
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solid liquid liquid extraction of porcine gastric mucins from homogenized animal material†
Veronika Schoemig ,
Eda Isik,
Lea Martin and
Sonja Berensmeier*
,
Eda Isik,
Lea Martin and
Sonja Berensmeier*
Bioseparation Engineering Group, Department of Mechanical Engineering, Technical University of Munich, Boltzmannstr. 15, D-85748 Garching, Germany. E-mail: s.berensmeier@tum.de
First published on 14th August 2017
Abstract
Mucins purified from porcine stomachs have gained importance in biomedical applications as they exhibit unique features such as hydrogel formation, lubricity and antivirality. Because commercially available porcine gastric mucins (PGM) are neither gel forming, nor do they reduce friction, a robust purification process for functional mucins from porcine tissue (mainly Muc5AC) is necessary. Based on our former investigations (V. J. Schömig, B. T. Käsdorf, C. Scholz, K. Bidmon, O. Lieleg and S. Berensmeier, RSC Adv., 2016, 6, 44932–44943), we further optimized the established purification process in terms of productivity and overall yield as well as ease of scalability. We therefore introduced a novel extraction method – solid liquid liquid extraction (SLLE) – as an early capture step from homogenized porcine stomach, which combines conventional solid liquid extraction with a second, immiscible solvent, to simultaneously delipidate the tissue and extract hydrophilic proteins into the polar phase. Using Design of Experiments (DoE) the parameters incubation time and ratio of solvent phases (hexane/water) were identified to 3 h and 1/15 for SLLE, respectively. PGM was collected in the polar phase and further purified by size exclusion chromatography and diafiltration. With the homogenization of porcine stomach and the introduction of the SLLE, up to 3570 mg mucin per stomach can be purified, resulting in 55 times more PGM compared to the former published reference process. The productivity of the process was up to 25 mg mucin per stomach and per h, representing an improved process productivity by a factor of 4. Lubricity and gel formation of the purified mucin were retained with the optimized extraction protocol.
1 Introduction
Mucins are high molecular weight glycoproteins and a component of mucus, a gel layer that protects inner parts of the body such as the gastrointestinal tract, genitals and the respiratory tract from shear forces and infection. Mucus consists of not only mucins, but it also contains a high portion of water (95%), lipids, salts, growth factors and other proteins.2 More than a dozen mucins have been identified, which are divided into membrane-bound and secreted mucins. Most of the secreted mucins are gel forming such as Muc5AC, which is common in the stomach, and Muc5B, which is found in the respiratory tract.3,4 Native gastric gel forming mucins offer unique properties, such as gel formation at acidic pH values,5,6 excellent lubricity7 and they act as a threshold against pathogens due to their physical and interaction-based barrier.Mucin monomers have a molecular weight of 640 kDa.8 However, because of inter- and intramolecular disulfide bonds, mucins are linked together to form polymers of up to 40 MDa.9–11 80% of the glycoprotein's molecular weight is assigned to carbohydrate moieties that make mucin mostly hydrophilic and play a major role in the protein's net negative charge and its functionality. Hydrophobic patches within the protein backbone contribute to the excellent adsorption onto hydrophobic surfaces12 and serve as the basis for the high lubricity of mucins.7,12–14 Another key feature is the gel formation of PGM at acidic conditions.5,6,11,15,16 Moreover, mucin distinguishes itself with interactions to molecules17 and charged particles.18 Also, the antiviral properties of PGM have been discussed19 and make this glycoprotein attractive for application in the medical field. Many of these functional properties have already been utilized in biomedical applications: layers of purified mucins decrease friction7,13,20 between materials making them a suitable coating for implants, and thermo-responsive mucin gels on wounds can reduce bacterial and viral infections.21 Commercially available mucins such as PGM type II and III (Sigma-Aldrich) lack these key properties, as has been shown previously.1,16,19 Even cell toxicity was observed for some commercial PGMs.19 Therefore, commercially available mucins from porcine stomach cannot be applied to biomedical purposes utilizing the above named key properties. The insufficient availability of functional mucins and the impossibility to produce mucins recombinantly in animal cells make an efficient purification of gel forming and lubricating mucins from tissue necessary.
While older publications mainly focused on the analysis of mucin structure and provided only small amounts of protein,9,22–24 no studies concentrated on high yields and productivities. In our former studies, a robust reference process (see Fig. 1, left) has already been established. There, the addition of protease inhibitors and buffer components, as well as the functionality and yields of the target protein, and process productivity were addressed.1 The mucus was manually scraped from porcine stomachs. The collected mucus was hydrated overnight in buffer, and three centrifugation steps followed to remove cells, cell debris and lipids, before the mucins were concentrated with crossflow filtration. Low molecular weight proteins and other small molecules were depleted using size exclusion chromatography (SEC). Next, salts were removed with diafiltration and PGM was formulated by lyophilization. With the reference process, up to 65 mg mucin per stomach and a productivity of approx. 6 mg mucin per stomach and per hour were achieved. However, the preparation of tissue by manual scraping of mucus was costly in terms of labor and time. Much PGM was lost during sample collection, and the successive centrifugation steps made further scale-up unrealistic. For this purpose, the aim of these studies was to optimize the capture steps during the purification of PGM in terms of (1) time-saving sample collection, (2) scalability of the capture steps, (3) improvement of yields and productivities and (4) preservation of functionality.
