
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Humidity-accelerated spreading of ionic liquids on a mica surface†
Zhantao Wang abc,
Fuxi Shid and
Cunlu Zhao*c
abc,
Fuxi Shid and
Cunlu Zhao*c
aKey Laboratory of Optoelectronic Devices and Systems of MOE, Institute of Optoelectronics, Shenzhen University, Shenzhen 518060, China
bInstitute for Advanced Study, Shenzhen University, Shenzhen 518060, China
cKey Laboratory of Thermo-Fluid Science and Engineering of MOE, School of Energy and Power Engineering, Xi'an Jiaotong University, Xi'an 710049, China. E-mail: mclzhao@mail.xjtu.edu.cn
dCollege of Mechanical and Electronic Engineering, Northwest A&F University, Yangling 712100, China
First published on 4th September 2017
Abstract
The role of relative humidity (RH) on the kinetic spreading and equilibrium contact angles of droplets of two [Rmim][NTf2] ionic liquids (ILs) on a mica surface was investigated using contact angle measurement and atomic force microscopy (AFM) imaging. Firstly, with contact angle measurement, we found that increasing RH accelerates the kinetic spreading and reduces the equilibrium contact angles of the droplets of two studied ILs on mica. Then by analyzing the mica surface with AFM imaging, we observed that the increasing RH leads to enhanced water vapor adsorption on mica and accordingly enhanced coverage of the IL precursor film on mica. It is essentially the enhanced coverage of the IL precursor film that induces the acceleration of IL droplet spreading and the reduction of their equilibrium contact angles. The analysis of experimental results also suggests that the surface diffusion coefficients of IL precursor films as well as the equilibrium contact angles of IL droplets have a threshold at a RH of ∼70%, which indicates the formation of a complete water monolayer on mica. Such a water layer facilitates the formation of a uniform IL precursor film on the mica surface, which would modify the surface energy of mica and thus the contact angle of IL droplets on mica. The findings from the present investigation are instrumental in the fundamental understanding of the wetting behavior of ionic liquids in humid environments and also offer practical insights into applications of ionic liquids in lubrication and electrochemical energy systems.
1. Introduction
Room temperature ionic liquids (ILs) are liquid salts composed solely of ions.1 Their low vapor pressure and high thermal and electrochemical stability showed promise for using them as green solvents,2,3 in lubrication4,5 and electrochemical energy systems (e.g., super capacitors, fuel cells and batteries).6,7 Many of the aforementioned applications are interface dominated, in which the wetting properties of ionic liquids on a solid surface play a critically important role.8 The statics9,10 and dynamics11–13 of the wetting behaviour of ionic liquids on solid surfaces have been studied only in a few cases, leaving a number of questions unanswered in the literature.8 Some new observations such as asymmetric electrowetting13 and time-dependent contact angles14,15 still await for further investigations. Although the fundamental causes of the time-dependent contact angles at some solid–liquid interfaces are not fully explained yet,16,17 previous reports indicated that molecular (ad)sorption and ‘overturning’ at the solid–liquid interface are important causes for such phenomena,18,19 and the solvation of the top adsorbed layers on the solid surface by water were shown to be key for molecular reorientation and subsequent change in the apparent contact angle.16,20,21 In the case of ionic liquids, it has been shown that the time-dependent contact angle of some imidazolium ILs on mica was caused by a thin precursor film extending from the droplet via surface diffusion process, the development of such precursor film involves molecular reorientation and packing process, similar to that of self-assembled monolayers (SAMs) of organic molecules on solid surface.14By investigating the effect of water on the interfacial layering and spreading of a group of hydrophobic ILs on mica, we proposed that the precursor film in the peripheral of the ILs droplet on mica could be an extension of the interfacial layering of ILs between the droplet and the mica surface.15 Although molecular solvation layers of ILs have been extensively studied at various solid surfaces,22–31 the fundamental causes of the spontaneous layering is not clear, reported studies so far indicated that confinement, surface charge and water content are all important factors.1,32–36 The effect of water on the interfacial nanostructure32,33,37,38 and wetting behaviour15 of ILs has been of growing interest in recent years. However, the interactions of water with ILs are complicated,39 and its effect on the solvation layers34 and surface tensions40 of ILs appear to be not monotonic. Trace amounts of water can enhance ion layering on mica by facilitating K+ dissociation,41,42 but excessive amount of water were shown to disrupt the interfacial layering of ILs due to both packing and capillary effects,15,37,38,43 and the details seem to depend on identity of individual ILs.
When the system involving an IL droplet spreading on solid surface like mica is placed in a humid environment, the water effects have two forms: water absorbed in the IL and water adsorbed on the mica surface. These two forms can have different effects on the wetting behaviour of ILs. Langmuir pioneered the research on water vapour adsorption and showed that adsorbed water can change the surface energy of solid surfaces through masking effect.44 Zisman investigated the effect of humidity on the wetting and spreading of hydrophobic liquids on surfaces of borosilicate glass, quartz, and sapphire, and the results suggested that adsorbed water can lower the wettability of these substrate by the studied liquids.45,46 However, latter research by Cazabat et al. using ellipsometry47,48 found that humidity facilitates the spontaneous spreading of tiny droplet of PDMS on oxidized silicon wafers. Zeid and Brutin studied the effect of relative humidity on the spreading dynamics of blood drops on glass and show that the drop spreading is marked by fast and slow regimes,49 however, even the slow spreading regime in their study is much faster than the contact angle relaxation of ILs on mica.14 All these studies suggest that the humidity effect is specific to droplet material, and the exact mechanism behind the humidity effect is different for different droplet materials. This paper studied the effect of humidity on the kinetic wetting (namely contact angle relaxation) of ILs on mica. By recording the contact angle relaxation using sessile drop methods and characterizing the mica surface using atomic force microscopy (AFM), the effect of water adsorption on the precursor film formation and macroscopic spreading of ILs is discussed and highlighted. We identified that the enhanced spreading of IL droplet on mica under increased humidity is driven by the enhanced IL precursor film due to water adsorption on mica; while water absorbed by the ILs from humid surroundings plays a minor role. In addition, we revealed for the first time that although increasing the humidity tend to increases the adhesion and friction between hydrophilic surfaces in ILs,37,50 it actually enhances the spreading of these ILs on mica. This intriguing finding presents an important step toward a deeper understanding of the IL–water interactions on mica, and offers implications to application of ILs as lubricants.
