
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced flame-retardancy and controlled physical properties of flexible polyurethane foams based on a shear-responsive internal network†
Jingyi Rao *,
María P. Fernández-Ronco,
Michel Vong and
Sabyasachi Gaan
*,
María P. Fernández-Ronco,
Michel Vong and
Sabyasachi Gaan
Laboratory of Advanced Fibers, Functional Materials, Swiss Federal Laboratories for Materials Science and Technology, Lerchenfeldstrasse 5, 9014, St. Gallen, Switzerland. E-mail: jingyirao@gmail.com
First published on 12th September 2017
Abstract
Solid additives are commonly used in manufacturing flexible polyurethane foams (FPUFs) to incorporate novel functionality for various application purposes. However, the viscosity change as a result of solid addition in the FPUF formulation can adversely alter the physical and mechanical properties of foam materials. Here, we report a shear-responsive internal network based on –P–N–H⋯O– interfacial hydrogen bonds between the polyether–polyol chain and a solid flame-retardant (FR) of 6,6′-(ethane-1,2-diylbis-(azanediyl))-bis-9,10-dihydro-9-oxa-phosphaphenanthrene-10-oxide (EDAB-DOPO) and its utilization in FPUFs. This interfacial interaction can stabilize the FR-polyol suspensions against sedimentation and it can be destroyed by shear force before foaming to control the fluid viscosity and thus control the physical–mechanical properties of FPUFs. The excellent dispersibility of FR particles from suspensions is well preserved in FPUFs by the optimized processes, which significantly enhances the flame retardancy of FPUFs with low FR content to achieve a HF1 rating in UL 94 test. The design of a shear-responsive internal network between solid additives and a polymer matrix provides a simple and practical method for producing functional foam composites.
Introduction
Flexible polyurethane foams (FPUFs) are one of the most popular and most versatile materials due to their lightweight porous structure, high surface area, and the availability of large amounts of polar and non-polar groups.1 In industry, FPUFs are usually produced by mixing two or more liquid streams, based on the polymerization of polyols and isocyanates. The isocyanate component is separately pumped into the reaction tank to meet the polyol stream, which is usually of a more complex composition, containing a blend of catalysts, surfactants, blowing agents and so on.2,3 In order to enhance the material performance of FPUFs to fulfil various application requirements, a commonly used strategy is to pre-disperse functional additives in polyols before the polymerization reaction.However, incorporating additives, especially solids, in polyols as suspension precursors is a challenge in the foaming process. So far, various solid additives/fillers, including multi-walled carbon nanotubes (MWNTs), clay, graphenes, carbon black, aluminum trihydroxide (ATH), melamine and etc., have been prepared as suspensions in polyols to improve the flame retardancy or mechanical properties of FPUFs.4–8 Poor stability of solid-polyol suspensions leads to defective products and severe damage to foam manufacturing facilities. The surface chemistry of solid fillers has thus to be modified to increase the suspension stability, usually by multi-step processes, e.g. involving coupling/dispersing agents, surface functionalization reactions, or the co-solvent-assisted method.9–12 The enhanced surface interaction between solids and polyols can stabilize the suspension, but also influences the ordering of the hard segments (urea) during foaming and therefore affects the mechanical performance of foamed materials.11–13 Moreover, the notable viscosity difference between solid-polyol suspensions and pristine polyols during the manufacturing process could induce strong impact on the cell growth, cell geometry, and thus the physical structure of the foams.11,14,15 Intensive formulation work for FPUFs with solid additives has to be carried out to manage the balance between functional performance and appropriate physical–mechanical properties.
Shear-responsive behavior, prominently the shear thinning, has been well investigated for colloidal suspensions in polymer processing, food industry, and bio-applications.16–19 Such suspensions are usually stabilized at low stresses by a delicate network based on repulsive electrostatic interaction, supramolecular interaction, or steric force. However, this internal structure can be destroyed by the external forces (shear) acting upon the suspension when processing takes place. When the external forces overcome the cohesion force of the suspension network, evidenced by the yield point, the fluid viscosity will break down and suspended particles will tend to sediment. Therefore, to incorporate this shear-responsive character to solid-polyol suspensions is highly interesting for the manufacturing of solid-incorporated FPUFs. By determining the yield point, stable solid-polyol suspensions can be prepared for long term storage against sedimentation and flocculation. The suspension viscosity, which can be well controlled by shear stress to eliminate the influence in the cell growth process during foaming, can manipulate the cellular structure of foamed materials.
In this work, a shear-responsive internal network in flexible PUFs preparation based on the surface hydrogen bonding between a solid FR and polyether–polyol chain is studied. The solid FR, 6,6′-(ethane-1,2-diylbis-(azanediyl))-bis-9,10-dihydro-9-oxa-phospha-phenanthrene-10-oxide (EDAB-DOPO), is selected here as a model FR system as it exhibits the highest thermal stability, the best flame retardant efficiency, and lowest cytotoxicity, among all the DOPO-phosphonamidates and commercially available FRs (TCPP, DOPO, and Exolit® OP 560).20–23 More importantly, NH groups from the EDAB-DOPO particle surface can act as the binding sites to form hydrogen bonding interactions with the oxygen groups of polyether–polyol chains (Scheme 1). By decreasing particle sizes and increasing the surface area of solid FR, the interfacial interaction between particle and polyol medium can be significantly strengthened. Polyether–polyol molecules bridge with neighboring particles by the surface hydrogen interaction, resulting in an internal network and excellent suspension stability (Scheme 2). Such internal network in FR-polyol suspension can be destroyed by applying shear stresses, which has been systematically studied. The shear-responsive behavior of the FR-polyol suspension can be utilized to control the fluid viscosity during the foaming step, and further to control the cellular structure and mechanical properties of FR-incorporated FPUFs. After the optimization of the suspension and foam preparation process, excellent dispersibility of EDAB-DOPO particles is well preserved in FPUFs, which significantly enhances the flame retardancy of materials. The design of a shear-responsive internal network between solid additives and polymer matrix offers a simple and practical method for producing functional foam composites with consistent properties.