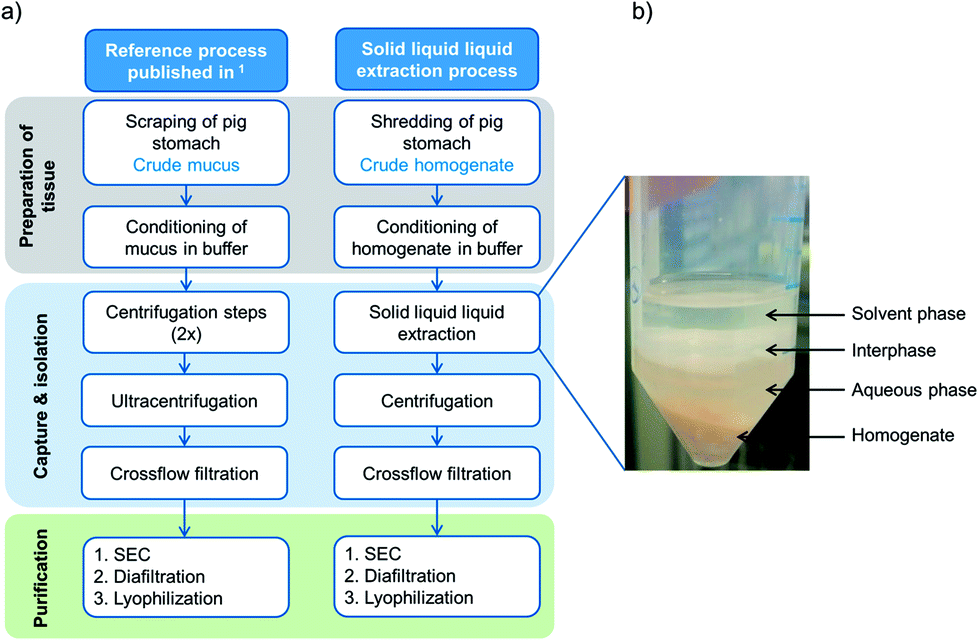 | ||
| Fig. 1 (a) Comparison of the reference protocol1 with the advanced extraction process, using solid liquid liquid extraction (SLLE) as one capture step after homogenization of whole porcine stomachs. (b) Illustration of solvent extraction from porcine homogenate with both liquid phases, aqueous and solvent phases, respectively. After agitation, a lipid rich interphase is formed. | ||
One promising possibility is the solvent-based extraction of mucins from homogenized raw stomachs as an early capture step (see Fig. 1a, right). Solvent extraction from solid phases is mostly used in the food industry to obtain sugar from sugarcane, produce instant coffee or tea25 or extract fats from plants.26,27 In the pharmaceutical industry, organic solvents are used to extract pharmacologically active compounds such as vitamins, antibiotics and steroids.26,28,29 Yet, solvent extraction is only seldom applied to remove undesired lipids and thereby further purify proteins from a complex tissue.30 However, the extraction simplifies the collection of biological tissue in the early purification process and can save expensive and time consuming process steps. For extraction, sample collection and manual scraping by hand is replaced by homogenizing entire porcine stomachs. For further capture of the target protein from the homogenized tissue, we introduced a novel extended extraction method called solid liquid liquid extraction (SLLE). For this method, the conventional solid–liquid extraction is advanced and an additional phase, which is immiscible with the other liquid phase, is added. Thereby, three phases coexist: the solid phase, which incorporates the target protein and lipids among other compounds, a polar solvent and a nonpolar solvent. Within one process step, undesired hydrophobic compounds are discarded into the hydrophobic phase (delipidation) and hydrophilic complexes such as (glyco-)proteins (e.g. Muc5AC) diffuse into the aqueous phase. The process is easily scalable and two centrifugation steps used in the reference process1 can be omitted. Thereby, process productivity is enhanced.
In the following, the optimization of mucin purification with SLLE as an alternative capture step is demonstrated and compared to Schömig et al.1 Porcine stomach is homogenized and delipidated with organic solvents, while compounds such as glycoproteins (Muc5AC) are collected in the hydrophilic phase. The parameters incubation time and ratio of organic and inorganic solvents for the delipidation and extraction of glycoproteins, respectively, are examined using statistical design. Further purification of Muc5AC from the aqueous phase is conducted analogously to the reference process, consisting of SEC and diafiltration. In order to represent a more convenient alternative to former capture steps,1 we demonstrate the improvement of yield and productivity as well as the preservation of the protein's functional properties.
2 Experimental
2.1 Materials
Hexane, Schiff's stain and periodic acid (1%) were obtained from Carl Roth (Karlsruhe, Germany). Antibodies against Muc5AC (ABIN966608) and the secondary antibody (HRP conjugated, ABIN237501) were purchased from antibodies-online.de. Agarose was obtained from Cleaver Scientific (Rugby, UK). Column material Sepharose 6 Fast Flow was obtained from GE Healthcare, UK.2.2 Methods
After thawing the material at 4 °C, 1.4 g homogenate were weighted in a 50 mL sample tube and mixed with hexane/water ratios between 3/2 and 1/15. The final volume was 8 mL. The samples were mixed with stirring bars (1200 rpm) at 4 °C for 15 min to 24 h. Subsequently, the samples were centrifuged for 30 min at 4 °C and 3200 × g (Heraeus™ Megafuge™ 16R, Thermo Fisher Scientific GmbH) and the upper solvent phase and lipid phase were removed. The lower aqueous solvent phase was collected and analyzed for glycoproteins (response Y1) using PAS assay, and Muc5AC (response Y2) using an indirect ELISA. Based on the responses, the models were approximated with quadratic functions describing the surface of the model (eqn (1))31
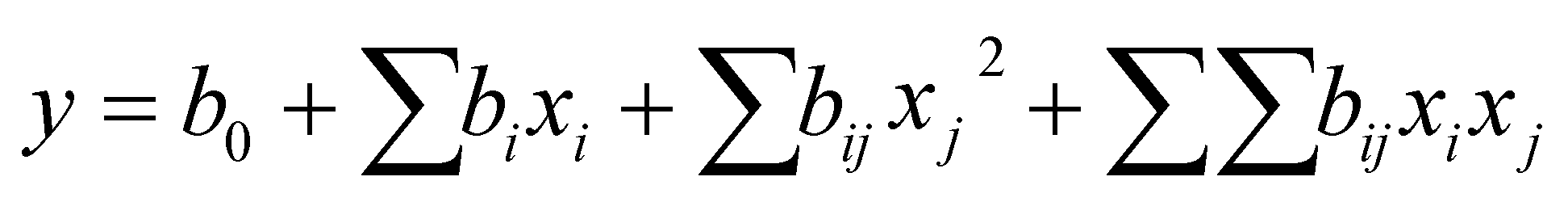 | (1) |
2.3 Analytical methods
2.4 Characterization
3 Results & discussion