2. Experimental section
Muscovite mica (ProSciTech, grade V-1, 12.5 mm disk) was chosen as the substrate in this study for its atomic flatness. In our experiments, the mica disks were cleaved by inserting the sharp tip of a pair of tweezers at the side of the mica disk. Contact mode AFM scanning indicates that the mica surface has a RMS roughness less than 0.2 nm over a 1 μm × 1 μm scanning area. The ILs chosen in this study were 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([emim][NTf2]), 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([bmim][NTf2]). Contact angles were measured using the SCA 20 system of Data Physics.To investigate the effects of relative humidity (RH) on the relaxation of droplets, a 0.2 L air-tight quartz cell (see Fig. S1†) was used. Saturated salt solutions were used to change and maintain the RH inside the cell at certain levels51 (see Table S1†). In our experiments, freshly cleaved mica disks were pre-exposed to each level of RH for ∼14 hours, such a long exposure time would be sufficient for the mica surface to be equilibrated with the vapor phase. Then the IL drops were deposited on these pre-equilibrated substrates and the contact angle was measured using the sessile drop method. The temperature was maintained at 22 ± 1 °C in the quartz cell. RH was varied from 20% to 90% in these experiments.
The MultiMode 8 AFM from Veeco Instrument was used in tapping mode for characterization of the mica surface. TAP150A (Bruker) cantilevers were used with a resonant frequency of 150–200 kHz and a spring constant of 5–10 N m−1. The nominal tip radius is 8 nm. The AFM scanning resolution is 512 pixels per line.
The pre-wetting was achieved by dipping the freshly cleaved mica surface into the pure [bmim][NTf2] and removing the substrate for AFM imaging and sessile drop measurement (see ESI† material for more information).
To check the effect of RH on the water concentration inside the IL, [bmim][NTf2] was exposed to the RH of 37% and 90% in the quartz cell for varying period of time. The change of water concentration with time was monitored using Karl Fischer titration. The surface tension of the IL with different water concentration was measured using the pendent drop method.
3. Results and discussion
After an IL drop was deposited on the pre-equilibrated mica surface in the quartz cell of different RH, the variation of its contact angle with time was recorded. The relaxation of the contact angle with time can be characterized by the initial contact angle (measured immediately after the initial viscous relaxation of the droplet, i.e. several seconds), the ‘final’ contact angle c, and a relaxation time τ. τ can be determined by fitting the time-dependent contact angle with an exponential decay function θt = ae−t/τ + c,14 where t is time and a and c are the fitting parameters.The contact angle relaxation of both ILs against time and under various RH levels was plotted in Fig. 1. For [emim][NTf2], it took less than 25 min to obtain a final (stable) contact angle; for [bmim][NTf2] the relaxation time min falls within 45 min. When RH was increased from 20% to 90%, the relaxation time decreased by more than one order of magnitude, to less than 3 min for both ILs. For reference, the advancing and receding contact angle of [bmim][NTf2]on mica was measured using sessile drop methods (Fig. S2a†). The time dependence of the advancing and receding contact angle was also recorded and the result was plotted in Fig. S2b.† It is seen that the receding contact angle (RCA) decreased significantly with time, while the change in the advancing contact angle (ACA) was much less. The reduction of the RCA with time is very likely to be caused by the re-orientation of the IL molecules adsorbed on the mica surface. As the NTf2 ILs are hydrophobic and the mica surface is hydrophilic, water molecules tend to move from the bulk of the IL to the interface to form a water layer, such process causes the IL ions near interface to slowly reorder themselves, which we believe to be causing the receding contact angle to decrease with time.38 On the other hand, the ACA was measured by increasing the droplet volume, while the contact line expands on the mica spot that was partially covered by IL precursor film, these islands of IL film are isolated and uniform molecular reorientation is not very likely, therefore the values of ACA are more stable.