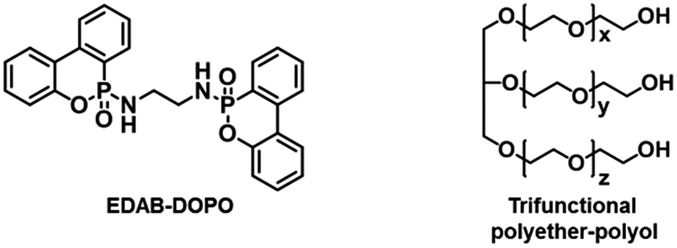 | ||
| Scheme 1 Chemical structures of EDAB-DOPO and polyether–polyol. | ||
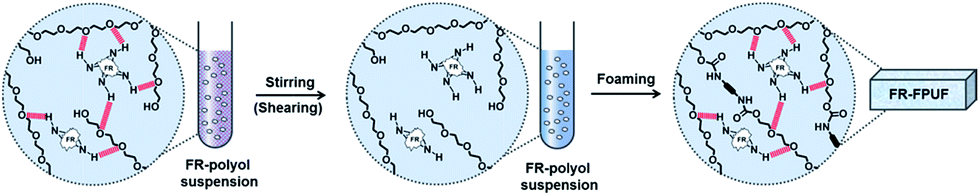 | ||
| Scheme 2 The illustration of shear-responsive internal network in FR-polyol suspensions and FR-FPUFs. | ||
Experimental section
Materials
The solid flame retardant, 6,6′-(ethane-1,2-diylbis-(azanediyl))-bis-9,10-dihydro-9-oxa-phosphaphenanthrene-10-oxide (EDAB-DOPO), was synthesized following the literature procedure.22 A tri-functional polyether polyol kindly supplied by Dow Chemical was used in this study (VORANOL WK 3138, OH value: 56 KOH per g, Mw: 3000 g mol−1). The following chemicals were used in the PU foam formulation without further purification (kindly supplied by FoamPartner, Fritz Nauer AG, Switzerland): TDI 80 (a 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 mixture of 2,4-toluene diisocyanate and 2,6-toluene diisocyanate), demineralized water, Kosmos EF (60% water solution of tin(II) ricinoleate, Evonik Industries AG), Tegoamin ZE 1 (an amine catalyst, Evonik Industries AG), STABI AKTIV (a silicone surfactant, Momentive Performance Materials GmbH).
:20 mixture of 2,4-toluene diisocyanate and 2,6-toluene diisocyanate), demineralized water, Kosmos EF (60% water solution of tin(II) ricinoleate, Evonik Industries AG), Tegoamin ZE 1 (an amine catalyst, Evonik Industries AG), STABI AKTIV (a silicone surfactant, Momentive Performance Materials GmbH).
Preparation of FR-polyol suspensions
Three types of FR-polyol suspension, normal-suspensions (N), well-suspensions (W), and pre-stirred well-suspensions (W*) were prepared by mixing 100 g of polyol with required amount of EDAB-DOPO via different procedure.Synthesis of FR-FPUFs
All foams were prepared according to the standard formulation (Table S1 in ESI†), based on polyether polyol (VORANOL WK 3138) and toluene diisocyanate (TDI 80).Characterization
Sedimentation velocities of suspension samples were determined by a LUMiSizer analytical photocentrifuge (LUM GmbH, Berlin, Germany) at 20 °C using 2 mm path length polyamide cells.24–27 The measurements were conducted on freshly-prepared FR-polyol suspensions using centrifugation rates of 200–4000 rpm at 20 °C. The transmission profiles were recorded every 20 min continually for 40 h, where the intersection at about 108 mm corresponds to the filling height in sample cuvettes. The position of the cuvette bottom was set at 130 mm. FR-polyol suspensions were diluted to 0.05% to evaluate the particle size distribution. At this concentration, hindered settling and multiple scattering are irrelevant.26,27The solubility of EDAB-DOPO in VORANOL WK 3138 polyol was measured by a Bruker AV400 MHz spectrometer. A clear liquid was separated by centrifugation and filtration of W-6% suspension to remove the undissolved solid particles. 100 mg of the collected liquid was fully dissolved in 600 μL d-DMSO and measured by NMR at 338 K. No characteristic signal from EDAB-DOPO can be observed in the 1H and 31P NMR spectra (data not shown). The solubility of EDAB-DOPO in polyol is thus less than 200 mg L−1 at 338 K, which is the detection limit of the NMR analysis for the FR-polyol suspension system.
ATR-IR measurements of suspensions and FPUFs were performed using a Brucker Tensor-27 spectrometer with a MCT detector in the range of 600–4000 cm−1 at 2 cm−1 resolution. The final spectrum is the mean of three replicates at random area. Each spectrum was collected over 256 scans.
