
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Detoxification of organophosphates using imidazole-coated Ag, Au and AgAu nanoparticles†
Valmir B. Silvaa,
Thenner S. Rodrigues b,
Pedro H. C. Camargob and
Elisa S. Orth*a
b,
Pedro H. C. Camargob and
Elisa S. Orth*a
aDepartamento de Química, Universidade Federal do Paraná, CP 19032, 81531-980 Curitiba, PR, Brazil. E-mail: elisaorth@ufpr.br
bInstituto de Química, Departamento de Química Fundamental, Universidade de São Paulo, Av. Prof. Lineu Prestes 748, SP 05508-900, São Paulo, Brazil
First published on 18th August 2017
Abstract
Organophosphate (OP) detoxification is a worldwide problem due to the high stability of P–O bonds. Here, we designed several imidazole-coated nanocatalysts targeted towards the cleavage of OP and thus their detoxification. Specifically, Ag, Au and bimetallic AgAu nanoparticles (NPs) supported on SiO2 were functionalized with methimazole (MTZ), which comprises thiol and imidazole groups, foreseeing enabling freely available imidazole groups. Raman analyses indicate that MTZ interacts preferably via its sulfur atom over Au NPs and via the nitrogen of the imidazole ring over Ag NPs. The MTZ-derived nanocatalysts were effective towards OP cleavage. For instance, rate enhancements of 108 fold were obtained for the toxic pesticide Paraoxon, compared to the uncatalyzed reaction. Interestingly, Au-derived nanocatalysts were significantly more effective since the imidazole group is free to react with the OP, which is not possible when Ag–N interactions take place.
Introduction
Organophosphates (OP) are intrinsically related to many fundamental biological functions, such as biosynthesis, signaling, regulation, and energy transduction.1 Moreover, they are responsible for the preservation of genetic information since they constitute DNA and RNA strands.2 Actually, one-third of all biological proteins are phosphorylated at some time, evidencing the importance of phosphoryl transfer. OP are mostly known for their high stability.3 For example, the hydrolysis of DNA is estimated take in over 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 years.4 Since enzymes are very successful in promoting phosphorylation in biological systems,2 they have bioinspired many mimicking studies. The downside of OP's stability is that they have been used as a scaffold for many chemical warfare and pesticides (Scheme 1), knowingly to have high toxicity, sometimes even being lethal.5 Not surprising, much effort has been directed towards the development of efficient methods of destruction and detection of these toxic OP, focusing on deactivating unused (probably prohibited) stockpiles and monitoring abusive use of agrochemical and/or chemical attacks5,6
000 years.4 Since enzymes are very successful in promoting phosphorylation in biological systems,2 they have bioinspired many mimicking studies. The downside of OP's stability is that they have been used as a scaffold for many chemical warfare and pesticides (Scheme 1), knowingly to have high toxicity, sometimes even being lethal.5 Not surprising, much effort has been directed towards the development of efficient methods of destruction and detection of these toxic OP, focusing on deactivating unused (probably prohibited) stockpiles and monitoring abusive use of agrochemical and/or chemical attacks5,6
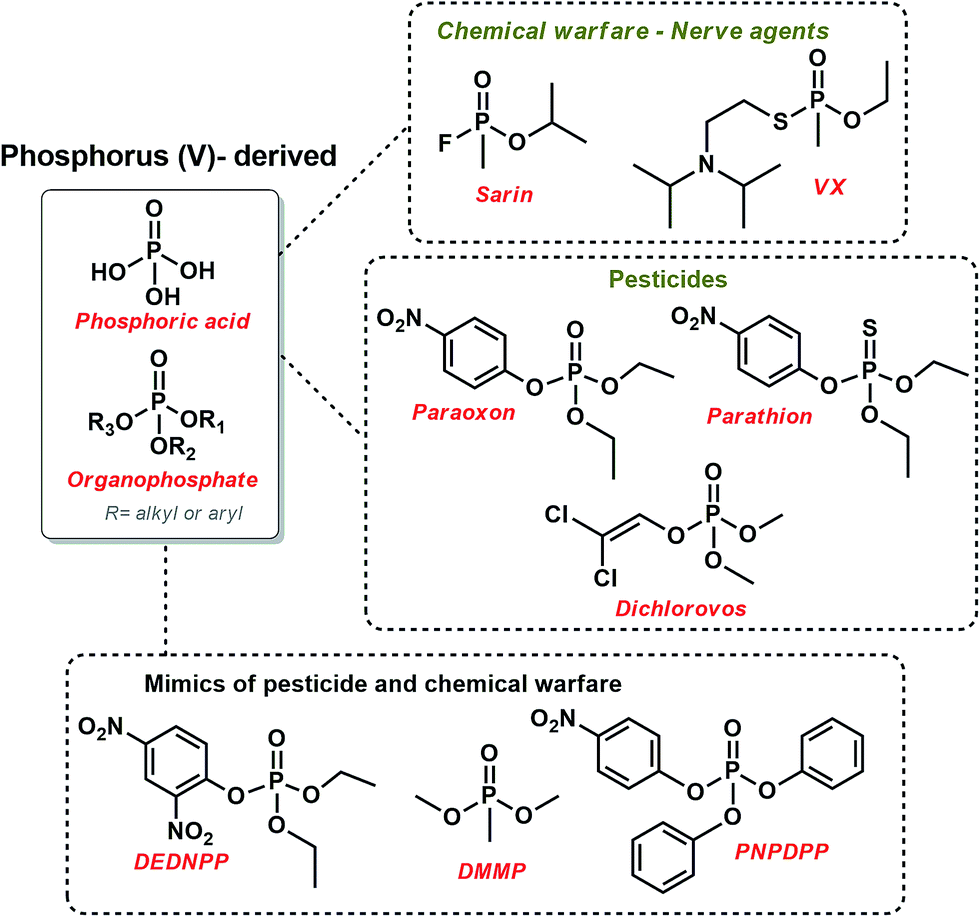 | ||
| Scheme 1 The structure of some relevant organophosphates used as chemical warfare and pesticides, along with some mimics of toxic OP. | ||
It is noteworthy that poisoning by pesticides is a serious worldwide public concern.7 As a matter of security, many studies in this field have used structural mimics of pesticides and chemical warfare that usually have lower toxicity and higher reactivity so that the reaction can be feasibly followed. For example, dimethyl methylphosphonate (DMMP), p-nitrophenyl diphenylphosphate (PNPDPP),5 and diethyl-2,4-dinitrophenyl phosphate (DEDNPP)8–10 have been explored, as shown in Scheme 1.