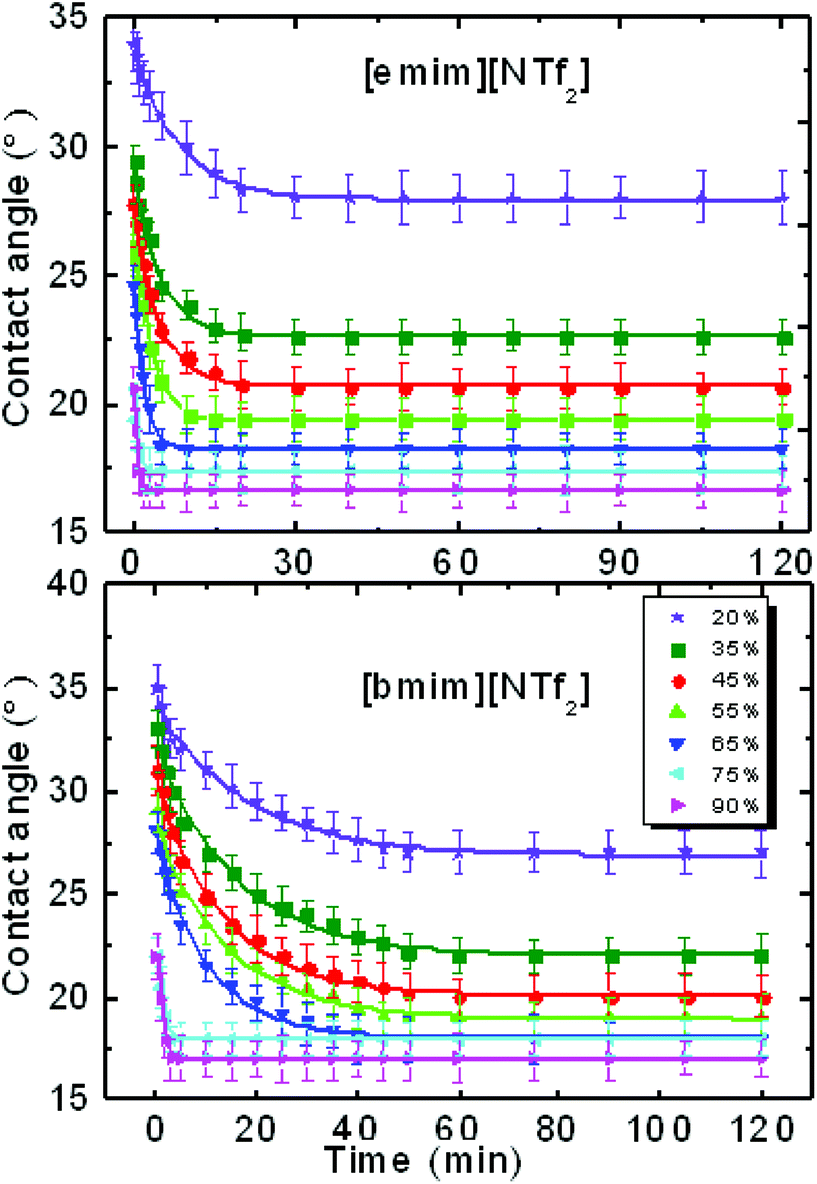 | ||
| Fig. 1 Contact angle relaxation of [emim][NTf2] and [bmim][NTf2]-ILs against time (min) on mica at various levels of relative humidity. | ||
The observation here differs from that by Belman et al.21 due to the difference of the system setup. Here we used freshly cleaved mica and in ref. 21 there is already a physically adsorbed octadecylamine or chemically adsorbed octadecanethiol film on the solid substrate. A thin film of IL is left behind after retracting the IL droplet from the mica surface using a syringe, which produced the pre-wetting effect. This is to be discussed in detail in the following section.
We have proposed in previous report that the contact angle relaxation of ILs was mainly caused by the thin precursor film spreading ahead of the bulk of the droplet, and showed that the propagation of the precursor film obeys the law of surface diffusion,14,48,52 this has recently been re-confirmed by Liu et al.13 We also proposed that the evolution of the precursor film follows a process that resembles the self-assembled monolayers, which involves adsorption and molecular reorientation as suggested by Ulman et al.17 Here by using tapping mode AFM we imaged the surrounding area of a [bmim][NTf2] droplet on mica after initial spreading of ∼30 min. The scanning was done at the relative humidity of 38% and 51% respectively. It is seen from Fig. 2a and b that the area in the vicinity of the contact line was covered by patches of thin film with thickness of ∼1.7 nm, suggesting that the patches are actually one ion pair of [bmim][NTf2].26 When RH was increased from 38% (Fig. 2a) to 51% (Fig. 2b), the area fraction of the IL precursor film on mica increased from ∼50% to 72%, suggesting that the water adsorption enhanced the precursor film formation of ILs.41 We argue that since the bulk of the droplet tightly follows the spreading of the precursor film before reaching an equilibrium contact angle, the macroscopic spreading of the droplet can actually be described by the law of surface diffusion. Two supplementary experiments was conducted to check this assumption, namely AFM imaging of the mica surface pre-wetted by one IL, and the spreading on such a pre-wetted mica surface by droplet of the same IL (Fig. S3†).
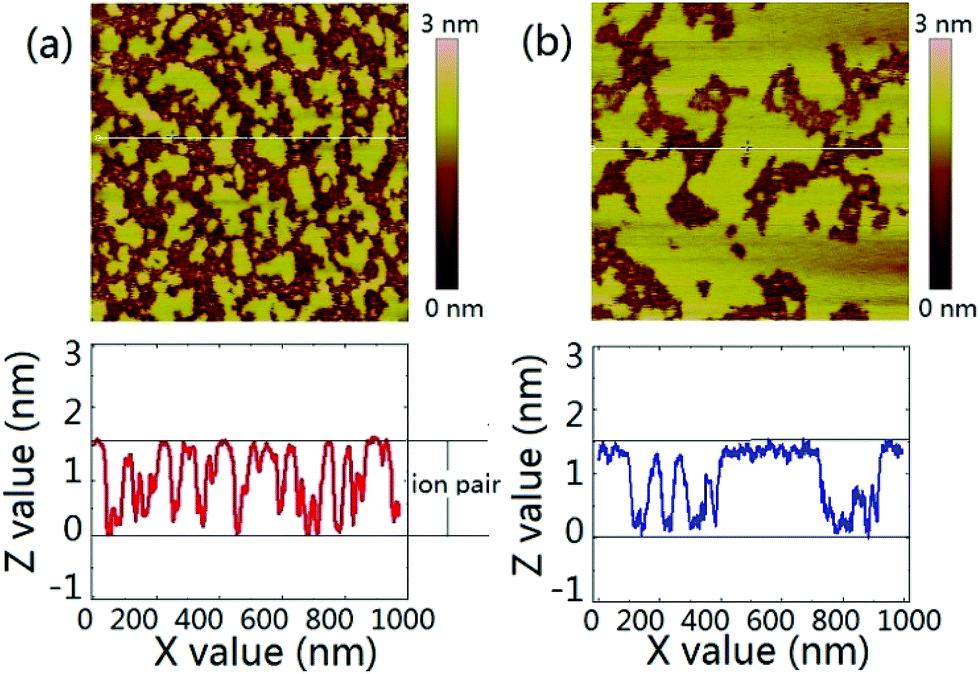 | ||
| Fig. 2 Tapping mode AFM image of the vicinity of the three phase contact line of a macroscopic droplet on mica surface at (a) RH of 38% and (b) RH of 51%. The image size is 1 μm. A line scan below each image shows the profiles of the partial monolayer of [bmim][NTf2]. | ||
From the tapping mode AFM image (Fig. S4a†) we see clearly that apart from some tiny residual droplets (R < 1 mm), the whole surface is actually covered by a thin film of ∼3 nm, which is equal to the thickness of two ion pairs of [bmim][NTf2].26 By tracking the contact angle evolution of a same type of IL droplet on the pre-wetted mica surface, we observe that the equilibrium contact angle of ∼22 degrees was obtained instantly (<20 s) on the pre-wetted mica surface and there is no contact angle relaxation after the inertia spreading (Fig. S4b† and inset). This observation has two implications: the first is that the droplet of [bmim][NTf2] does not wet the pre-wetted mica surface completely but has a finite contact angle; the second is that once the mica surface is pre-wetted by the IL thin film, the equilibration of the contact angle should be instant. Since spreading of the apparent triple line follows the propagation of the precursor film,15 the spreading kinetics of the droplet essentially reflects the extending of the precursor film,48,52,53 which was described as
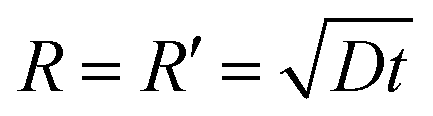 | (1) |
In order to quantify the kinetics of spreading, the droplets radii were extracted from the sessile drop images during contact angle relaxation. The variation of droplet radius R (mm) against the square root of time 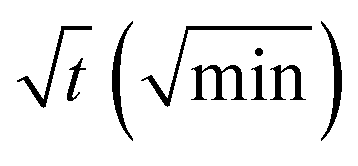 was plotted together (Fig. S5†). The droplet radii increased approximately linearly with