Rheological data were collected by a stress-controlled rotational rheometer MCR301 (Anton Paar, Buchs, Switzerland) using a plate–plate (N) or cone–plate (W) geometry with 1 mm or 0.1 mm gap, respectively. To determine the flow curves of suspensions, a constant stress-ramp at 20 °C was applied, where the shear-stress was increased at a constant rate from 0.01 to 100 Pa and maintained for 10 seconds. All samples were pre-sheared at 5 s−1 for 30 seconds and then equilibrated for 1 min at 20 °C.
Foam samples were embedded in epoxy resin and cut by microtome to give thin slices (0.5 mm thick), which can be observed by Keyence VHX-1000 optical microscope under transmission mode. The cellular morphologies of foam samples in the rising direction were investigated by a field-emission scanning electron microscopy (FE-SEM, Philips XL30). Samples were freeze-fractured in liquid nitrogen and the fracture surfaces were sputter-coated with 15 nm of Au/Pt before observation. Image analysis was performed on the SEM micrographs using ImageJ to obtain the average cell size and cell density.
The apparent density of foams was measured according to ISO 845 standard, with specimen bar cut to the dimensions of 150 × 50 × 13 mm3. The tensile strength and elongation of foam materials were determined in conformity with ISO 1798 standard. The compression stress was examined accordingly to ISO3386-1 standard, using specimens with dimensions of 50 × 50 × 10 mm3. Airflow was measured according to ISO 9237 standard. The tests were conducted on Zwick Roell testing machine in FoamPartner at Wolfhausen, Switzerland.
UL94 horizontal burning test was carried out to evaluate the flame retardancy of FPUFs according to ASTM D 4986 standard. The test specimen of 150 × 50 × 13 mm3 was oriented in a horizontal position. The flame was applied to the free end of the specimen for 60 s and then removed, while cotton was placed under the specimen. The 150 mm long test specimen is marked at the 25 mm, 60 mm, and 125 mm positions and the burning rate is measured over a 100 mm span. The appropriate material classifications are provided according to the procedure described in the literature.28
Results and discussion
Characterization of FR-polyol suspensions
As a solid flame retardant (FR), EDAB-DOPO shows poor solubility in VORANOL WK 3138 polyol (<200 mg L−1 at 338 K) according to the NMR analysis. In order to incorporate such solid FR and enhance the flame retardancy of FPUFs, the preparation of FR-polyol suspensions has to be optimized and investigated. Hence, two types of FR-polyol suspensions with a fixed EDAB-DOPO concentration of 6 wt% (N-6% and W-6%) were prepared by different methods and quantitatively characterized by the analytical centrifugation technique. As shown in Fig. 1a and b, the evolution of transmission profiles was recorded at 20 °C under the RCF (relative centrifugal force) of 1930g for 40 h from freshly-prepared suspension samples.27 The first profile was taken after 10 s and exhibits low transmissions (∼0%) along the sample length for all suspensions. N-6%, which is produced by a mechanical homogenizer, shows fast sedimentation with polydispersed speeds and produces 0.2 cm of sediment layer at the end of the centrifugal test (Fig. 1a). The analysis shows an average particle size of 4.65 μm with a broad size distribution (Fig. 1c and Table 1). The presence of polydispersed FR particles and large agglomerates in N-6% is confirmed by the optical microscopic image (Fig. S1a in ESI†). On the other hand, by employing a two-step milling process, W-6% shows sharp fronts moving towards the cell bottom (Fig. 1b). Compared to N-6%, thicker sediment layer of 1.3 cm is generated in W-6%, which indicates the sedimentation was not yet completed even after 40 h of centrifugation. The transmittance profiles are evenly spaced with smaller distance, which reveal that FR particles were dispersed in relatively mono-dispersed size and settled with considerably slower velocity. An average particle diameter of 2.15 μm (Fig. 1d and Table 1) and absence of larger agglomerates in optical microscopic image confirm the excellent homogeneity of FR particles in W-6% (Fig. S1b in ESI†). Therefore, W-6%, which is produced by the milling technique, possesses smaller particle size and thus more surface area of FR particles than N-6%.