, 2005, 26, 6736–6742 CrossRef CAS PubMed.
- X. Wei, C. Gong, M. Gou, S. Fu, Q. Guo, S. Shi, F. Luo, G. Guo, L. Qiu and Z. Qian, Int. J. Pharm., 2009, 381, 1–18 CrossRef CAS PubMed.
- S. Zhou, X. Deng and H. Yang, Biomaterials, 2003, 24, 3563–3570 CrossRef CAS PubMed.
- S. Shima and H. Sakai, Agric. Biol. Chem., 1977, 41, 1807–1809 CAS.
- S. C. Shukla, A. Singh, A. K. Pandey and A. Mishra, Biochem. Eng. J., 2012, 65, 70–81 CrossRef CAS.
- I.-L. Shih, M.-H. Shen and Y.-T. Van, Bioresour. Technol., 2006, 97, 1148–1159 CrossRef CAS PubMed.
- Z. Xu, Z. Xu, X. Feng, D. Xu, J. Liang and H. Xu, Appl. Microbiol. Biotechnol., 2016, 100, 6619–6630 CrossRef CAS PubMed.
- Y.-Q. Li, Q. Han, J.-L. Feng, W.-L. Tian and H.-Z. Mo, Food Control, 2014, 43, 22–27 CrossRef CAS.
- S. Shima, H. Matsuoka, T. Iwamoto and H. Sakai, J. Antibiot., 1984, 37, 1449–1455 CrossRef CAS PubMed.
- R. Ye, H. Xu, C. Wan, S. Peng, L. Wang, H. Xu, Z. P. Aguilar, Y. Xiong, Z. Zeng and H. Wei, Biochem. Biophys. Res. Commun., 2013, 439, 148–153 CrossRef CAS PubMed.
- L. Zhang, R. Li, F. Dong, A. Tian, Z. Li and Y. Dai, Food Chem., 2015, 166, 107–114 CrossRef CAS PubMed.
- W.-R. Li, X.-B. Xie, Q.-S. Shi, H.-Y. Zeng, Y.-S. OU-Yang and Y.-B. Chen, Appl. Microbiol. Biotechnol., 2010, 85, 1115–1122 CrossRef CAS PubMed.
- C. Beer, R. Foldbjerg, Y. Hayashi, D. S. Sutherland and H. Autrup, Toxicol. Lett., 2012, 208, 286–292 CrossRef CAS PubMed.
- J. Lou, G. Chu, G. Zhou, J. Jiang, F. Huang, J. Xu, S. Zheng, W. Jiang, Y. Lu, X. Li, Z. Chen and J. He, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2010, 697, 55–59 CrossRef CAS PubMed.
- W. Chen, P. Zhong, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2013, 169, 171–179 CrossRef CAS PubMed.
- J. Huang, C. Bonduelle, J. Thévenot, S. Lecommandoux and A. Heise, J. Am. Chem. Soc., 2012, 134, 119–122 CrossRef CAS PubMed.
- Y. Xi, T. Song, S. Tang, N. Wang and J. Du, Biomacromolecules, 2016, 17, 3922–3930 CrossRef CAS PubMed.
- J.-Y. Je and S.-K. Kim, Biochim. Biophys. Acta, Gen. Subj., 2006, 1760, 104–109 CrossRef CAS PubMed.
- M. A. Fox, J. E. Thwaite, D. O. Ulaeto, T. P. Atkins and H. S. Atkins, Peptides, 2012, 33, 197–205 CrossRef CAS PubMed.
- O. Wiarachai, N. Thongchul, S. Kiatkamjornwong and V. P. Hoven, Colloids Surf., B, 2012, 92, 121–129 CrossRef CAS PubMed.
- V. Sambhy, M. M. MacBride, B. R. Peterson and A. Sen, J. Am. Chem. Soc., 2006, 128, 9798–9808 CrossRef CAS PubMed.
- J. Johansson, G. H. Gudmundsson, M. n. E. Rottenberg, K. D. Berndt and B. Agerberth, J. Biol. Chem., 1998, 273, 3718–3724 CrossRef CAS PubMed.
- D. Pranantyo, L. Q. Xu, E.-T. Kang, M. K. Mya and M. B. Chan-Park, Biomacromolecules, 2016, 17, 4037–4044 CrossRef CAS PubMed.
- Y. Cong, T. Xia, M. Zou, Z. Li, B. Peng, D. Guo and Z. Deng, J. Mater. Chem. B, 2014, 2, 3450–3461 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra, GPC trace, TEM images of vesicles. See DOI: 10.1039/c7ra07102g |
| This journal is © The Royal Society of Chemistry 2017 |