was plotted together (Fig. S5†). The droplet radii increased approximately linearly with 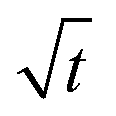 before it levels off, which resembles the spreading behaviour of precursor films through surface diffusion.47,48 The calculated surface diffusion coefficient D was plotted in Fig. 3. From Fig. 3, it is seen that D is between 10−11 m2 s−1 and 10−9 m2 s−1. These values are of the same order of magnitude as those reported in the literature for PDMS precursor films (spreading of microscopic droplets) on various substrates.52 This is the first time that the surface diffusion mechanism was verified for characterization of the kinetics of the contact angle relaxation. D increases slowly before RH reaches 65%. However, it increased by approximately one order of magnitude (10 times and 8 times for [emim][NTf2] and [bmim][NTf2] respectively) when RH was increased from 35% to 75%, this is followed by another notable increase when RH reaches 90%. Worth being noted is that Cazabat et al. suggested that the dependence of the diffusion coefficient of precursor film spreading on hydrophilic and hydrophobic substrates ought to be differentiated, because hydrophobic substrates adsorbs negligible amount of water even at high RH levels, which make the observed spreading largely independent of the friction coefficient.53 As suggested by previous literature, the difference between the surface diffusion coefficient of the two ILs was probably caused by the difference between the molecular weight and viscosity of the ILs.47,54
before it levels off, which resembles the spreading behaviour of precursor films through surface diffusion.47,48 The calculated surface diffusion coefficient D was plotted in Fig. 3. From Fig. 3, it is seen that D is between 10−11 m2 s−1 and 10−9 m2 s−1. These values are of the same order of magnitude as those reported in the literature for PDMS precursor films (spreading of microscopic droplets) on various substrates.52 This is the first time that the surface diffusion mechanism was verified for characterization of the kinetics of the contact angle relaxation. D increases slowly before RH reaches 65%. However, it increased by approximately one order of magnitude (10 times and 8 times for [emim][NTf2] and [bmim][NTf2] respectively) when RH was increased from 35% to 75%, this is followed by another notable increase when RH reaches 90%. Worth being noted is that Cazabat et al. suggested that the dependence of the diffusion coefficient of precursor film spreading on hydrophilic and hydrophobic substrates ought to be differentiated, because hydrophobic substrates adsorbs negligible amount of water even at high RH levels, which make the observed spreading largely independent of the friction coefficient.53 As suggested by previous literature, the difference between the surface diffusion coefficient of the two ILs was probably caused by the difference between the molecular weight and viscosity of the ILs.47,54
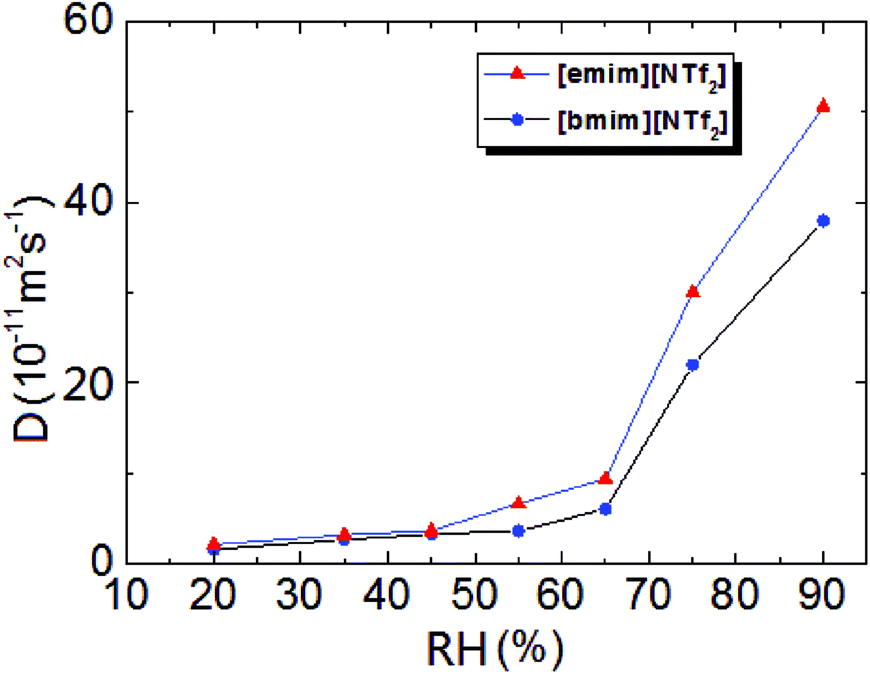 | ||
| Fig. 3 Surface diffusion coefficient of [emim][NTf2] and [bmim][NTf2] as a function of relative humidity. | ||
The exact mechanism of the enhanced spreading of liquid droplets on solids under humid environment is still open to debate.48 However, since mica is a hydrophilic substrate, we attribute the quickened spreading partially to the reduction of the friction coefficient of solid surfaces, as suggested by previous report.48,55 Because the conformational freedom and the mobility of adsorbed water increases dramatically at high RH,56,57 the friction coefficient on mica can be reduced by as much as 50% when RH is increased from 20% to 80%.58