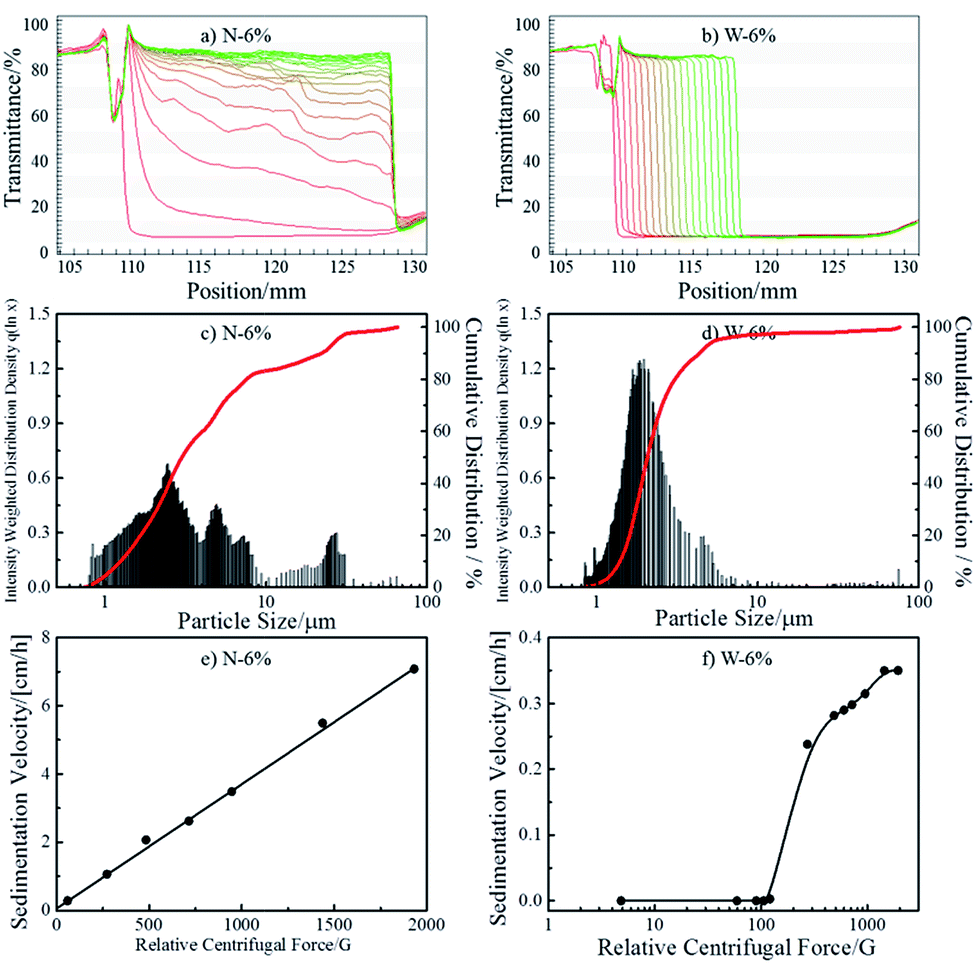 | ||
| Fig. 1 Recorded evolution (from left to right) of time dependent transmission profiles of FR-polyol suspensions: N-6% (a) and W-6% (b). Profiles are taken every 20 min at 4000 rpm (1930g) for 40 h, where the first profile was taken after 10 s for all suspension samples. The red line was the first recorded profile, which then shifted into the green line during measurements. x-axis: position at the rotor. The particle size distribution of FR-polyol suspensions: N-6% (c) and W-6% (d). Sedimentation velocity of FR-polyol suspensions in dependence on relative centrifugal force (RCF): N-6% (e) and W-6% (f). | ||
| FR-polyol suspensions | Db [μm] | PSDb | db [cm] | RCFcgb | Yield pointc [Pa] | |
|---|---|---|---|---|---|---|
| Method 1 | Method 2 | |||||
| a The W*-6% sample was not analyzed by LUMiSizer, because particle agglomeration aggravates over time after pre-stirring.b D: average particle size; PSD: particle size distribution; d: sediment layer thickness; RCFc: no sedimentation behavior was detected under the RCFc value; obtained by LUMiSizer.c Method 1: strain/shear stress curve method; method 2: Herschel–Bulkley fitting method; obtained by rheometer. n.d. Not described. | ||||||
| N-6% | 4.65 | 1.84 | 0.2 | —n.d. | —n.d. | 0.0279 |
| W-6% | 2.15 | 1.30 | 1.3 | 100 | 0.4080 | 0.6971 |
The suspension stability was evaluated by measuring sedimentation velocities at different RCF values.29 Typical linear regression behaviour has been found from N-6% as shown in Fig. 1e. This result reveals that dispersed FR particles behave as predicted by classical latex suspensions and no structural alteration of the suspension occurs during accelerated stability testing.24,27,30 By extrapolating from the fitted results, the sedimentation velocity of N-6% can be estimated as 0.04 cm h−1 at the earth's gravity (1g). However, in W-6%, no sedimentation behavior is detected under RCF 100g (Fig. 1f). It shows excellent dispersion stability under normal storage condition, which is often found in suspensions containing internal networks.24,25 Above the critical RCF of 100g, sedimentation occurs sluggishly, which indicates that the stable network in suspension is gradually destroyed due to the increasing centrifugal force.
Rheology study was performed to further investigate the suspension properties. As can be seen from the different slope experienced by viscosity and strain curves with increased stress (Fig. S2 in ESI†), the yield point of N-6% cannot be determined or is lower than 0.1 Pa, whereas W-6% shows a measurable yield point. Two different methods were employed to calculate the yield point of W-6%: (1) 0.41 Pa, based on the delimitation of the linear-elastic deformation region from the strain/shear stress curve (method 1 in Table 1, Fig. 2b), and (2) 0.70 Pa, based on the correlation of the flow curve by means of Herschel–Bulkley model (method 2 in Table 1, Fig. S2d in ESI†). Below the yield point, the suspension structure can return to its former shape if stress is removed, i.e. elastic deformation behavior occurs. When external forces are stronger than the internal interaction (above the yield point), suspension starts to flow and sedimentation of particles can occur.