In Schömig et al.1 we addressed the rising demand for functional mucins for biomedical applications and established a purification process that offered purified gel forming and lubricating PGM. To further enhance the productivity and increase the overall recovery of PGM, the process was optimized by altering the harvest and by introducing a novel process step, the solid liquid liquid extraction (SLLE). The extraction process differs from the reference process in several important characteristics: the tissue is prepared by shredding the whole stomach instead of scraping the interior mucosa. Thereby, mucins produced inside the goblet cells are released and can also be recovered. Time consuming centrifugation steps that were needed in the reference process to discard cells, cell debris and lipids are replaced. Instead, the technique of SLLE enables the simultaneous separation of cell debris as well as delipidation by organic solvents and hydration of polar glycoproteins into the aqueous phase. Also, the scale-up of SLLE is easily applicable in contrast to (ultra-)centrifugation. After SLLE, the aqueous phase is injected onto a SEC column and the sample further diafiltrated and lyophilized as established earlier. A comparison between both processes is given in Fig. 1.3.1 Design of Experiments
In pilot experiments, various organic polar and nonpolar solvents were screened in order to examine their compatibility with the target protein. Hexane was the most suitable solvent, as it is immiscible with water and gel formation was not affected (see ESI Fig. S1†). Using statistical design, the influence of the incubation time and the ratio of hexane/water on the delipidation and diffusion of glycoproteins and Muc5AC out of the homogenate was evaluated. These parameters were chosen to be optimized since delipidation is strongly influenced by time.34 Also, the ratio of solvent phases and thus the ratio of each solvent to the sample is known to have an effect on the yield during extraction.35 While also the temperature influences the extraction, we deliberately set the temperature constant to 4 °C. The reasons for this were the following: (1) at elevated temperatures proteins are more susceptible to denaturation in the presence of organic solvents36 and (2) the absence of protease inhibitors could not be completely precluded and possible enzymatic cleavage was thereby set to a minimum. The total volume was kept constant at 8 mL because this ensured the same turbulence in each experimental set-up and enabled a comparison between experiments. This in turn made a constant mass of sample (1.4 g) necessary. Optimization was conducted using the software Design Expert® with a Response Surface Method (RSM) and Face Centered Composite Design (CCF). The time of extraction (A) was varied between 0.25 and 6 h. The ratio of hexane/water (B) was investigated between 1/15 and 3/2. Mixing with a magnetic stirrer was set to 1200 rpm. The masses of glycoproteins (Y1) and Muc5AC (Y2) in the aqueous phase were chosen as responses. The resulting models are shown as 3D contour plots in Fig. 2. Experiments were conducted randomly to reduce systematic errors. The center point was repeated five times, while the axial points were repeated twice.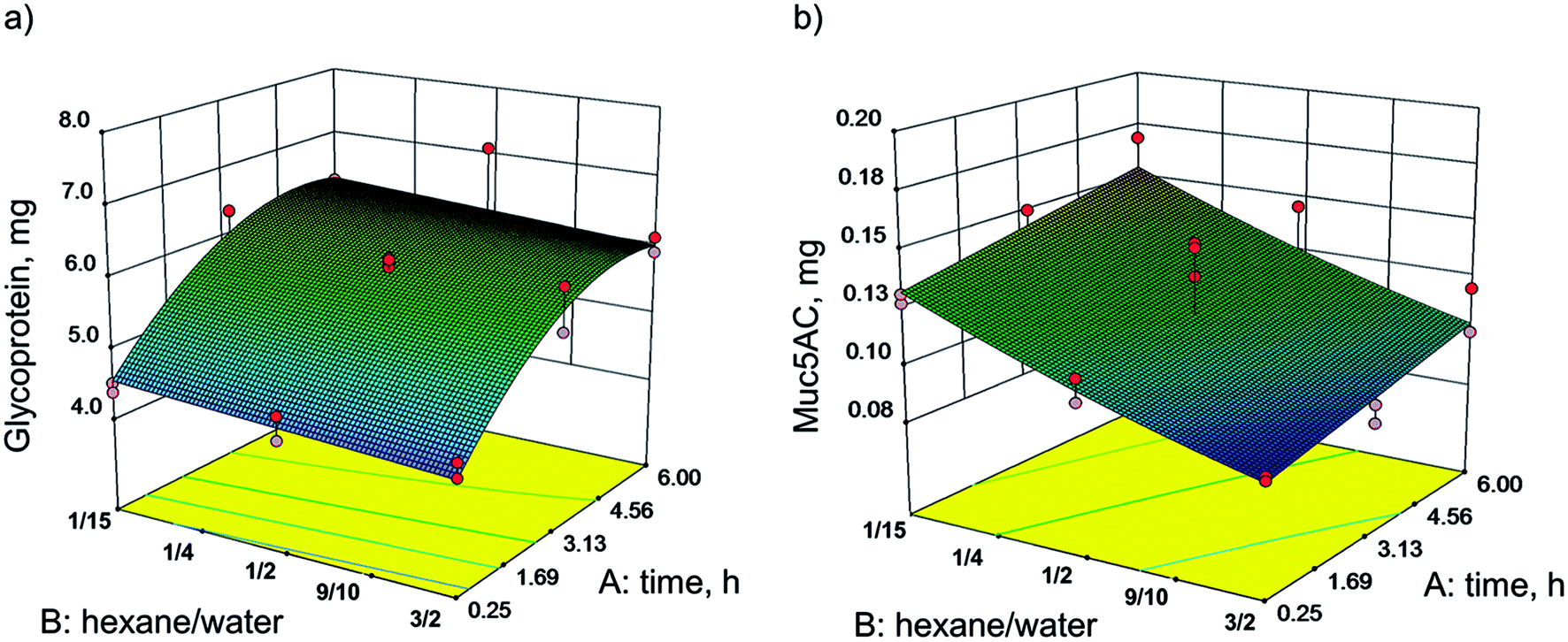 | ||
| Fig. 2 Influence of the ratio of hexane/water and incubation time on (a) the mass of glycoproteins and (b) the mass of Muc5AC in the aqueous phase after SLLE of 1.4 g stomach homogenate, respectively. The total volume was 8 mL throughout the experiments. The data were interpreted using Design of Experiments, and assimilated with significant models. The glycoprotein concentration was determined using PAS assay, and Muc5AC was determined with ELISA. Each model is based on 21 experiments. | ||
The mass of glycoproteins, analyzed with PAS assay, was 7.08 mg at its maximum and 4.22 mg at its minimum (Fig. 2a). The mass of Muc5AC, determined by ELISA, varied between 0.09 and 0.17 mg (Fig. 2b).
For both responses, the masses of glycoproteins (Y1) and Muc5AC (Y2), significant models were generated following the quadratic functions given in eqn (2) and (3), respectively. The evaluation of statistical relevance within the experimental space was conducted using the software Design Expert®. The validity of the models was confirmed by validation at three random points within the model space. ANOVA tables with p-values and lack of fit values of both models as well as the model validation are provided in the ESI (Tables S1–S5).†
| Y1 = 5.89 + 0.87A + 0.15B − 0.61A2 | (2) |
| Y2 = 0.12 + 0.011A + 0.024B + 2.616 × 10−4AB + 2.957 × 10−3B2 | (3) |
The mass of glycoproteins in the aqueous phase was strongly influenced by the extraction time. After 3 h more than 6 mg were extracted. No further increase was observed within the investigated range. However, the glycoprotein mass in the aqueous phase was not significantly changed with varying ratios of hexane/water. The highest yield was reached with the lowest amount of hexane (ratio of hexane/water 1/15). In contrast, the mass of Muc5AC showed a strong dependency on the ratio. The lower the ratio, the more Muc5AC was detected in the aqueous phase. The highest amount measured was 0.17 mg at 1/15 hexane/water and 6 h of incubation.