We have reported the formation of IL precursor films through extended layering on mica surface and attributed the contact angle relaxation to the propagation of such film.14 Gong et al. suggested that whether ILs can develop extended layering on mica surface is largely dependent on the amount of water adsorbed on the mica surface.41 Under higher RH levels IL droplet tend to develop precursor film more easily because the adsorbed water molecules facilitates the exchange between the cations of ionic liquid and K+ ions on the mica surface.41 Since water coverage on mica increases with the RH levels and the precursor film fraction of IL on mica scales with the amount of adsorbed water or the water area fraction, the precursor film coverage should also increase with RH. AFM imaging of water adsorption on mica is frequently subject to interference of the scanning tips.59 However, Xu et al. has obtained very neat image of water adsorption on mica using graphene templating to seal the water molecules on mica.60 The fact that the area fraction of the precursor films in Fig. 2a (∼50%) corresponds to the water layer fraction from ref. 60 at the same relative humidity appears to support such conjecture. The developed precursor film will produce a pre-wetting effect on mica surface similar to that displayed in Fig. S4.† A detailed AFM imaging of the IL precursor film coverage on mica under varying humidity is beyond the scope of this investigation.
Adsorbed water can significantly alter the surface energy of mica surface and this has been characterized by ref. 61. Similarly, the adsorbed precursor film of IL is also expected to change the surface energy of mica and cause a variation of the contact angle. When adsorbed water facilitates the IL droplet to form extended patches of monolayer on mica surface at the RH <70%, the IL droplet would contact ‘patched’ heterogeneous mica surface. Based on the results in Fig. 2 and those from ref. 41, we assume that the surface coverage of the precursor film scales with the water monolayer coverage on mica. Therefore, the contact angle of the same IL on the ‘patched’ mica surface should decrease with the area fraction of the IL precursor film. On such heterogeneous mica surface, the final contact angle of the studied ILs are expected to obey Cassie's equation.62
The value of cos(θ) was plotted against RH in Fig. 4, and the results indicate that the cos(θ) increases linearly with RH before the contact angle levelled off at RH ∼70%. The turning point of cos(θ) (RH of ∼65%) also agrees with the suggested RH level of water monolayer formation.61
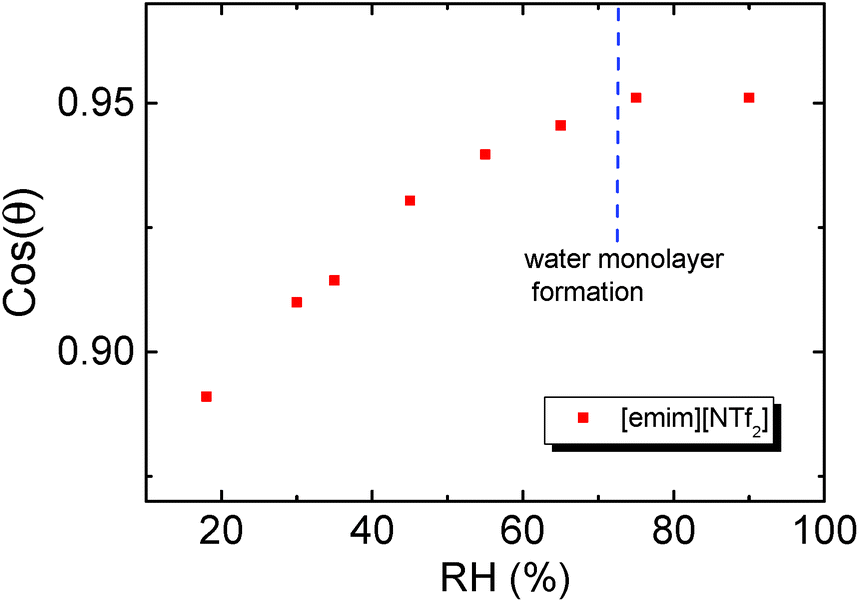 | ||
| Fig. 4 Variation of cosine of contact angle (θ) of [emim][NTf2] on mica with RH. | ||
Worth noting is that we reported previously the molecular-layering of water-saturated ILs at interfaces with mica was disrupted and significantly weakened due to the hydrogen bond between water molecules and the IL ions. As a result, the contact angle relaxation was also reduced.15 Here by turning our focus from water-inside the ILs to the water adsorbed on the mica surface (between IL droplet and mica), the effect of water on the interfacial phenomena of ILs appear to be strikingly different. Since ionic liquids are hygroscopic and they absorb water from air, the increase of water concentration in the ionic liquids under a humid environment can lead to potential change to the mica–IL interaction.33,37 It is important to know the variation of water concentrations under different RH levels. Therefore we measured the change of water concentration of [bmim][NTf2] exposing to both ambient (38%) and high (90%) levels of RH, the results from Karl Fischer titration were shown in Fig. 5. In our experiments, it took less than one hour for the two ILs to reach an equilibrium contact angle at RH of ∼40% (∼25 min for [emim][NTf2] and 50 min [bmim][NTf2]). Their water mole fraction increased to 0.03–0.04 by exposing to the RH of 38% for 60 min. It is seen from Fig. 1 that the spreading time of the ILs droplet decreases sharply at higher humidity, which means less time for taking up water within the spreading time. The water-vapor adsorption on the mica surface and the subsequent contact angle relaxation (spreading) is much faster than water absorption by the ILs at corresponding RH levels. Therefore, the increase of water concentration in the ILs within the spreading time is not significant at high RH level. For example, at higher RH (90%), it takes less than 5 minutes for [emim][NTf2] and [bmim][NTf2] to reach a final contact angle and the increase of water mole fraction within such a short time is less than 0.03.