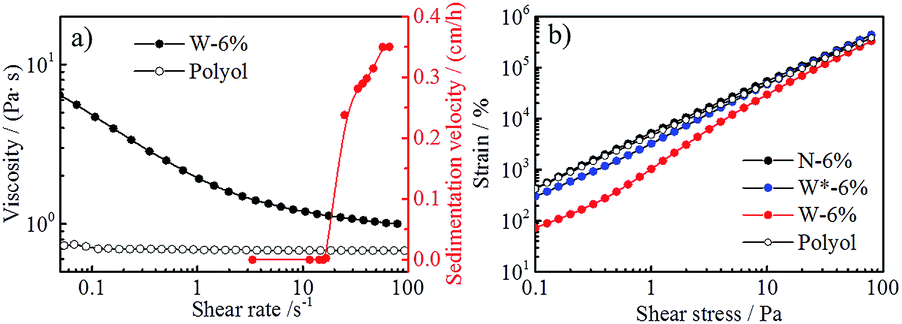 | ||
| Fig. 2 The comparison of shear rate on the evolution of viscosity and sedimentation velocity of W-6% suspension and pure polyol (a), and the strain/shear stress curves of different suspensions to determine the yield point by method 1 (b). | ||
By converting the RCF value to shear rate, an initial comparison of the viscosity evolution with the sedimentation velocity of W-6% is shown in Fig. 2a. It turns out that results from the analytical centrifugation test and the rheology measurement are comparable, despite the intrinsic differences of both characterization methods. At shear rates higher than 10 s−1, viscosity value of W-6% suspension is significantly reduced and approaches the viscosity of the pure polyol, while the sedimentation velocity is also increased. This phenomenon indicates that possible network structures in the suspension can be broken down by applying shear stresses or centrifugal forces.
Hence, a stirring step of 9000 rpm (shear rate of 150 s−1, above the yield point) was applied on W-6% for 3 min to prepare W*-6%, in order to further understand the impact of shear stress on the suspension stability. The delimitation of the linear-elastic deformation in Fig. 2b shows that W*-6% suspension presents a rather similar behavior to N-6%, but reduced stability in comparison to W-6%. The appearance of a yield point at 0.30 Pa for W*-6% (method 2, Fig. S2d in ESI†) can probably be attributed to the destabilization of its internal structure, which is partially broken down by the 9000 rpm stirring.
Interfacial hydrogen bonding interaction
In order to investigate the internal network in suspensions, ATR-IR spectra of polyol, EDAB-DOPO, and FR-polyol suspension samples were thus recorded. An adsorption peak at 3170 cm−1 is observed in the spectra of pristine EDAB-DOPO solid, which corresponds to the secondary N–H stretching signal (Fig. 3a).31,32 The N–H peak can be also found in N-6% but significantly decreases in W-6%. Moreover, the signal of the P–N–C bond at 750 cm−1, which is clearly observed in EDAB-DOPO solid and N-6% suspension,33 shifts to 756 cm−1 in W-6% (Fig. 3b). These changes in IR signals are due to the change of chemical environment on amine groups of EDAB-DOPO particles and most probably the existence of hydrogen interaction.34–38 According to the chemical structure of suspension components, the hydrogen bonds should be formed between the amide groups from EDAB-DOPO surface and the oxygen of the polyether–polyol chain (Scheme 1).15,39–42 Compared to N-6%, W-6% suspension has smaller particle size and thus larger surface area, which provides more binding sites to form –P–N–H⋯O– interactions in the solid-polyol interface. Polyether–polyol molecules bridge with the neighbor particles by the interfacial hydrogen interaction, resulting in an internal network in W-6% suspension and excellent suspension stability (Scheme 2).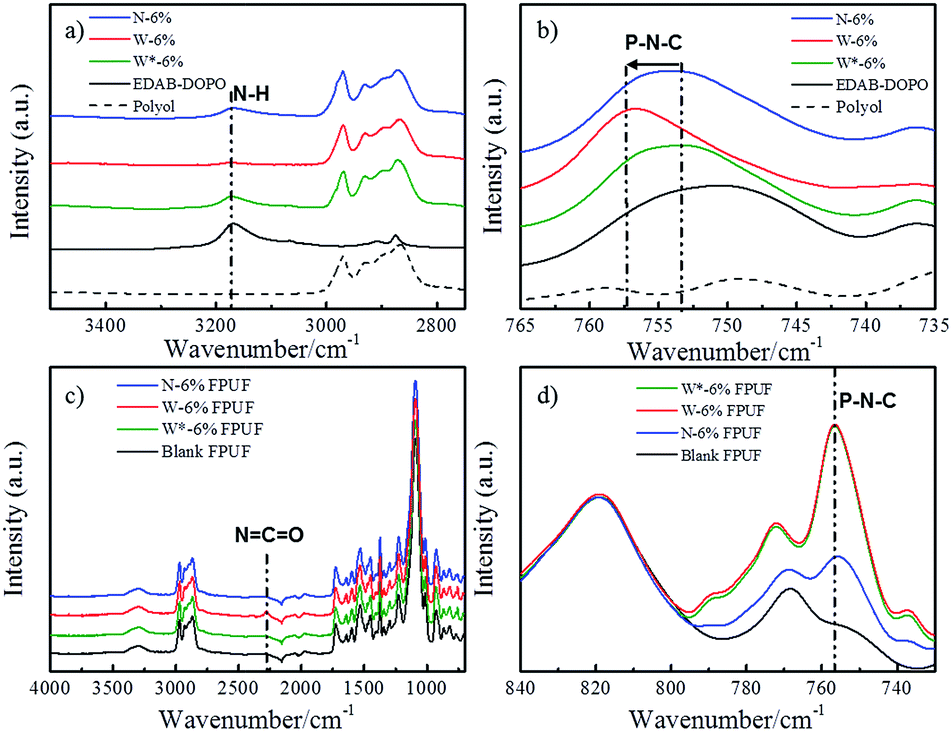 | ||
| Fig. 3 ATR-IR spectra of polyol, EDAB-DOPO, and FR-polyol suspensions in N–H region (a) and in P–N–C region (b); FPUFs (c) and FPUFs in P–N–C region (d). | ||
After applying a stirring step at 9000 rpm (shear rate of 150 s−1, above the yield point), the signal changes of N–H and P–N–C bonds immediately appear in the IR spectra of W*-6% suspension, which reveal that the internal structure can be mechanically destroyed (Fig. 3a and b). FR agglomerates clearly appear according to the optical microscopic examination of W*-6% sample (Fig. S1c†). These results imply that, without the internal network, FR particles start to agglomerate, resulting in reduced surface area which provides less binding sites for stabilization. Instead of reforming interfacial hydrogen bonds, the agglomeration and thus sedimentation probably more dominate in a standing suspension after the removal of the internal network between FR particles and polyols. Therefore, in the foam preparation, a pre-stirring step, which is designed on all of the FR-polyol suspensions by a mechanical stirrer at a speed of 9000 rpm for 3 min, has to be conducted right before mixing with other components. Based on the rheology and IR studies, the suspension viscosity is thus forced to approach the pure polyol and the internal network can be broken down by the pre-stirring treatment.