In order to pursue the detoxification of OP, many bio-inspired models have been proposed as detoxifying agent or artificial enzymes targeting the phosphoryl cleavage.5 In this context, imidazole-derived catalysts have attracted great attention due to their presence in many enzymatic active sites and their chemical versatility,11 i.e., they can react as acid–base or nucleophilic catalysts under considerably mild conditions (pH ∼ 7).12,13 Another promising class of catalysts for OP cleavage involve nanoparticles (NPs),14 although there are still only a few reports in this field,5,15–20 in contrast to the many studies regarding NPs-based sensors for various OP.21
The physicochemical features of several NPs, such as shape and size, can be decisive for catalysis.22–24 A previous study showed that the reduction of 4-nitrophenol can be catalyzed by Ag and AgAu NPs alone, with higher reactivity for the later attributed to Au content and the increased surface area due to the hollow interiors.25 In another study, bimetallic nanoshells of AgAu, AgPd, and AgPt NPs were used as bio-metallo catalysts with dual activity for transesterification; and silane oxidation.26 Insofar, to the best of our knowledge, there are no studies correlating NPs features with its effect on OP degradation.
Herein, we coated silver (AgSiO2), gold (AuSiO2) and bimetallic nanoshells AgAuSiO2 NPs (supported on SiO2) with a thiol-substituted imidazole (methimazole, MTZ) in order to obtain nanocatalysts with freely available imidazole moieties. We choose MTZ due to the extraordinary catalytic activity known for imidazole groups (vide supra) and the thiol group that interacts favorably with NPs,20,27 hence anchoring efficiently imidazole moieties on the NPs surface. For this purpose, the NPs were firstly obtained with controlled shape and size following a previously reported procedure.28,29 Then, the NPs were functionalized with MTZ (Fig. 1). Finally, the nanocatalysts were evaluated toward OP degradation, using the simulant DEDNPP and the pesticide Paraoxon as model substrates.
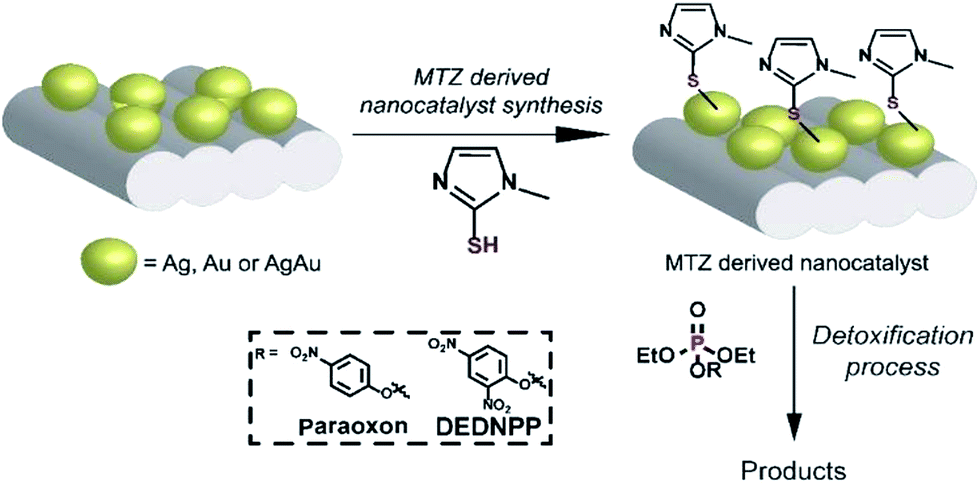 | ||
| Fig. 1 Functionalization of AuSiO2, AgSiO2 or AgAuSiO2 with MTZ. | ||
Experimental
Materials
MTZ Analytical grade silver nitrate (AgNO3, 99%, Sigma-Aldrich), polyvinylpyrrolidone (PVP, Sigma-Aldrich, M.W. 10000 g mol−1), polyvinylpyrrolidone (PVP, Sigma-Aldrich, M.W. 55000 g mol−1), ethylene glycol (EG, 99.8%, Sigma-Aldrich), tetrachloroauric(III) acid trihydrate (HAuCl4·3H2O, ≥99.9%, Sigma-Aldrich), silica (pore size 22 Å, 800 m2 g−1, CAS number 112926-00-8, Sigma-Aldrich), ethanol (Êxodo) were used as received.
Synthesis of Ag NPs29
Ag seeds were prepared by the polyol process. In a typical experiment, 5 g of polyvinylpyrrolidone (PVP, 10000 g mol−1) was dissolved in 37.5 mL of ethylene glycol. Then, AgNO3 (200 mg, 1.2 mmol) was added and mixed until the complete dissolution. The resulting solution was heated to 125 °C for 2.5 hours, leading to the appearance of a greenish-yellow color, allowed to cool down to room temperature, and diluted to 125 mL of water. After this step, 46 mL of the resulting suspension (0.2197 mg mL−1, determined by ICP) were washed twice with ethanol and three times with water by successive rounds of centrifugation at 15000 rpm and removal of the supernatant. After washing, the product was suspended in 10 mL of deionized water.
Synthesis of Au NPs30
The synthesis of Au nanospheres was performed by a seeded growth approach. In the first step, Au seeds were synthesized by adding 150 mL of a 2.2 mmol L−1 sodium citrate aqueous solution to a 250 mL round-bottom flask under magnetic stirring. This system was heated to 100 °C for 15 minutes. Then, 1 mL of a 25 mmol L−1 AuCl4−(aq) solution was added, and the reaction mixture was kept at 100 °C under vigorous stirring for 30 minutes. The obtained Au nanoparticles were employed as seeds for the synthesis of Au NPs having larger sizes by two successive steps of Au deposition. For the first deposition step, 1 mL of a 60 mmol L−1 sodium citrate solution was added to the same 250 mL round-bottom flask containing the Au NPs seeds under magnetic stirring at 100 °C for 5 minutes. Afterward, 1 mL of a 25 mmol L−1 AuCl4−(aq) solution was added to the reaction mixture containing the Au NPs seeds and the reaction mixture was carried out for another 30 min. Similarly, a second deposition step could be performed by adding another 1 mL of a 60 mmol L−1 sodium citrate solution and 1 mL of a 25 mmol L−1 AuCl4−(aq) solution to the reaction mixture obtained after the first deposition step, in which the Au NPs produced after the first deposition step served as seeds for further growth. After the reaction, the resulting suspension was allowed to cool down to room temperature. After this step, 88 mL of the resulting suspension (0.1146 mg mL−1, determined by ICP) were washed three times with water by successive rounds of centrifugation at 15000 rpm and removal of the supernatant. After washing, the product was suspended in 10 mL of deionized water.