It is obvious that the models of glycoprotein and Muc5AC yields did not correspond. For glycoproteins, the incubation time mattered. By rigorous stirring at 4 °C, the hydrophilic glycoproteins were hydrated and diffused into the water. The extraction time correlates with the diffusion of molecules due to a concentration gradient.37 After 3 h, a balance of glycoproteins in the aqueous phase seemed to be reached. The same tissue was extracted a second time. This time, more glycoproteins diffused into the water phase that had not been extracted in the previous extraction (data not shown). For the gel forming Muc5AC, more protein was detected with a higher volume of aqueous phase. This is in general observed for extractions, as the partition of a component between two phases can be enhanced by increasing the volume.26 Within this range of experimental set-up, no maximum of Muc5AC mass was detected. This led us to the hypothesis that more Muc5AC can be obtained with a higher volume of water corresponding to a further reduction of the hexane/water ratio, respectively. Maybe even a lamination with hexane can be successful in delipidating the tissue. This suggestion is part of future investigations.
The missing congruency can have several reasons. Glycoproteins were analyzed using the PAS assay. However, with this assay sugar residues can also be detected in addition to glycosylated proteins. These seemed to diffuse independently of the solvent/water ratio but with a longer incubation time. Glycoproteins and especially gastric mucins can be covalently and non-covalently attached to lipids.38–40 With the transition of lipids in the organic phase and thereby detachment from mucins, those hydrophilic mucins become more susceptible for the transition into the aqueous phase.
These results do not permit any conclusions about the functionality of purified mucin but solely describe the amount of glycoproteins and Muc5AC that can be obtained by delipidation of porcine stomach homogenate. The maximum mass of Muc5AC was determined for the hexane/water ratio of 1/15. In further purifications, extractions of 15 min, 3 h and overnight were conducted, and functionality in terms of viscoelasticity and tribology was compared as discussed in Chapter 3.3.
3.2 Purification of mucins with SLLE
The purification of Muc5AC was conducted based on the scheme depicted in Fig. 1. The aim of the advanced purification process was the increase of productivity with a high yield of gel forming Muc5AC and glycoproteins. After delipidation of 5.6 g homogenate (75% v/v) using the optimized parameters of DoE (hexane/water 1/15, incubation time 3 h), 20 mL aqueous phase with a total of 11 mg extracted Muc5AC were injected onto the SEC (CV = 167 mL). Due to the low volume, no pre-concentration using crossflow filtration was conducted. Fractions of 5 mL each were collected during SEC and analyzed for glycoproteins and Muc5AC (Fig. 3). Three distinctive peaks were eluted, in which glycoproteins were detected. The concentration of glycoproteins was highest in the first peak reaching a maximum of 0.35 mg mL−1. Muc5AC was solely detected in the first peak with up to 0.1 mg mL−1. The second and third peak showed a high concentration of general proteins, quantified by absorbance at 280 nm. In order to collect the least amount of contaminating proteins, fractions between 0.3 and 0.45 CV (corresponding to 9.5 mg Muc5AC) were pooled. The pooled sample was diafiltrated against ddH2O until the conductivity of the retentate was <100 μS cm−1 and the retentate was finally lyophilized.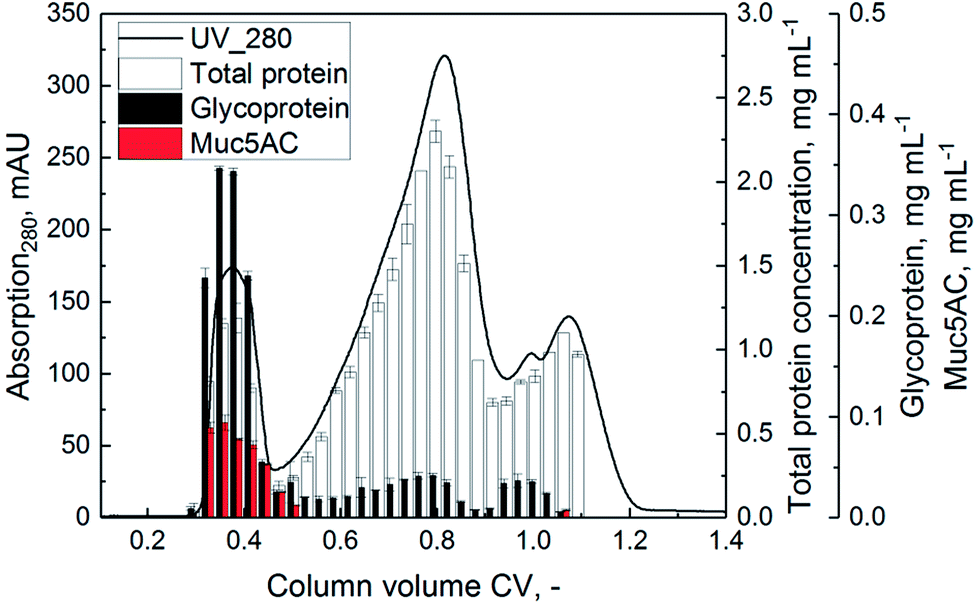 | ||
| Fig. 3 SEC chromatogram of the solvent phase after application of SLLE (5.6 g homogenate 75% v/v, hexane/water ratio 1/15, incubation time 3 h). Material: Sepharose 6 Fast Flow, molecular weight cutoff (MWCO): approximately 4 MDa, injection volume: 20 mL, flow rate: 30 cm h−1, column volume (CV) 167 mL, column diameter: 16 mm, running buffer: 10 mM sodium phosphate buffer with 170 mM NaCl, pH 7.0. All fractions of 5 mL each were analyzed in terms of total protein (280 nm, white), glycoprotein (PAS assay, black) and Muc5AC (ELISA, red). | ||
Only a small portion of low molecular weight sugar fragments was determined during the SEC run (Fig. 3). Therefore, a degradation of the target protein into sugar residues after delipidation with hexane (incubation time 3 h, solvent/water ratio 1/15) could be neglected.