Blank and FR-incorporated FPUFs were analyzed by ATR-IR to study the interfacial hydrogen bond interaction in foams. For the convenience of comparison, all the IR spectra were normalized at 820 cm−1 in Fig. 3c and d. This peak is attributed to the C–H bending peak of the aromatic ring from the TDI component and the same amount of TDI was added in all the foam formulations.43 With the pre-stirring step in foam preparation, the characteristic of IR spectra from all FR-incorporated FPUFs shows no significant change compared to the neat PU foam.44,45 However, the W-6% FPUF without pre-stirring shows a distinct stretching signal of N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO bond at 2270 cm−1 from the isocyanate component.38 This indicates that the interfacial hydrogen interaction of –P–N–H⋯O– can block some hydroxyl groups of polyol and interfere in the polymerization reaction. By applying the pre-stirring step before foaming (W*-6% FPUF), the incorporation of EDAB-DOPO particles doesn't affect the chemical structure of FPUFs anymore (Fig. 3c).
CO bond at 2270 cm−1 from the isocyanate component.38 This indicates that the interfacial hydrogen interaction of –P–N–H⋯O– can block some hydroxyl groups of polyol and interfere in the polymerization reaction. By applying the pre-stirring step before foaming (W*-6% FPUF), the incorporation of EDAB-DOPO particles doesn't affect the chemical structure of FPUFs anymore (Fig. 3c).
The characteristic peaks at 756 cm−1 were detected in all the FR-incorporated foam samples, which is in accordance with the signal change of –P–N–C– bond in W-suspensions (Fig. 3d). This infers that the –P–N–H⋯O– interaction between EDAB-DOPO particle surfaces and polyether segments can be rebuilt in PU foams after the polymerization of polyols and isocyanates (Scheme 2). Moreover, under the same FR concentration, W*-6% FPUF and W-6% FPUF show comparable intensity of adsorption peak at 756 cm−1, while N-6% FPUF displays much weaker signal. This result indicates that, after pre-stirring, dispersed FR particles in suspensions are immediately immobilized by the subsequent foaming process. No distinct agglomeration of EDAB-DOPO particles happened during foaming. Consequently, W*-6% FPUF, which preserves well dispersibility and larger surface area of FR particles from W-suspensions, shows stronger hydrogen interaction in foams than N-6% FPUF.
Characterization of FR-incorporated FPUFs
The cellular structure of FPUFs, which are prepared from FR-polyol suspensions, was examined by SEM at low magnifications (Fig. 4a and b). The foam density and cell size are summarized in Table 2. It can be seen that, after the pre-stirring step, FR-incorporated FPUFs possess open-cell structures with similar bulk density, airflow value, and cell morphology to neat FPUF (Fig. S3a in ESI†). On the contrary, the density and airflow permeability are both increasing in the W-6% FPUF with no pre-stirring, which is indicative of the population of cells with bigger size in the foam structure (Fig. S3b in ESI†). The viscosity difference between pure polyol and W-6% suspensions can be reduced by the pre-stirring step, which prevents the influence on the cell growth and cell opening process, and thus controls the cell geometry. However, in N-6% FPUF, polydispersed FR solids and big agglomerates are observed in the foam strut by optical microscope and high-magnification SEM examination (Fig. 4a and c), whereas FR particles are homogeneously dispersed in W*-6% FPUF (Fig. 4b and d). | ||
| Fig. 4 SEM images (a and b) and optical microscopic images (c and d) of N-6% FPUF and W*-6% FPUF. | ||
| FPUFs | ρfa [kg m−3] | Airflow [L m−2 s−1] | Dcellb [μm] | Ncellb [cm−2] | Ten strc [kPa] | Elongation [%] |
|---|---|---|---|---|---|---|
| a ρf: foam density.b Dcell: cell diameter; Ncell: cell number; obtained by ImageJ analysis.c Ten str: tensile strength. | ||||||
| Blank | 51.8 ± 0.5 | 180 ± 6 | 814 ± 3 | 192 ± 12 | 70 ± 4 | 124 ± 5 |
| N-6% | 51.2 ± 0.3 | 183 ± 12 | 791 ± 3 | 204 ± 25 | 59 ± 9 | 96 ± 12 |
| W-6% | 55.9 ± 0.3 | 241 ± 9 | 914 ± 3 | 153 ± 15 | 89 ± 6 | 136 ± 4 |
| W*-6% | 51.9 ± 0.4 | 186 ± 4 | 804 ± 3 | 197 ± 19 | 70 ± 5 | 125 ± 6 |
The mechanical properties of FPUFs are presented in Table 2. Among all FPUFs, N-6% FPUF shows lower tensile strength and elongation at break. The poor dispersibility and broad particle size distribution of solid FRs in the foam structure may create defects that promote the initiation and propagation of cracks as a result of the applied strength, decreasing both the tensile strength and the elongation at break. Without pre-stirring, W-6% FPUF sample shows relatively higher tensile strength and elongation at break, whereas comparable mechanical properties can be observed from blank FPUF and W*-6% FPUF. Hence, the physical–mechanical properties of FPUFs can be well controlled by the shear-responsive behavior of FR-polyol suspensions. The flammability of FPU foams was evaluated by the UL94 horizontal burning test, which is required for materials intended for applications in transportation and construction. A minimum of 5 specimens was burned to obtain the relevant classification.