Synthesis of AgAu nanoshells29
The synthesis of AgAu nanoshells was based on the galvanic replacement reaction between Ag nanospheres and AuCl4−(aq). In a typical procedure, a mixture containing 50 mL of PVP aqueous solution (0.1 wt%, M.W. 55000 g mol−1) and 10 mL of the as-prepared suspension containing the Ag nanospheres was stirred at 100 °C for 10 min in a 250 mL round-bottom flask. Then, 20 mL of AuCl4−(aq) (0.6 mmol L−1) was added dropwise and the reaction allowed to proceed at 100 °C for another 1 h. After that, the suspension was allowed to cool down to room temperature and 28 mL (0.0458 mg mL−1, determined by ICP) were washed twice with a supersaturated NaCl solution and three times with water by successive rounds of centrifugation at 15000 rpm and removal of the supernatant. After washing, the product was suspended in 10 mL of deionized water.
Synthesis of the SiO2 supported NPs28
The incorporation of Ag, Au, and AgAu nanostructures onto a silica matrix (1% wt, metal basis) were performed based on a wet impregnation approach. In a typical run, 10 mL of the suspensions containing the respective nanoparticles washed and concentrated were added dropwise to a beaker containing 1 g of commercial silica suspended in 20 mL of deionized water under magnetic stirring. The resulting mixture was stirred at room temperature for 24 h. After this step, the resulting suspensions were washed three times with distilled water and twice with ethanol by successive rounds of centrifugation at 7000 rpm and removal of the supernatant. The resulting catalysts were then treated at 120 °C for 2 h under air.Coating of the NPs with MTZ
The MTZ derived nanocatalysts were prepared as follows: 500 mg of the SiO2 supported AgSiO2, AuSiO2 or AgAuSiO2 NPs was added to a becker along with MTZ (26.0, 15.0 and 21.0 mg for AgSiO2, AuSiO2 and AgAuSiO2, respectively) in 50 mL of ethanol. The mixture was stirred during 24 h followed by centrifugation (12000 rpm) and separated. In the following, the material was washed with ethanol (three times), centrifugated, and the supernatant was discarded. The ethanol was evaporated at room temperature. The samples were denominated AgSiO2–MTZ, AuSiO2–MTZ and AgAuSiO2–MTZ, originated from AgSiO2, AuSiO2, and AgAuSiO2 NPs, respectively.
Characterizations
For the NPs, the scanning electron microscopy (SEM) images were obtained using a JEOL field emission gun electron microscope JSM6330F operated at 5 kV. The samples were prepared by drop-casting an aqueous suspension containing the nanostructures over a silicon wafer, followed by drying under ambient conditions. Transmission electron microscopy (TEM) images were obtained with a JEOL JEM1010 microscope operated at 80 kV. Samples for TEM were prepared by drop-casting an aqueous suspension of the nanostructures over a carbon-coated copper grid, followed by drying under ambient conditions. UV-vis spectra were obtained from aqueous suspensions containing the nanostructures with a Shimadzu UV-1700 spectrophotometer. The Ag and Au atomic percentages were measured by inductively coupled plasma optical emission spectrometry (ICP-OES) using a Spectro Arcos equipment. The X-ray diffraction (XRD) data were obtained using a Rigaku-Miniflex equipment, CuKα radiation. The diffraction pattern was measured in the range of 10–90° 2θ with a 1° min−1 angular speed scan. For the MTZ-derived NPs, Raman spectra were obtained with a Renishaw spectrophotometer using the 632.8 nm excitation laser, with 25 accumulations and 25–50% of the laser intensity.Catalytic studies of OP detoxification
For the catalytic studies, two OP were employed: DEDNPP and Paraoxon. Pseudo-first order conditions were maintained, where 30 mg of MTZ-derived nanocatalyst was added to 3 mL of a buffered solution (bicarbonate 0.2 mol L−1). The reaction started upon addition of 15 μL of DEDNPP or Paraoxon stock solution (5 × 10−3 mol L−1 in acetonitrile) and the medium was maintained under magnetic stirring (4 h and 15 days for DEDNPP and Paraoxon, respectively). The suspension was centrifugated (12000 rpm) and UV-vis spectra of the supernatant were recorded. The catalytic activity was calculated based on the product concentration, hence OP degradation by measuring the absorbance variation at 400 nm related to the products 4-nitrophenolate (PNP) or 2,4 dinitrophenolate (DNP), that originated from Paraoxon and DEDNPP, respectively (Scheme 2). The molar absorptivity (ε) used were 18380 and 12100 L mol−1 cm−1 for PNP and DNP, respectively.31,32 Considering pseudo-first order kinetics, already corroborated for these reactions in a separate study, the integrated rate law was used to obtain the pseudo-first order constant.