In order to determine the partial and overall yields as well as process productivity, samples were analyzed for protein content before and after each purification step. The results are summarized in Table 1.
| Process step | Yield,% | cbefore, mg mL−1 | cafter, mg mL−1 | Vbefore, mL | Vafter, mL | mbefore, mg | mafter, mg |
|---|---|---|---|---|---|---|---|
| SLLE 3 h | — | n.d. | 0.55 ± 0.02 | 30 | 30 | — | 13.7 ± 0.5 |
| SEC | 86.0 ± 4.2 | 0.55 ± 0.02 | 0.38 ± 0.01 | 20 | 25 | 11.0 ± 0.4 | 9.5 ± 0.3 |
| Diafiltration | 75.0 ± 2.6 | 0.38 ± 0.01 | 0.28 ± 0.01 | 25 | 22 | 8.3 ± 0.2 | 6.2 ± 0.1 |
| Lyophilization | — | 0.28 ± 0.01 | — | 21 | — | 6.0 ± 0.1 | 5.8 |
| Total | 65% | ||||||
The overall yield of Muc5AC was determined to be 65%, which represented an increase by a factor of two compared to the reference process. Due to solid compounds and lipids before the extraction process, no concentration could be determined using ELISA. Because of the small scale, no crossflow filtration was applied before injection onto SEC. Diafiltration was conducted with a 100 kDa membrane cassette.1 The low volume after SEC only permitted the use of centrifugal tubes (100 kDa membranes). In total, 5.8 mg PGM, corresponding to 3.2 mgmucin mLhomogenate−1 were purified from 4.2 g homogenate (100% v/v, 1.83 mL homogenate, respectively). With the complete injection of the aqueous phase, around 4.75 mgmucin mLhomogenate−1 can be obtained. With the homogenate of a complete stomach being around 750 mL, up to 3573 mg PGM can be collected from one stomach, which corresponds to a 55 fold increase of mucin per stomach compared to Schömig et al.1 with 65 mgmucin per stomach. By scaling up the amount of homogenate by a factor of 4 and applying subsequent crossflow filtration before SEC, we were able to increase productivity to 25 mgmucin per stomach per h compared to a productivity of 6 mgmucin per stomach per h in the reference process. The potential for even further increases is given. The number of process steps was reduced by a total of two centrifugation steps.
3.3 Characterization of purified mucin
In addition, the nativity of the purified mucin needed to be preserved in terms of viscoelasticity and lubricity in order to be of interest for biomedical applications. Therefore, the impact of delipidation on the quality of purified mucin for different extraction times (15 min, 3 h, overnight) and a constant hexane/water ratio (1/15) was investigated. Furthermore, the purified protein was screened for DNA and tested if the quality characteristics mentioned above are influenced by DNA. Mucins were purified analogous to the scheme depicted in Fig. 1 with the parameters mentioned.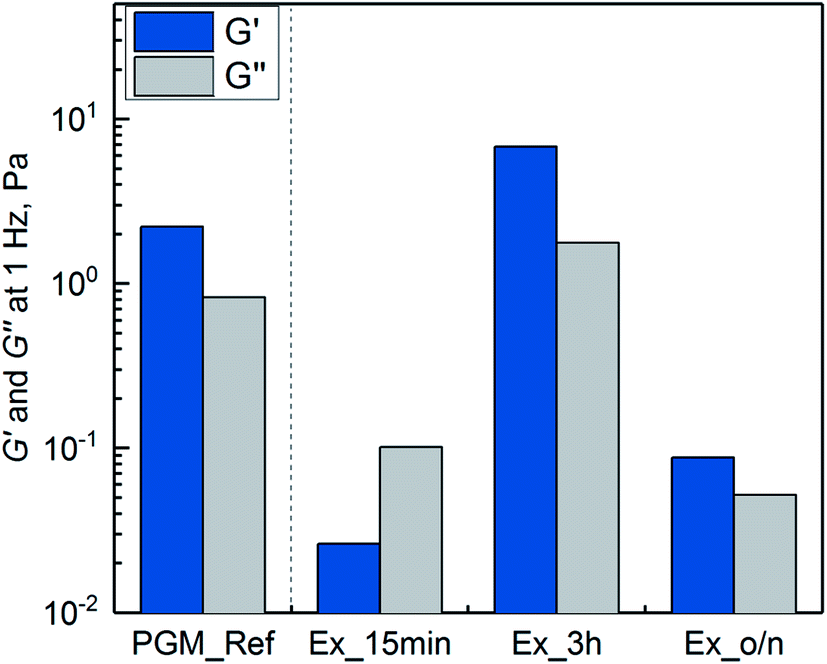 | ||
| Fig. 4 Storage modulus G′ and loss modulus G′′ at 1 Hz of purified mucins (1% w/v), using the SLLE as the capture purification step (extraction of 15 min, 3 h and overnight (o/n)). Mucin purified by the reference process (PGM_Ref) is shown for comparison. All measurements were conducted in 10 mM sodium phosphate buffer at pH 2 and room temperature. | ||
The missing gel formation after 15 minutes of SLLE can be attributed to the low recovery of general glycoproteins apart from Muc5AC (see Fig. 2) and to the lower ratio of extracted general glycoproteins to Muc5AC. With the established purification process, other high molecular weight mucins such as Muc5B, also present in stomachs, cannot be excluded. These can additionally contribute to gel formation but might not have been extracted after 15 min. Also, lipids that have not been extracted into hexane in this time span can be covalently attached to Muc5AC40,44–46 interfering with gel formation. However, gel formation seems not to be negatively influenced by lipids, on the contrary, a positive influence on mucin viscosity has been observed in previous studies.40,44–46 Extraction overnight did not result in gel forming mucin, either. This suggested that hydrophobic interactions with hexane did have a negative effect in the long run and eventually led to a disruption of inter- and intramolecular interactions needed for gel formation. Harsh conditions have been described to impair gel formation of mucins.16,47 SLLE for 3 h did result in a hydrogel, similar to the reference mucin. This might in reverse be due to a small portion of other gel forming glycoproteins such as Muc5B and lipids covalently attached to mucin as well as the shorter incubation with hexane.