Table 3 summarizes the fire results of the FR-incorporated FPUFs, which are prepared by FR-polyol suspensions with different FR content. To achieve the highest flame retardant rating of HF1 in UL94 HBF test of FPUFs, a minimum FR concentration of 6% is needed for W*-suspension procedure (Video 1–3 in ESI†). Above 6% FR content, a clear difference in the burning behavior of foams is found, i.e. the W*-FPUFs need lower FR concentration to achieve the HF1 rating (highest classification). The enhancement of the flame retardant property of FPUFs can be attributed to the preservation of excellent dispersibility of FR particles in foam structure.
Conclusions
A shear-responsive network between solid FR of EDAB-DOPO and polyether–polyol was studied and utilized in the preparation of flexible polyurethane foams (Scheme 2). The interfacial interaction is formed between the amide groups from EDAB-DOPO surface and the oxygen of the polyether–polyol chain. By employing a two-step of milling process, mono-dispersed size and excellent dispersibility of EDAB-DOPO particles are observed in W-suspensions. With larger surface area, the strength of interfacial interaction is significantly increased. An internal network based on interfacial hydrogen interaction is formed to stabilize the FR-polyol suspensions against sedimentation. By applying a shear force (pre-stirring) above the yield point before the foaming step, the internal structure can be destroyed to control the FR-polyol suspension viscosity, and further control the chemical structure and physical–mechanical properties of FPUFs. The excellent dispersibility of FR particles from suspensions is preserved in FPUFs by combining milling and pre-stirring process, which significantly enhance the flame retardancy of FPUFs with low FR content to achieve HF1 rating in UL 94 test. The design of shear-responsive internal network between solid additives and a polymer matrix provides a simple and practical method for producing functional foam composites.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Prof. Dr Manfred Heuberger, Milijana Jovic, Maria Isabel Butron Garcia, Dr Peter Schirg, Dr Daniel Rentsch, Dr Zoltan Balogh-Michels, Alain Zahn, Laurent Joye, and Fritz Nauer AG for technical supports and fruitful discussions.References
- H. W. Engels, H. G. Pirkl, R. Albers, R. W. Albach, J. Krause, A. Hoffmann, H. Casselmann and J. Dormish, Angew. Chem., Int. Ed., 2013, 52, 9422–9441 CrossRef CAS PubMed.
- X. D. Zhang, C. W. Macosko, H. T. Davis, A. D. Nikolov and D. T. Wasan, J. Colloid Interface Sci., 1999, 215, 270–279 CrossRef CAS PubMed.
- P. Cinelli, I. Anguillesi and A. Lazzeri, Eur. Polym. J., 2013, 49, 1174–1184 CrossRef CAS.
- G. Harikrishnan, T. U. Patro and D. V. Khakhar, Ind. Eng. Chem. Res., 2006, 45, 7126–7134 CrossRef CAS.
- J. J. Espadas-Escalante, F. Aviles, P. I. Gonzalez-Chi and A. Oliva, J. Cell. Plast., 2017, 53, 215–230 CrossRef.
- F. Saint-Michel, L. Chazeau and J. Y. Cavaille, Compos. Sci. Technol., 2006, 66, 2709–2718 CrossRef CAS.
- A. Konig, A. Malek, U. Fehrenbacher, G. Brunklaus, M. Wilhelm and T. Hirth, J. Cell. Plast., 2010, 46, 395–413 CrossRef.
- Y. L. Liu, J. Y. He and R. J. Yang, J. Mater. Sci., 2017, 52, 4700–4712 CrossRef CAS.
- M. Jimenez, N. Lesaffre, S. Bellayer, R. Dupretz, M. Vandenbossche, S. Duquesne and S. Bourbigot, RSC Adv., 2015, 5, 63853–63865 RSC.
- M. Bandarian, A. Shojaei and A. M. Rashidi, Polym. Int., 2011, 60, 475–482 CrossRef CAS.
- A. K. Appel, R. Thomann and R. Mulhaupt, Polymer, 2012, 53, 4931–4939 CrossRef CAS.
- Y. Xu, M. Chen, X. Ning, X. L. Chen, Z. D. Sun, Y. H. Ma, J. Yu, Z. B. Zhang, X. J. Bo, L. Yang and Z. N. Chen, J. Therm. Anal. Calorim., 2014, 115, 689–695 CrossRef CAS.
- M. Peng, M. X. Zhou, Z. J. Jin, W. W. Kong, Z. B. Xu and D. Vadillo, J. Mater. Sci., 2010, 45, 1065–1073 CrossRef CAS.