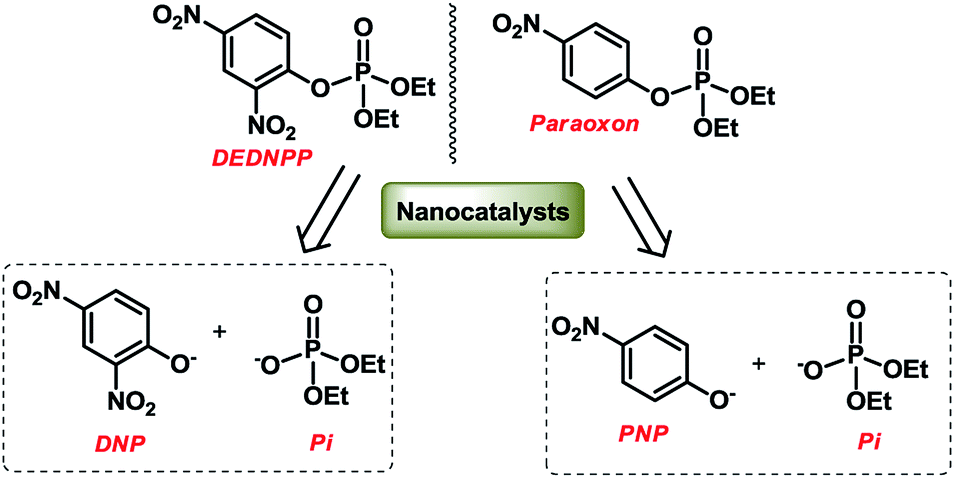 | ||
| Scheme 2 Cleavage reaction of DEDNPP and Paraoxon in the presence of the nanocatalysts. | ||
Results and discussion
Characterization of the SiO2 supported NPs
Our studies started with the synthesis of Ag nanospheres, Au nanospheres, and AgAu nanoshells as shown in Fig. 2A–C, respectively. Ag nanospheres (Fig. 2A) were obtained by a one-step polyol approach and were 35 ± 4 nm in diameter.33 On the other hand, Au nanospheres (Fig. 2B) were obtained by a seeded growth approach, in which Au nanospheres obtained by citrate reduction of AuCl4−(aq) acted as template for additional Au deposition and growth leading to the formation of nanoparticles displaying larger diameters (36 ± 3 nm) that were similar to the Ag nanospheres.30 For the AgAu nanoshells, the obtained Ag nanospheres were employed as sacrificial templates in the galvanic replacement reaction using AuCl4−(aq), which represents an effective strategy for the synthesis of hollow nanostructures displaying controllable properties in a single-step.29,34 As depicted in Fig. 2C, the formation of the hollow interiors is confirmed by the brighter mass-thickness contrast at the center of the AgAu nanoshells, which were 36 ± 4 nm in diameter, in agreement with the use of Ag nanospheres as templates. Interestingly, all obtained nanostructures displayed spherical shapes and relatively monodisperse sizes, which allowed us systematically investigate their catalytic performances as a function of the compositions and structures. The metal at% in the nanostructures were determined by ICP-OES analyses and corresponded to 100% of Ag, 100% of Au, and 49% of Ag + 51% of Au for Ag, Au, and AgAu, respectively. | ||
| Fig. 2 (A–C) SEM images of Ag nanospheres (A), Au nanospheres (B), and AgAu nanoshells (C). The insets correspond to their corresponding TEM images (the scale bars in the insets correspond to 20 nm). (D–F) SEM images of (D) AgSiO2, (E) AuSiO2, and (F) AgAuSiO2 materials that were obtained from wet impregnation of the samples (A–C) onto a commercial SiO2 support. | ||
After the synthesis of the Ag, Au, and AgAu nanostructures, we turned our attention to their incorporation onto the surface of a commercial SiO2 in order to produce powdered catalysts, which are strongly desired in terms of heterogeneous catalysis due to their relatively easy separation, purification, and reuse. In addition, the uniform incorporation of NPs over solid supports have also been demonstrated as a promising strategy to prevent the particle agglomeration during the catalytic processes.28 Fig. 2D–F show SEM images for the produced catalysts after the NPs incorporation onto the SiO2 support. Interestingly, for all nanomaterials, an excellent dispersion of metal constituents was observed over the entire SiO2 surface and no significant agglomeration and changes on the morphologies were detected, indicating the robustness of our employed wet impregnation method to produce solid metal/SiO2 catalysts. Here, the metal loading corresponded to 1 wt% (Fig. 2F). The solid catalysts were denoted AgSiO2 (Fig. 2D), AuSiO2 (Fig. 2E), and AgAuSiO2.
Fig. 3 shows the UV-vis extinction spectra for the obtained Ag and Au nanospheres and AgAu nanoshells. The Ag and Au nanospheres displayed clear extinction peaks centered at ≈ 410 and 520 nm assigned to the dipolar mode of their surface plasmon resonance excitation29,30 On the other hand, a red-shift and broadening of the SPR in the AgAu nanoshells was observed compared to the Ag nanospheres employed as templates. This behavior can be associated with the Au deposition as well as the formation of the hollow interiors in during the galvanic reaction.25
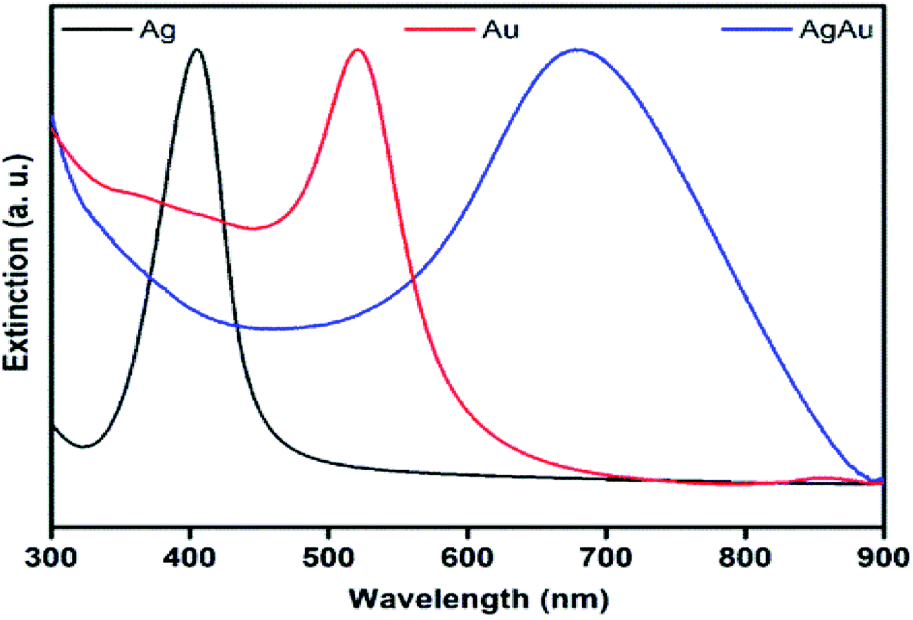 | ||
| Fig. 3 UV-vis extinction spectra recorded from aqueous suspensions containing Ag nanospheres (black trace), Au nanospheres (red trace), and AgAu nanoshells (blue trace). | ||
The formation of Ag, Au, and AgAu was also monitored by XRD analyses (Fig. 4), in which the XRD patterns also confirmed the formation of Ag (Fig. 4A), Au (Fig. 4B) and AgAu (Fig. 4C) nanostructures without the presence of any significant crystalline impurities. However, as both Ag and Au has very similar lattice constants, the XRD diffraction peaks for these two species could not be resolved under our experimental conditions.