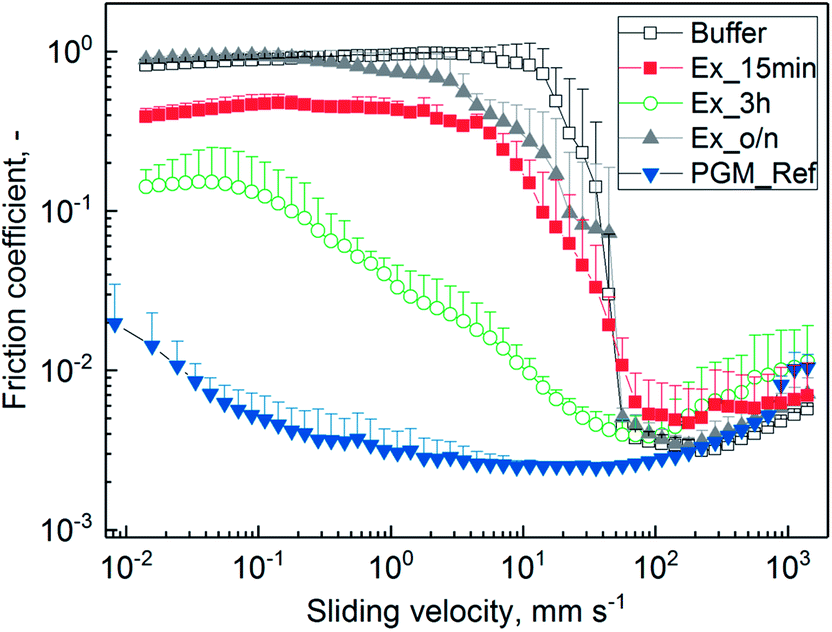 | ||
| Fig. 5 Lubricity between steel/PDMS of 0.1% (w/v) mucins purified by SLLE and hydrated in 20 mM HEPES buffer pH 7.4. The extraction was conducted for 15 min (Ex_15 min), 3 h (Ex_3 h) and overnight (Ex_o/n). Mucin purified with the reference process (PGM_Ref)1 and buffer are depicted for comparison. Friction coefficients were recorded over a sliding speed range of 10−2 to 103 mm s−1. Error bars represent analytical triplicates. | ||
The missing lubricity for Ex_15 min might be due to insufficient hydration in the boundary layer, which is needed for lubrication.12,13 However, a difference was observed compared to pure buffer. It has been discussed that covalently bound lipids that have not been depleted during purification can lower friction.48–54 Also, intact sugar residues are needed for lubricity of mucin.7,12,13 With the overnight incubation, the glycoproteins were exposed to hexane for a longer time, and glycoproteins were fragmented as observed from SEC chromatograms (see ESI Fig. S2 and S3†). Therefore, reduced hydration and decreased lubrication might have occurred.13 Incubation for 3 h resulted in a reasonable decrease in friction. Since the yield of glycoproteins was at its maximum after 3 h (see Fig. 2) without any observable fragmentation during SEC, the hydrating effect was retained. Not-depleted lipids might have additionally added lubricity to purified PGM.55 The process needs to be optimized in terms of robustness, as functionality was not confirmed in all cases. This makes a thorough characterization of mucin structure and impact factors for its key properties necessary. However, influence factors have not been completely elucidated so far.6,43,56
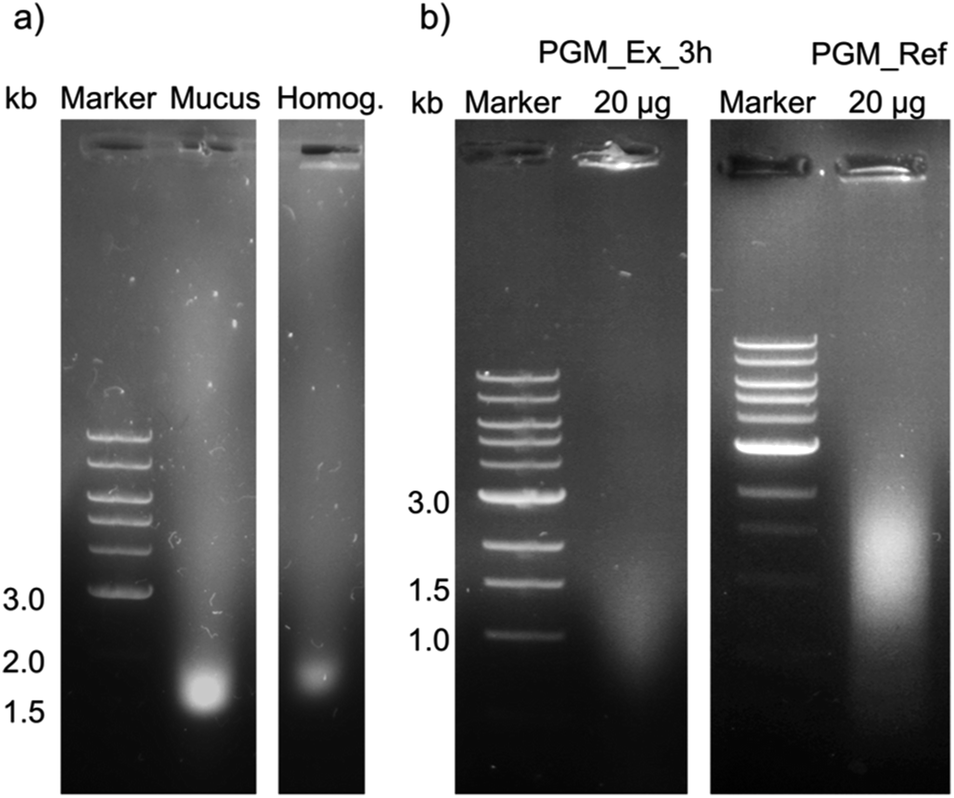 | ||
| Fig. 6 A 0.8% (w/v) agarose gel of (a) mucus and homogenate as well as (b) purified mucins for the analysis of DNA contamination. (a) Mucus and homogenate (Homog.) after hydration in 5 mL 10 mM sodium phosphate buffer pH 7.0 with 170 mM NaCl before further purification. (b) Left: mucin purified by SLLE with an incubation time of 3 h (PGM_Ex_3 h). Right: mucin purified with the reference process1 (PGM_Ref). 6 μL of 1 kb DNA marker and 24 μL samples, and 20 μg of purified mucins, respectively, were injected into the gel pockets. | ||
DNA residues were detected in mucus, homogenate and purified mucin. Due to the degradation of genomic DNA, the bands were broad and not defined. There was a distinct band for non-purified mucus (a), while a weak band was identified for the homogenate. The amount of DNA residues in purified reference mucin was higher compared to mucin from the extraction process (b). Furthermore, quantitative analysis of the DNA content with the Eppendorf BioSpectrometer® basic confirmed the higher concentration of DNA in scraped mucus compared to shredded crude homogenate as well as a higher purity of purified PGM_Ex_3 h in contrast to PGM_Ref in terms of nucleic acids (for detailed information see ESI Table S6†). These results indicated that the white smear observed in extracted mucin solutions was not attributed to an increased DNA concentration. The reason for the lower content of nucleic acids after shredding instead of scraping the stomach might be due to the following: (1) homogenization for 10 min did not break up cells, or only did so partially, while scraping destroyed epithelial cells. (2) Hydration in buffer overnight, as was the case in the reference process, could have promoted the diffusion of DNA polymers out of the mucus into the buffer. In contrast to that, after 3 h not all nucleic acids were extracted from the homogenate. (3) DNA polymers interact with cationic lipids,59–61 leading to the diffusion of the created hydrophobic complex into nonpolar solvents. (4) Accumulation of DNA polymers in the interphase is also often described for the extraction of DNA.62
As expected, nucleic acids were not completely removed by filtration using 100 kDa membranes and subsequent SEC.57 In order to minimize the DNA content, CsCl gradient centrifugation can be applied23,24,57 or DNA can be precipitated.24 Despite a DNA content of around 15% in purified PGM_Ref, gel formation and lubricity were reported. Moreover, the addition of nucleic acids (150 μg mL−1 fish sperm DNA) to non-gel forming mucin (Muc_Ex_o/n) increased viscoelasticity (ESI Fig. S4†).63 Additional DNA, however, did not influence lubricity (ESI Fig. S5†). In conclusion, the rise of friction by a factor of 10 could not be explained with a high content of nucleic acids. On the contrary, the amount of DNA residues was reduced with the novel SLLE process.