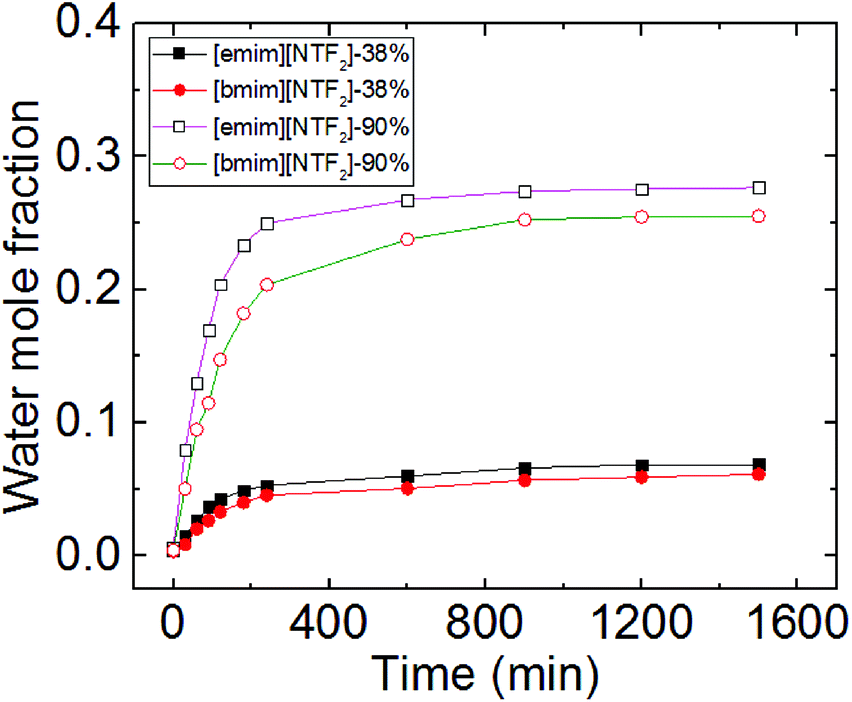 | ||
| Fig. 5 Increase of water mole fraction in [emim][NTf2] and [bmim][NTf2] under RH of 38% and 90%. | ||
The surface tension of the [bmim][NTf2] ILs of different water concentration was measured using pendent drop method and was shown in Fig. 6. The results indicates that the surface tension (γ) of the [Rmim][NTf2] IL is hardly affected by the presence of water, this is consistent with previous findings by Freire et al.40 and Liu et al.,13 although the ILs investigated here are slightly different. Therefore, we conclude that the absorption of water at the IL–air interface does not cause a notable change in the surface tension of the studied ILs. Although ILs is likely to take up water molecules from humid air, it does not cause significant interfacial effect within the time of contact angle relaxation.15 Therefore, in summary, from this experimental study on the effect of humidity on the IL droplet_on_mica system, both the changes in the spreading velocity and the final contact angle were caused by the water adsorbed on the mica surface, the water taken by the ILs from the surrounding humid environment seem to have a minor effect on the wetting behaviour of ILs.