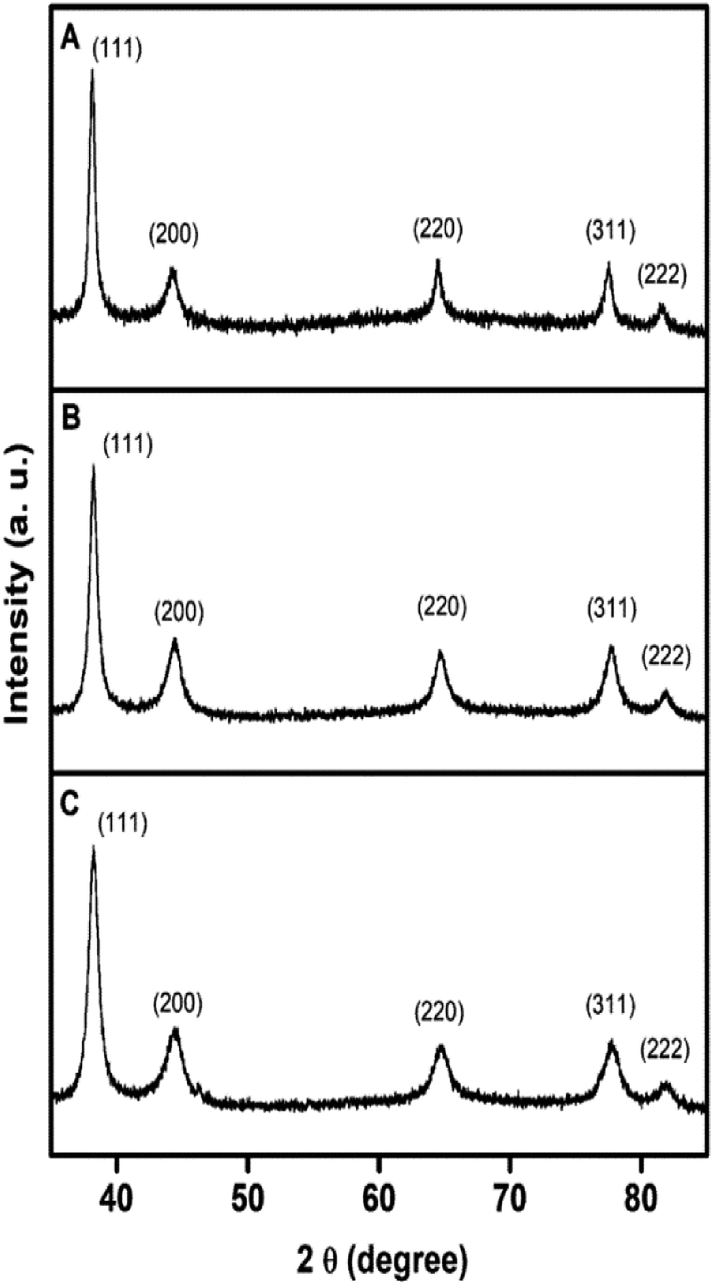 | ||
| Fig. 4 XRD profiles of (A) Ag nanospheres, (B) Au nanospheres, and (C) AgAu nanoshells. | ||
Characterization of the MTZ-derived NPs coated material
Regarding the MTZ-derived NPs, to the best of our knowledge, this represents the first report using MTZ on NPs supported on SiO2. There are other studies that report other thiol-substituted imidazole for coating NPs, evidencing that the thiol group interacts with the NPs, enabling freely available imidazole moieties. For example, in a previous study, authors report a dipeptide-functionalized thiol with terminal histidine and carboxylate groups that was used to passivate Au NPs. The material was successfully prepared and used as catalysts in acyl transfer reactions, although the NPs nature was not the focus of study.27 In another report, N-methylimidazole derivative functionalized Au NPs were obtained similarly via thiol anchoring on NPs surface, which afterward was employed in the cleavage of 2,4-dinitrophenyl acetate.20 It is noteworthy, that we also attempted to anchor MTZ on NPs (Ag, Au, and AgAu) not supported on SiO2, but functionalization was followed by aggregation of the particles.MTZ is a known drug (commercially Tapazol) used for the treatment of hyperthyroidism.35 MTZ has a tautomeric equilibrium of thiol–thione as shown in Scheme 3. The concentration of each species depends on the pH, although in general, the thione species predominates. For example, at pH 9, the percentage of thione is 65%.36 Another important feature of MTZ is its pKa of 11.38.35
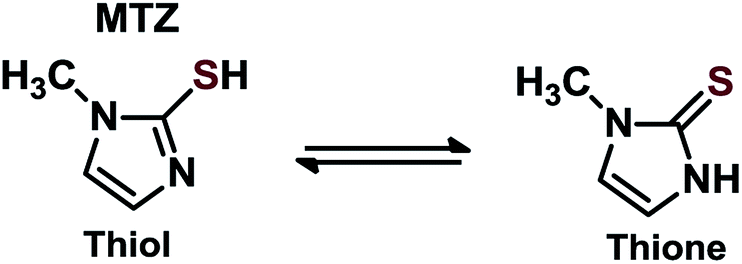 | ||
| Scheme 3 Tautomeric equilibrium for MTZ. | ||
The MTZ-derived NPs were characterized by Raman spectroscopy. Indeed, Raman analysis of MTZ is quite difficult due to its tautomeric and ionic equilibrium, but overall results evidence that the NPs were successfully functionalized. Raman spectra are presented in Fig. 5 including spectrum of pure MTZ for comparison and additionally all spectra all plotted together in Fig. S1.† The Raman spectra for the NPs prior to the MTZ functionalization (red trace on Fig. 5) show bands that were characteristic of PVP (in the case of AgSiO2 and AgAuSiO2), citrate (in the case of AuSiO2) and SiO2 (spectra for pure SiO2 and PVP are shown in the ESI†).37,38 These species are present at the surface of the NPs as they were employed as stabilizers during the synthesis of each respective material. It is important to note that all the nanoparticles were washed by successive rounds of centrifugation and removal of the supernatant after their synthesis and isolation, material. Although this process removes the reducing agents, unreacted precursors, and the excess of stabilizers that are employed during the synthesis, stabilizer molecules remain at the surface of the NPs. Their detection in the Raman experiments is due to the surface-enhanced Raman scattering (SERS) effect as enabled by Ag, Au, and AgAu NPs.