- H. Gui, X. H. Zhang, Y. Q. Liu, W. F. Dong, Q. G. Wang, J. M. Gao, Z. H. Song, J. M. Lai and J. L. Qiao, Compos. Sci. Technol., 2007, 67, 974–980 CrossRef CAS.
- X. Cao, L. J. Lee, T. Widya and C. Macosko, Polymer, 2005, 46, 775–783 CrossRef CAS.
- Y. Q. Zhao, J. B. Beck, S. J. Rowan and A. M. Jamieson, Macromolecules, 2004, 37, 3529–3531 CrossRef CAS.
- E. Brown, N. A. Forman, C. S. Orellana, H. J. Zhang, B. W. Maynor, D. E. Betts, J. M. DeSimone and H. M. Jaeger, Nat. Mater., 2010, 9, 220–224 CAS.
- R. Stanway, Mater. Sci. Technol., 2004, 20, 931–939 CrossRef CAS.
- X. Cheng, J. H. McCoy, J. N. Israelachvili and I. Cohen, Science, 2011, 333, 1276–1279 CrossRef CAS PubMed.
- C. Hirsch, B. Striegl, S. Mathes, C. Adlhart, M. Edelmann, E. Bono, S. Gaan, K. A. Salmeia, L. Hoelting, A. Krebs, J. Nyffeler, R. Pape, A. Burkle, M. Leist, P. Wick and S. Schildknecht, Arch. Toxicol., 2017, 91, 407–425 CrossRef CAS PubMed.
- S. Gaan, S. Y. Liang, H. Mispreuve, H. Perler, R. Naescher and M. Neisius, Polym. Degrad. Stab., 2015, 113, 180–188 CrossRef CAS.
- N. M. Neisius, M. Lutz, D. Rentsch, P. Hemberger and S. Gaan, Ind. Eng. Chem. Res., 2014, 53, 2889–2896 CrossRef CAS.
- A. Konig and E. Kroke, Fire Mater., 2012, 36, 1–15 CrossRef.
- D. Lerche and T. Sobisch, J. Dispersion Sci. Technol., 2011, 32, 1799–1811 CrossRef CAS.
- T. Sobisch and D. Lerche, Colloids Surf., A, 2008, 331, 114–118 CrossRef CAS.
- T. Detloff, T. Sobisch and D. Lerche, Powder Technol., 2007, 174, 50–55 CrossRef CAS.
- D. Lerche, J. Dispersion Sci. Technol., 2002, 23, 699–709 CrossRef CAS.
- D. K. Chattopadhyay and D. C. Webster, Prog. Polym. Sci., 2009, 34, 1068–1133 CrossRef CAS.
- R. M. Lueptow and W. Hubler, J. Biomech. Eng.-T ASME, 1991, 113, 485–491 CrossRef CAS.
- B. Lamas, B. Abreu, A. Fonseca, N. Martins and M. Oliveira, J. Colloid Interface Sci., 2012, 381, 17–23 CrossRef CAS PubMed.
- J. E. Stewart, J. Chem. Phys., 1959, 30, 1259–1265 CrossRef CAS.
- R. A. Heacock and L. Marion, Can. J. Chem., 1956, 34, 1782–1795 CrossRef CAS.
- K. Wazarkar, M. Kathalewar and A. Sabnis, Prog. Org. Coat., 2015, 87, 75–82 CrossRef CAS.
- L. Andrews and R. Lascola, J. Am. Chem. Soc., 1987, 109, 6243–6247 CrossRef CAS.
- X. Li, L. Liu and H. B. Schlegel, J. Am. Chem. Soc., 2002, 124, 9639–9647 CrossRef CAS PubMed.
- J. Joseph and E. D. Jemmis, J. Am. Chem. Soc., 2007, 129, 4620–4632 CrossRef CAS PubMed.
- M. Solimannejad and S. Scheiner, J. Phys. Chem. A, 2007, 111, 4431–4435 CrossRef CAS PubMed.
- Y. Liu, W. Liu, H. Li, J. Liu and Y. Yang, J. Phys. Chem. A, 2006, 110, 11760–11764 CrossRef CAS PubMed.
- J. Graton, M. Berthelot and C. Laurence, J. Chem. Soc., Perkin Trans. 2, 2001, 2130–2135 RSC.
- D. J. Millen and G. W. Mines, J. Chem. Soc., Perkin Trans. 2, 1977, 73, 369–377 CAS.
- H. U. Ung, A. R. Moehlig, S. Khodagholian, G. Berden, J. Oomens and T. H. Morton, J. Phys. Chem. A, 2013, 117, 1360–1369 CrossRef CAS PubMed.
- E. Zagar and J. Grdadolnik, J. Mol. Struct., 2003, 658, 143–152 CrossRef CAS.
- H. Han, S. S. Li, X. L. Zhu, X. B. Jiang and X. Z. Kong, RSC Adv., 2014, 4, 33520–33529 RSC.
- M. J. Elwell, A. J. Ryan, H. J. M. Grunbauer and H. C. VanLieshout, Polymer, 1996, 37, 1353–1361 CrossRef CAS.
- M. J. Elwell, A. J. Ryan, H. J. M. Grunbauer and H. C. VanLieshout, Macromolecules, 1996, 29, 2960–2968 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The formulation of FPUFs, additional optical microscopic images of FR-polyol suspensions, rheological data of FR-polyol suspensions, additional SEM images of FPUFs, and UL 94 burning test videos of FPUFs. See DOI: 10.1039/c7ra07083g |
| This journal is © The Royal Society of Chemistry 2017 |