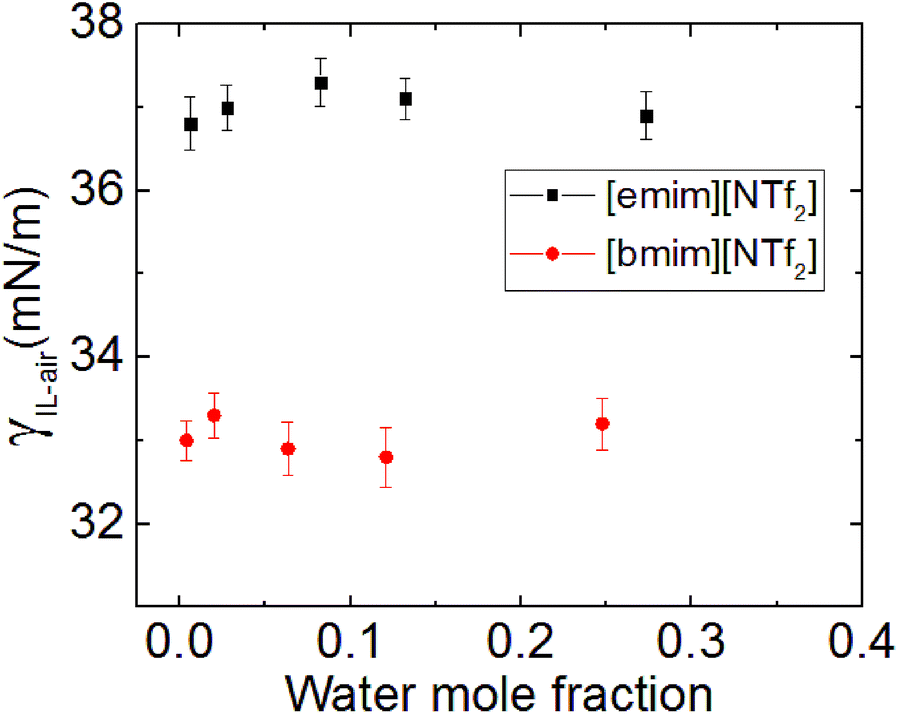 | ||
| Fig. 6 Surface tension of [emim][NTf2] and [bmim][NTf2] ILs against their water concentration. | ||
4. Conclusions
The role of water vapor adsorption on the kinetic spreading and contact angles of two [Rmim][NTf2] ILs on mica surface has been investigated under varying levels of relative humidity. It was found that increasing RH significantly accelerated the spreading of the studied IL droplets on mica and also reduced their equilibrium contact angles. This is attributed to the effect of adsorbed water. Water vapor adsorption enhances IL precursor film formation, as revealed by AFM imaging. We propose that water vapor adsorption on mica modifies the mica surface into a heterogeneous substrates and the coverage of the ILs precursor film increases with RH, while equilibrium contact angles of the investigated ILs decrease with RH. Both the surface diffusion coefficients and the equilibrium contact angle have a threshold at the RH of ∼70%, which is believed to be caused by the formation of a complete water monolayer on mica under such condition. This study could offers fundamental insights to applications of ILs in lubrication and electrochemical energy systems.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors would like to thank Craig Priest for helpful discussions. This project was also supported financially by the Top Young Talent Support Plan of Xi'an Jiaotong University and the Australian Research Council Discovery Project Scheme.References
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115, 6357–6426 CrossRef CAS PubMed
.
- T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS PubMed
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed
- M.-D. Bermúdez, A.-E. Jiménez, J. Sanes and F.-J. Carrión, Molecules, 2009, 14, 2888 CrossRef PubMed
- C. Ye, W. Liu, Y. Chen and L. Yu, Chem. Commun., 2001, 2244–2245 RSC
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, P. Simon and C. A. Angell, Energy Environ. Sci., 2014, 7, 232–250 CAS
- I. Delcheva, J. Ralston, D. A. Beattie and M. Krasowska, Adv. Colloid Interface Sci., 2015, 222, 162–171 CrossRef CAS PubMed
- L. Gao and T. J. McCarthy, J. Am. Chem. Soc., 2007, 129, 3804–3805 CrossRef CAS PubMed
- H. Liu and L. Jiang, Small, 2016, 12, 9–15 CrossRef CAS PubMed
- S. Millefiorini, A. H. Tkaczyk, R. Sedev, J. Efthimiadis and J. Ralston, J. Am. Chem. Soc., 2006, 128, 3098–3101 CrossRef CAS PubMed
- M. Paneru, C. Priest, R. Sedev and J. Ralston, J. Am. Chem. Soc., 2010, 132, 8301–8308 CrossRef CAS PubMed
- Z. Liu, T. Cui, G. Li and F. Endres, Langmuir, 2017 DOI:10.1021/acs.langmuir.7b00082
- Z. Wang and C. Priest, Langmuir, 2013, 29, 11344–11353 CrossRef CAS PubMed
- Z. Wang, H. Li, R. Atkin and C. Priest, Langmuir, 2016, 32, 8818–8825 CrossRef CAS PubMed
- C. Lam, R. Wu, D. Li, M. Hair and A. Neumann, Adv. Colloid Interface Sci., 2002, 96, 169–191 CrossRef CAS PubMed
- S. D. Evans, R. Sharma and A. Ulman, Langmuir, 1991, 7, 156–161 CrossRef CAS
- I. Langmuir, Science, 1938, 87, 493–500 CAS
- P. N. Yiannos, J. Colloid Sci., 1962, 17, 334–347 CrossRef CAS
- G. Vigil, Z. Xu, S. Steinberg and J. Israelachvili, J. Colloid Interface Sci., 1994, 165, 367–385 CrossRef CAS
- N. Belman, K. Jin, Y. Golan, J. N. Israelachvili and N. S. Pesika, Langmuir, 2012, 28, 14609–14617 CrossRef CAS PubMed
- T. Cremer, M. Stark, A. Deyko, H. P. Steinruck and F. Maier, Langmuir, 2011, 27, 3662–3671 CrossRef CAS PubMed
- F. Endres, Phys. Chem. Chem. Phys., 2012, 14, 5008–5009 RSC
- Y. Liu, Y. Zhang, G. Wu and J. Hu, J. Am. Chem. Soc., 2006, 128, 7456–7457 CrossRef CAS PubMed
- M. Mezger, H. Schroder, H. Reichert, S. Schramm, J. S. Okasinski, S. Schoder, V. Honkimaki, M. Deutsch, B. M. Ocko, J. Ralston, M. Rohwerder, M. Stratmann and H. Dosch, Science, 2008, 322, 424–428 CrossRef CAS PubMed
- R. Singh Payal and S. Balasubramanian, ChemPhysChem, 2012, 13, 1764–1771 CrossRef CAS PubMed