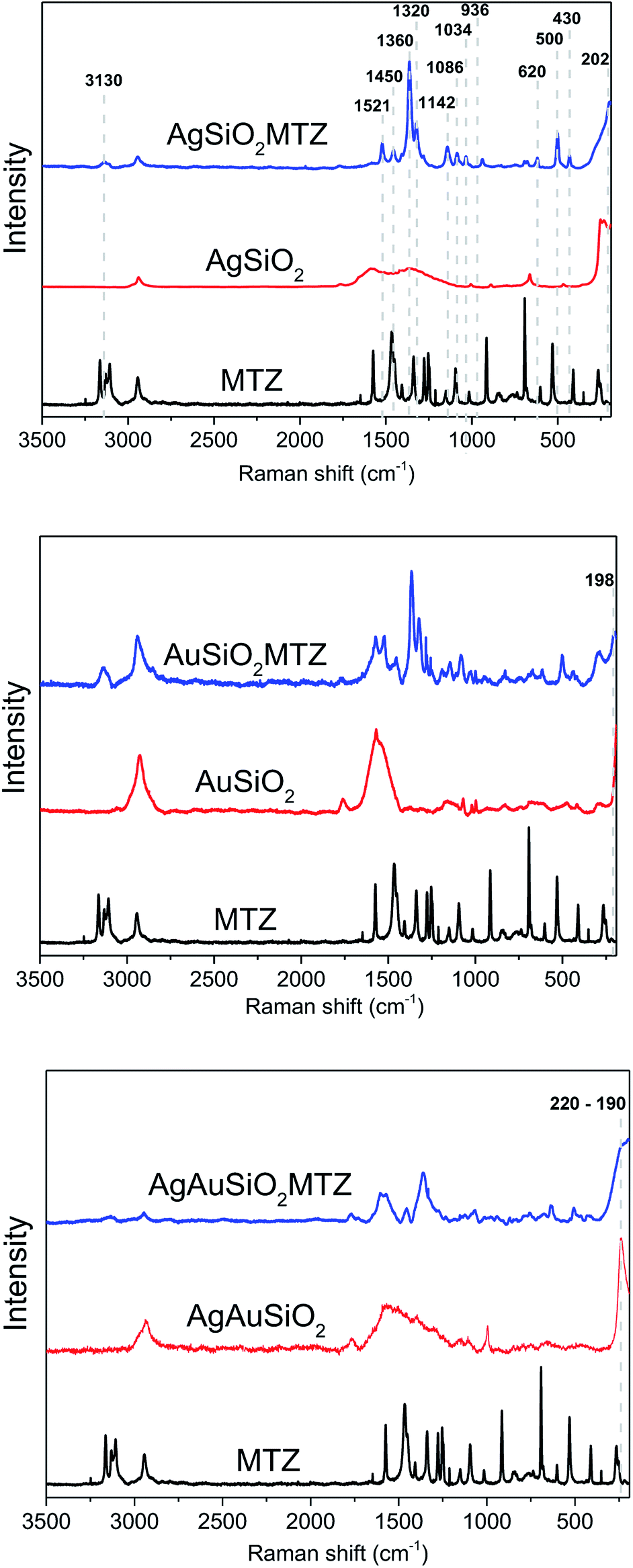 | ||
| Fig. 5 Ordinary Raman and SERS spectra (l = 632.8 nm) for pure MTZ, bare NPs, and MTZ-functionalized Ag, Au and AgAu NPs. | ||
Upon functionalization (AgAuSiO2–MTZ, AgSiO2–MTZ and AuSiO2–MTZ in Fig. 5) it is possible identify some characteristic bands of MTZ that are identified in the first spectrum:17,35,36 (i) at 3130 cm−1 due to ring C–H stretching; (ii) at 1521 cm−1 due to C–C and C–N stretching, along with ring C–H bending; (iii) at 1450 cm−1 attributed to C–S and ring C–N stretching, in addition to ring C–H bending; (iv) at 1360 cm−1 (most intense band) referent to C–N ring stretching with contribution of C–H bending; (v) at 1320 cm−1 attributed to ring C–N stretching, ring bending and ring CH–NH bending; (vi) 1142 cm−1 referent to C–S and ring C–N stretching, along with ring C–H bending; (vii) at 1086 cm−1 due to ring C–N stretching and ring C–H bending; (viii) at 1034 cm−1 attributed to ring bending, ring C–H bending, methylene C–H bending (ix) at 936 cm−1 referent to ring bending, ring C–N stretching and ring CH–NH bending; (x) at 620 cm−1 due to out-of-plane ring and CH–NH bending; (xi) at 500 and 430 cm−1 due to out-of-plane ring bending. It is noticeable that spectrum for pure MTZ presents more bands than discussed above and in addition some bands of MTZ are shifted when comparing coated materials to pure MTZ. Actually, these detected differences for the SERS spectra from the functionalized materials and ordinary Raman from pure MTZ are characteristic of many molecules and indicate that the functionalization was successful as previously observed.35,36,39 Moreover, the bands observed herein indicate that MTZ is present as thione and thiol/thiolate,35,36 although it should be noted that despite the high pKa for MTZ, previous studies show that the thiolate species is preferable for interacting with Ag and Au NPs.35
Regarding the nature of the interaction between NPs and MTZ, previous studies report that MTZ interacts with Au NPs preferably via the thiol group.35 On the other hand, with Ag NPs, there are some reports that claim a preferable interaction via the nitrogen atom of the imidazole group,36 although another study evidences that Ag NPs can also interact with thiol group35 or with both groups concomitantly.39 In fact, when analyzing Raman spectrum for AuSiO2–MTZ, there is a band at 198 cm−1, not present in the non-coated material. Despite the typical bands of SiO2 in this region, they don't seem to overlap. This new band has been attributed in the literature to Au–S stretching, indicating that for this material, MTZ interacts preferably via the thiolate group.35 In the case of AgSiO2–MTZ, the band at 202 cm−1, inexistent without MTZ, can be attributed to Ag–N stretching,36 indicating that for this material MTZ interacts preferably via the imidazole ring through the lone pair of the nitrogen atom. Finally, for AgAuSiO2–MTZ, the spectrum is noisier, with a broad band in the region of 220–190 cm−1 that could be attributed to both Ag–S and Au–N interactions.
It should be noted that despite the similarities in the bands attributed to Au–S and Ag–N, the other possible interactions Au–N and Ag–S are known to occur at higher wavenumbers (Au–N ∼225 cm−1 and Ag–S ∼240 cm−1).40,41 Taken together, Raman analysis confirms that MTZ was anchored on the NPs and that for AuSiO2–MTZ the interaction seems to occur through the imidazole ring (Ag–N). For AgSiO2–MTZ, this occurs via the thiolate group (Au–S). For AgAuSiO2–MTZ, there could be a combination of these interactions. In addition, the following catalytic studies agree with the interaction proposed since due to the lower catalytic activity of AgSiO2–MTZ.