4 Conclusion
In this study, a purification process for porcine gastric mucins1 was further optimized in terms of productivity, yield and ease of scalability. Therefore, pig stomachs were homogenized to facilitate the collection of sample. A novel solvent extraction method, namely solid liquid liquid extraction (SLLE) was introduced, which combines the delipidation of lipid rich homogenized tissue using hexane with the extraction of (glyco-)proteins into the polar phase. SLLE is thus a promising alternative to non-scalable centrifugations in the early purification phase. With the new process step, process productivity and overall yields were significantly increased. Gel formation of purified mucin was retained and lubricity was improved compared to commercially available mucins. The process unit of SLLE can easily be transferred to similar high molecular weight proteins. In order to retain the functional characteristics of future target proteins, the compatibility towards organic solvents needs to be investigated for each protein individually. Alternatively, less toxic solvents and ionic liquids might be promising for the purification of glycoproteins from animal tissue. These will be the subject of future investigations.Conflicts of interest
There are no conflicts of interest to declare.Abbreviations
| BSA | Bovine serum albumin |
| CV | Column volume |
| Muc5AC | Mucin type 5AC |
| PGM | Porcine gastric mucin |
| SEC | Size exclusion chromatography |
| SLLE | Solid liquid liquid extraction |
Acknowledgements
We thank Prof. Lieleg, professor of Biomechanics, and his research group “Biological Hydrogels” (in particular Benjamin Kaesdorf) at the Technical University of Munich (IMETUM) for kindly providing us with purified PGM and assistance with rheology and tribology measurements. The project was funded by the Dean's Innovation Fond of the Mechanical Engineering Department of the Technical University of Munich.Notes and references
- V. J. Schömig, B. T. Käsdorf, C. Scholz, K. Bidmon, O. Lieleg and S. Berensmeier, RSC Adv., 2016, 6, 44932–44943 RSC.
- A. Allen, The structure and function of gastrointestinal mucus, in Basic Mechanisms of Gastrointestinal Mucosal Cell Injury and Protection, Baltimore, 1981 Search PubMed.
- A. P. Corfield, Biochim. Biophys. Acta, Gen. Subj., 2015, 1850, 236–252 CrossRef CAS PubMed.
- L. Tailford, E. Crost, D. Kavanaugh and N. Juge, Front. Genet., 2015, 6, 1–18 CAS.
- R. Bansil and B. S. Turner, Curr. Opin. Colloid Interface Sci., 2006, 11, 164–170 CrossRef CAS.
- K. R. Bhaskar, D. H. Gong, R. Bansil, S. Pajevic, J. A. Hamilton, B. S. Turner and J. T. LaMont, Am. J. Physiol., 1991, G827–G832 CAS.
- J. M. Coles, D. P. Chang and S. Zauscher, Curr. Opin. Colloid Interface Sci., 2010, 15, 406–416 CrossRef CAS.
- R. Mejías-Luque, L. Cobler and C. de Bolós, Atlas of Genetics and Cytogenetics in Oncology and Haematology, 2011, vol. 14, pp. 566–569 Search PubMed.
- A. J. Libao-Mercado and C. F. M. de Lange, Livest. Sci., 2007, 109, 141–144 CrossRef.
- B. J. Van Klinken, A. W. Einerhand, H. A. Buller and J. Dekker, Anal. Biochem., 1998, 265, 103–116 CrossRef CAS PubMed.
- G. J. Strous and J. Dekker, Crit. Rev. Biochem. Mol. Biol., 1992, 27(1/2), 57–92 CrossRef CAS PubMed.
- S. Lee, M. Müller, K. Rezwan and N. D. Spencer, Langmuir, 2005, 21, 8344–8353 CrossRef CAS PubMed.
- T. Crouzier, K. Boettcher, A. R. Geonnotti, N. L. Kavanaugh, J. B. Hirsch, K. Ribbeck and O. Lieleg, Adv. Mater. Interfaces, 2015, 2, 1500308 CrossRef.
- L. Shi and K. D. Caldwell, J. Colloid Interface Sci., 2000, 224, 372–381 CrossRef CAS PubMed.
- R. Bansil, J. Celli, B. Chasan, S. Erramilli, Z. Hong, N. H. Afdhal, K. R. Bhaskar and B. S. Turner, MRS Proceedings, 2005, 897, 0897-J02-04 CrossRef.
- J. Kočevar-Nared, J. Kristl and J. Šmid-Korbar, Biomaterials, 1997, 18, 677–681 CrossRef.
- S. C. Baos, D. B. Phillips, L. Wildling, T. J. McMaster and M. Berry, Biophys. J., 2012, 102, 176–184 CrossRef CAS PubMed.
- O. Lieleg, I. Vladescu and K. Ribbeck, Biophys. J., 2010, 98, 1782–1789 CrossRef CAS PubMed.
- O. Lieleg, C. Lieleg, J. Bloom, C. B. Buck and K. Ribbeck, Biomacromolecules, 2012, 13, 1724–1732 CrossRef CAS PubMed.
- I. C. Hahn Berg, L. Lindh and T. Arnebrant, Biofouling, 2004, 20, 65–70 CrossRef CAS PubMed.
- C. Nowald, A. Penk, H.-Y. Chiu, T. Bein, D. Huster and O. Lieleg, Macromol. Biosci., 2016, 16, 567–579 CrossRef CAS PubMed.
- M. Faure, D. Moënnoz, F. Montigon, L. B. Fay, D. Breuillé, P. A. Finot, O. Ballèvre and J. Boza, Anal. Biochem., 2002, 307, 244–251 CrossRef CAS PubMed.