- A. Deyko, T. Cremer, F. Rietzler, S. Perkin, L. Crowhurst, T. Welton, H.-P. Steinrück and F. Maier, J. Phys. Chem. C, 2013, 117, 5101–5111 CAS
- R. Atkin and G. G. Warr, J. Phys. Chem. C, 2007, 111, 5162–5168 CAS
- D. A. Beattie, R. M. Espinosa-Marzal, T. T. M. Ho, M. N. Popescu, J. Ralston, C. J. E. Richard, P. M. F. Sellapperumage and M. Krasowska, J. Phys. Chem. C, 2013, 117, 23676–23684 CAS
- S. Bovio, A. Podestà, C. Lenardi and P. Milani, J. Phys. Chem. B, 2009, 113, 6600–6603 CrossRef CAS PubMed
- R. S. Anaredy and S. K. Shaw, Langmuir, 2016, 32, 5147–5154 CrossRef CAS PubMed
- H. W. Cheng, P. Stock, B. Moeremans, T. Baimpos, X. Banquy, F. U. Renner and M. Valtiner, Adv. Mater. Interfaces, 2015, 2, 1500159 CrossRef
- H.-W. Cheng, J.-N. Dienemann, P. Stock, C. Merola, Y.-J. Chen and M. Valtiner, Sci. Rep., 2016, 6, 30058 CrossRef CAS PubMed
- M. A. Gebbie, A. M. Smith, H. A. Dobbs, G. G. Warr, X. Banquy, M. Valtiner, M. W. Rutland, J. N. Israelachvili, S. Perkin and R. Atkin, Chem. Commun., 2017, 53, 1214–1224 RSC
- L. R. Griffin, K. L. Browning, S. M. Clarke, A. M. Smith, S. Perkin, M. Skoda and S. E. Norman, Phys. Chem. Chem. Phys., 2017, 19, 297–304 RSC
- S. Perkin, M. Salanne, P. Madden and R. Lynden-Bell, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E4121 CrossRef CAS PubMed
- R. M. Espinosa-Marzal, A. Arcifa, A. Rossi and N. D. Spencer, J. Phys. Chem. C, 2014, 118, 6491–6503 CAS
- K. Sakai, K. Okada, A. Uka, T. Misono, T. Endo, S. Sasaki, M. Abe and H. Sakai, Langmuir, 2015, 31, 6085–6091 CrossRef CAS PubMed
- Y. Wang, H. Li and S. Han, J. Phys. Chem. B, 2006, 110, 24646–24651 CrossRef CAS PubMed
- M. G. Freire, P. J. Carvalho, A. M. Fernandes, I. M. Marrucho, A. J. Queimada and J. A. Coutinho, J. Colloid Interface Sci., 2007, 314, 621–630 CrossRef CAS PubMed
- X. Gong, A. Kozbial and L. Li, Chem. Sci., 2015, 6, 3478–3482 RSC
- H. K. Christenson and N. H. Thomson, Surf. Sci. Rep., 2016, 71, 367–390 CrossRef CAS
- J. A. Smith, O. Werzer, G. B. Webber, G. G. Warr and R. Atkin, J. Phys. Chem. Lett., 2009, 1, 64–68 CrossRef
- I. Langmuir, J. Am. Chem. Soc., 1918, 40, 1361–1403 CrossRef CAS
- M. K. Bernett and W. A. Zisman, J. Colloid Interface Sci., 1969, 29, 413–423 CrossRef CAS
- E. G. Shafrin and W. A. Zisman, J. Am. Ceram. Soc., 1967, 50, 478–484 CrossRef CAS
- M. Valignat, S. Bardon, S. Villette and A. Cazabat, Fluid Phase Equilib., 1998, 150, 615–623 CrossRef
- S. Villette, M. Valignat, A. Cazabat, L. Jullien and F. Tiberg, Langmuir, 1996, 12, 825–830 CrossRef CAS
- W. Bou-Zeid and D. Brutin, Colloids Surf., A, 2014, 456, 273–285 CrossRef CAS
- A. Arcifa, A. Rossi, R. M. Espinosa-Marzal and N. D. Spencer, ACS Appl. Mater. Interfaces, 2016, 8, 2961–2973 CAS
- A. Wexler and S. Hasegawa, J. Res. Natl. Bur. Stand., 1954, 53, 19–26 CrossRef CAS
- M. N. Popescu, G. Oshanin, S. Dietrich and A. Cazabat, J. Phys.: Condens. Matter, 2012, 24, 243102 CrossRef CAS PubMed
- F. Tiberg and A.-M. Cazabat, Europhys. Lett., 1994, 25, 205 CrossRef CAS
- M. Voué, M. Valignat, G. Oshanin and A. Cazabat, Langmuir, 1999, 15, 1522–1527 CrossRef
- M. A. Chowdhury and M. M. Helali, Tribol. Interface Eng. Ser., 2006, 39, 958–962 CAS
- D. R. Kinney, I.-S. Chuang and G. E. Maciel, J. Am. Chem. Soc., 1993, 115, 6786–6794 CrossRef CAS
- J. Lancaster, Tribol. Interface Eng. Ser., 1990, 23, 371–389 CAS
- A. Malani and K. Ayappa, J. Phys. Chem. B, 2009, 113, 1058–1067 CrossRef CAS PubMed
- C. Spagnoli, K. Loos, A. Ulman and M. K. Cowman, J. Am. Chem. Soc., 2003, 125, 7124–7128 CrossRef CAS PubMed
- K. Xu, P. Cao and J. R. Heath, Science, 2010, 329, 1188–1191 CrossRef CAS PubMed
- T. E. Balmer, H. K. Christenson, N. D. Spencer and M. Heuberger, Langmuir, 2008, 24, 1566–1569 CrossRef CAS PubMed
- A. Cassie and S. Baxter, Trans. Faraday Soc., 1944, 40, 546–551 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07077b |
| This journal is © The Royal Society of Chemistry 2017 |