FTIR analyses were also carried out (shown in ESI†) but didn't show any evident differences, probably due to the low degree of functionalization and also to the known low intensity of the bands of imidazole and thiol moieties. The MTZ molecule is only detected by Raman spectroscopy as a result of the SERS effect. Due to the low amount of metal (∼1% in mass) and given that the MTZ is anchored solely on the nanoparticle, the overall amount of MTZ is extremely low to detect or differentiate by FTIR.
Catalytic activity on the detoxification of OP
The catalytic activity of the MTZ-derived materials was evaluated in the cleavage reactions of OP, thus its detoxification. Firstly, as a proof-of-concept, we choose one class of material, derived from Au, to react with the model OP (i.e. simulant) DEDNPP. This is a common strategy before applying with real life OP due to their higher toxicity. In addition, DEDNPP is more reactive than Paraoxon, hence its reaction can be followed on a shorter time scale. Upon confirmation of the catalytic activity with DEDNPP, the nanocatalysts are then evaluated in the reaction with Paraoxon. Fig. 6 presents the rate constants at different pH values for the cleavage of DEDNPP with AuSiO2-derived catalysts. Constants solely with SiO2 under the same conditions is also shown along with the reaction in water (spontaneous).42,43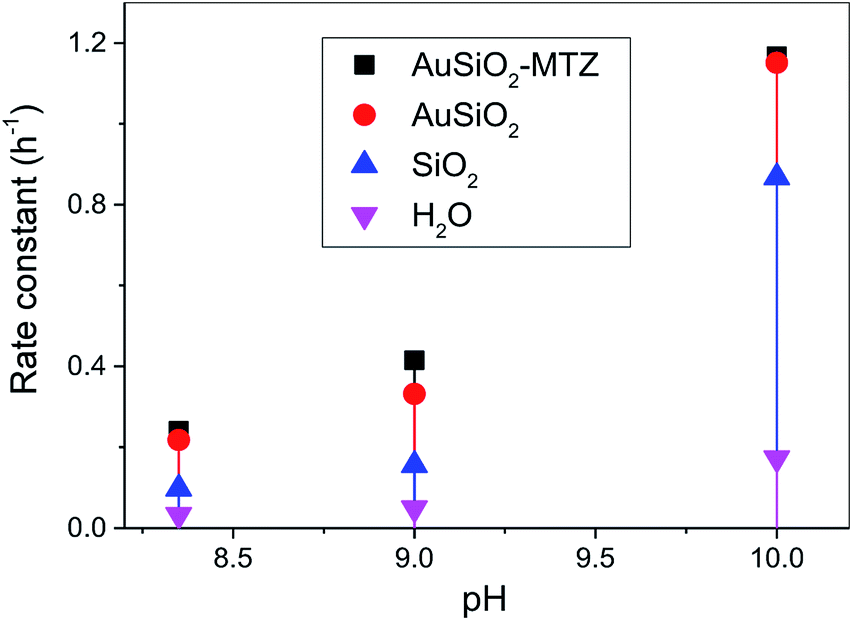 | ||
| Fig. 6 pH effect rate constant for the reaction of Au-derived NPs with DEDNPP at 20 °C. Reaction with SiO2 and in water is shown for comparison.42 | ||
Firstly, it should be noted that reactions with solely MTZ is not shown, since its comparison is not straightforward because of its homogenous reaction nature (herein is heterogeneous) and to the difficulty of estimating the amount of MTZ coating the NPs. Therefore we cannot obtain the rate constant of pure MTZ in the same concentration coating the NPs, hence would lead to erroneous underestimated comparison. In addition, we could not evaluate the reactions of bare NPs since they aggregate. Results show that AuSiO2–MTZ is, in fact, effective in cleaving DEDNPP, specially at pH 9.0, since it differentiates from the non-coated material (AuSiO2). At lower pH, the MTZ-derived material shouldn't exhibit any activity due to the lower reactivity of imidazole moiety.42 On the other hand, at pH 10, the reaction is very effective in all cases due to the contribution of the alkaline hydrolysis (reaction in water). Reactions solely with SiO2 evidenced for pH 8 and 9, similar constants to the spontaneous reaction, although at pH 10, its higher activity can be attributed to hydroxide species on the surface.44 Insofar, the pH of interest to carry out further studies with the pesticide was 9.0, since the MTZ-derived material showed higher reactivity (although discrete) than their non-functionalized analogous.
Considering the positive results with DEDNPP, the nanocatalysts were then evaluated in the reactions with the highly toxic pesticide Paraoxon at pH 9. Table 1 presents the rate constants obtained given as a function of the mass of metal for the reactions of Paraoxon with all the materials: coated and non-coated with MTZ. In general, catalytic rate constants are given as a function of the reactive group of the catalysts, although in this case, it was not possible to calculate the concentration of MTZ in the materials obtained, low values are expected. One approach is to obtain the ratio of the rate constants and the total amount of catalysts, but since in the present study it was impregnated with SiO2, we found more convenient to consider the amount of metal that is responsible for interacting with the MTZ molecules. Nevertheless, not all NPs surface may be covered by MTZ, hence the rate constant considering solely the amount of MTZ reacting should be even higher. Overall, Table 1 evidences high rate constants for Paraoxon were among the highest reported in the literature. For example, previous studies with thiol-based nanocatalysts derived from graphene oxide show rate constants of 2.13 g−1 h−1 (considering the amount of the reactive group thiolate) with Paraoxon.10 In another study, Ag, and cobalt (Co) NPs were coated with imidazole derived fatty acids that give rate constants for Co-derived material ∼1.3 g−1 h−1 (considering the amount of metal).16 A magnetic nanocatalyst of iron oxide with supported amino acids (Asp–His) was also used to cleave Paraoxon with a rate constant of 0.30 g−1 h−1 (considering the amount of Fe2O3–Asp–His).18
| Rate constant, g−1 h−1 | |
|---|---|
| a pH 9.0, 20 °C. Rate constants are given as a function of the mass of metal in the nanocatalysts. Reactions solely with SiO2, where similar to the analogous spontaneous reaction at pH 9.0. | |
| AgAuSiO2 | 4.10 |
| AgSiO2 | 4.03 |
| AuSiO2 | 4.58 |
| AgAuSiO2–MTZ | 13.10 |
| AgSiO2–MTZ | 3.76 |
| AuSiO2–MTZ | 9.52 |
While only discrete enhancements were observed for the MTZ-derived materials towards DEDNPP in comparison to its non-functionalized material, the difference was substantially different with Paraoxon (for Au- and AgAu-based materials). This is not unprecedented since Paraoxon is much less reactive than DEDNPP. In fact, as mentioned, DEDNPP is a simulant of OP such as Paraoxon and is used to verify possible catalytic effects, since the response is fast.