- M. Mantle and A. Allen, Biochem. J., 1981, 195, 267–275 CrossRef CAS PubMed.
- T. Marshall and A. Allen, Biochem. J., 1978, 173, 569–578 CrossRef CAS PubMed.
- A. Ibarz and G. V. Barbosa-Canovas, Unit operations in food engineering, CRC Press, Boca Raton, Florida, 2002 Search PubMed.
- A. B. de Haan and H. Bosch, Industrial separation processes: Fundamentals, De Gruyter, 2013 Search PubMed.
- M. R. Ladisch, Bioseparations engineering: principles, practice, and economics, Wiley, Canada, 2001 Search PubMed.
- R. G. Harrison, P. Todd, D. P. Petrides and S. R. Rudge, Bioseparations science and engineering, Oxford University Press, USA, 2015 Search PubMed.
- K. Sambamurthy, Pharmaceutical Engineering, New Age International (P) Limited, 2007 Search PubMed.
- K. Schügerl, Solvent extraction in biotechnology – recovery of primary and secondary metabolites, Springer-Verlag, Berlin, Heidelberg, 1994 Search PubMed.
- R. L. Mason, R. F. Gunst and J. L. Hess, Statistical design and analysis of experiments, John Wiley & Sons, Inc., 2003 Search PubMed.
- M. Kilcoyne, J. Q. Gerlach, M. P. Farrell, V. P. Bhavanandan and L. Joshi, Anal. Biochem., 2011, 416, 18–26 CrossRef CAS PubMed.
- K. Boettcher, S. Grumbein, U. Winkler, J. Nachtsheim and O. Lieleg, Rev. Sci. Instrum., 2014, 85, 093903 CrossRef CAS PubMed.
- B. E. Cham and B. R. Knowles, J. Lipid Res., 1976, 17, 176–181 CAS.
- T. Pérez-Palacios, J. Ruiz, I. M. P. L. V. O. Ferreira, C. Petisca and T. Antequera, Meat Sci., 2012, 91, 369–373 CrossRef PubMed.
- J. C. Janson, Protein Purification: Principles, High-Resolution Methods, and Applications, John Wiley & Sons, New York, 1998 Search PubMed.
- J. M. Aguilera, in Extraction optimization in food engineering, ed. C. Tzia and G. Liadakis, Marcel Dekker, Inc, New York, 2003 Search PubMed.
- D. H. Gong, B. Turner, K. R. Bhaskar and J. T. Lamont, Am. J. Physiol., 1990, 259, G681–G686 CAS.
- B. L. Siomiany, V. L. N. Murty, S. R. Carter and A. Slomiany, Digestion, 1986, 34, 275–280 Search PubMed.
- A. Slomiany, H. Witas, M. Aono and B. L. Slomiany, J. Biol. Chem., 1983, 258, 8535–8538 CAS.
- X. Cao, R. Bansil, K. R. Bhaskar, B. S. Turner, J. T. LaMont, N. Niu and N. H. Afdhal, Biophys. J., 1999, 76, 1250–1258 CrossRef CAS PubMed.
- A. Maleki, G. Lafitte, A. L. Kjoniksen, K. Thuresson and B. Nystrom, Carbohydr. Res., 2008, 343, 328–340 CrossRef CAS PubMed.
- J. P. Celli, B. S. Turner, N. H. Afdhal, R. H. Ewoldt, G. H. McKinley, R. Bansil and S. Erramilli, Biomacromolecules, 2007, 8, 1580–1586 CrossRef CAS PubMed.
- A. Slomiany, Z. Jozwiak, A. Takagi and B. L. Slomiany, Arch. Biochem. Biophys., 1984, 229, 560–567 CrossRef CAS PubMed.
- J. Sarosiek, A. Slomiany, A. Takagi and B. L. Slomiany, Biochem. Biophys. Res. Commun., 1984, 118, 523–531 CrossRef CAS PubMed.
- V. L. N. Murty, J. Sarosiek, A. Slomiany and B. L. Slomiany, Biochem. Biophys. Res. Commun., 1984, 121, 521–529 CrossRef CAS PubMed.
- O. Svensson and T. Arnebrant, Curr. Opin. Colloid Interface Sci., 2010, 15, 395–405 CrossRef CAS.
- J. Klein, Friction, 2013, 1, 1–23 CrossRef CAS.
- T. A. Schmidt, N. S. Gastelum, Q. T. Nguyen, B. L. Schumacher and R. L. Sah, Arthritis Rheum., 2007, 56, 882–891 CrossRef CAS PubMed.
- R. Goldberg, A. Schroeder, G. Silbert, K. Turjeman, Y. Barenholz and J. Klein, Adv. Mater., 2011, 23, 3517–3521 CrossRef CAS PubMed.
- R. Goldberg and J. Klein, Chem. Phys. Lipids, 2012, 165, 374–381 CrossRef CAS PubMed.
- A. M. Trunfio-Sfarghiu, Y. Berthier, M. H. Meurisse and J. P. Rieu, Tribol. Int., 2007, 40, 1500–1515 CrossRef CAS.
- M. Daniel, Wien. Med. Wochenschr., 2014, 164, 88–94 CrossRef PubMed.
- A. Boţan, L. Joly, N. Fillot and C. Loison, Langmuir, 2015, 31, 12197–12202 CrossRef PubMed.
- S. Bhuyan, S. Sundararajan, L. Yao, E. G. Hammond and T. Wang, Tribol. Lett., 2006, 22, 167–172 CrossRef CAS.
- R. Bansil, J. P. Celli, J. M. Hardcastle and B. S. Turner, Front. Immunol., 2013, 4, 310 Search PubMed.
- E. Chantler, Mucus in Health and Disease—II, Springer, US, 2012 Search PubMed.
- J. M. Creeth, Br. Med. Bull., 1978, 34, 17–24 CrossRef CAS PubMed.
- F. M. P. Wong, D. L. Reimer and M. B. Bally, Biochemistry, 1996, 35, 5756–5763 CrossRef CAS PubMed.
- P. Harvie, F. M. Wong and M. B. Bally, Biophys. J., 1998, 75, 1040–1051 CrossRef CAS PubMed.
- D. Matulis, I. Rouzina and V. A. Bloomfield, J. Am. Chem. Soc., 2002, 124, 7331–7342 CrossRef CAS PubMed.
- P. Chomczynski and N. Sacchi, Anal. Biochem., 1987, 162, 156–159 CrossRef CAS PubMed.
- R. A. Cone, Adv. Drug Delivery Rev., 2009, 61, 75–85 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06594a |
| This journal is © The Royal Society of Chemistry 2017 |