A more detailed analysis of Table 1 shows that upon functionalization of AuSiO2 with MTZ, the rate constant nearly doubles. In the case of AgAuSiO2, the analogous MTZ nanocatalyst increases more significantly the rate constant. In contrast, with AgSiO2, functionalization slightly decreases the rate constant. This behavior agrees with previous Raman analyses that indicate that for Ag-derived materials, MTZ interacts via the nitrogen while Au-based materials prefer the sulfur atom. Hence, the effect of MTZ on Au- and AgAu-based nanocatalysts is different from that of Ag-based materials. This can be explained by the expected mechanism for reactions of imidazole with OP, shown in Fig. 7. Based on previous mechanistic studies,45 the nitrogen of the imidazole ring attacks the phosphorus atom, thus cleaving the OP and leading to a phosphorylated intermediate and the phenolic product –OR. In the following, the unstable intermediate is hydrolyzed giving inorganic phosphate and regenerating the MTZ-derived nanocatalyst. This mechanism has been broadly accepted for imidazole mediated reactions and occurs analogously in many enzymatic active sites, therefore the nanocatalysts can be considered as artificial enzymes. Further, the proposed mechanism is consistent with the recycling of the nanocatalyst. Indeed, the UV-vis spectra taken after the reactions don't show any lixiviation of MTZ or the NPs, since the only band that appears in solution is of the products.
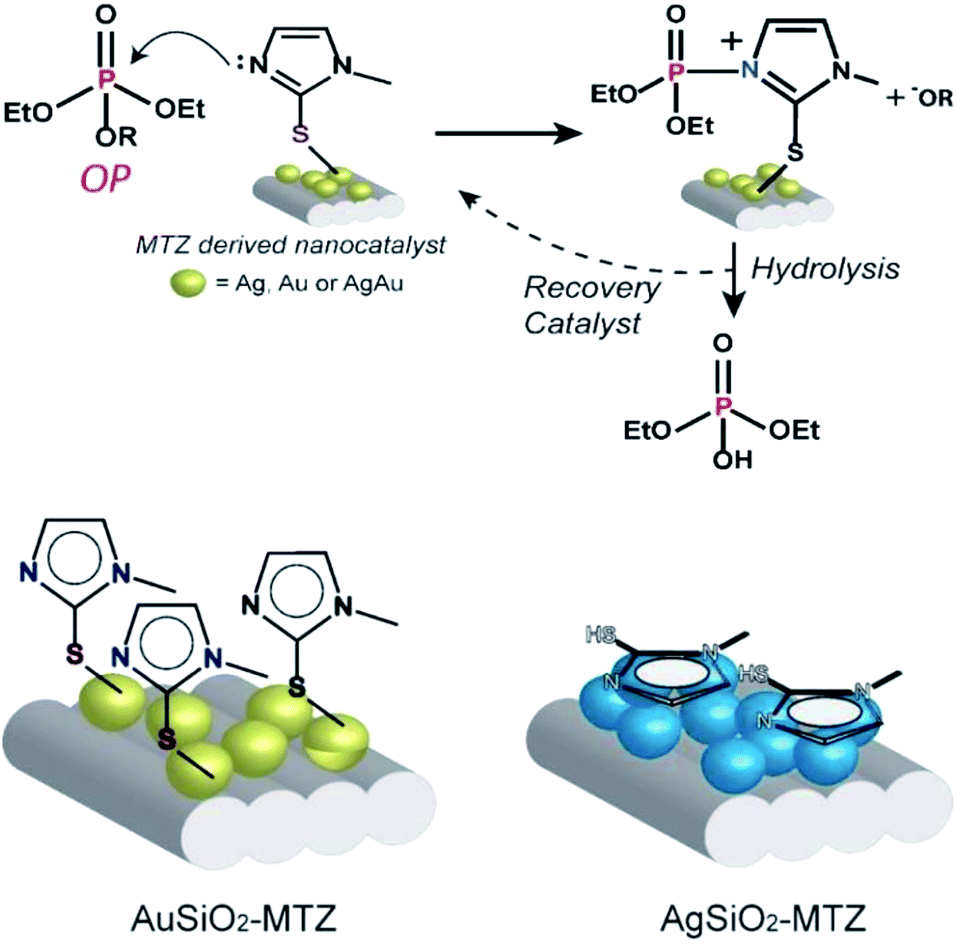 | ||
| Fig. 7 The mechanism proposed for the reaction of the OP with the nanocatalysts and interaction of MTZ with Au and Ag-derived materials. | ||
Correlating the mechanism with the proposed interaction for AuSiO2–MTZ (or AgAuSiO2–MTZ) and AgSiO2–MTZ (vide Raman), we can observe that, while Au–S guarantees freely available imidazole groups, Ag–N, doesn't (shown in Fig. 7). Therefore, AgSiO2–MTZ doesn't exhibit significant catalytic activity in contrast to AuSiO2 due to the type of interaction that directly disables the mechanistic preference.
Regarding the catalytic activity for the NPs without MTZ, this can be attributed to the Lewis acidity of the metal which interacts favorably with the phosphoryl oxygen, leading to a more electrophilic phosphorus center, as observed for ester cleavage (phosphate or acyl).15,16
Fig. 8 presents the rate enhancements of the nanocatalysts with Paraoxon when compared to its spontaneous reactions (kH2O = 1.20 × 10−7 h−1 or 4.0 × 10−8 g−1 h−1),45 evidencing that the materials proposed are highly reactive towards OP detoxification. Interestingly, AgAuSiO2–MTZ is the most efficient nanocatalyst. This could be related to another previous study that concluded that bimetallic AgAu NPs (without SiO2) are better catalysts toward 4-nitrophenol reduction than Au or Ag NPs. This effect was attributed to a possible higher surface area, resulting from the formation of porous/hollow walls.25
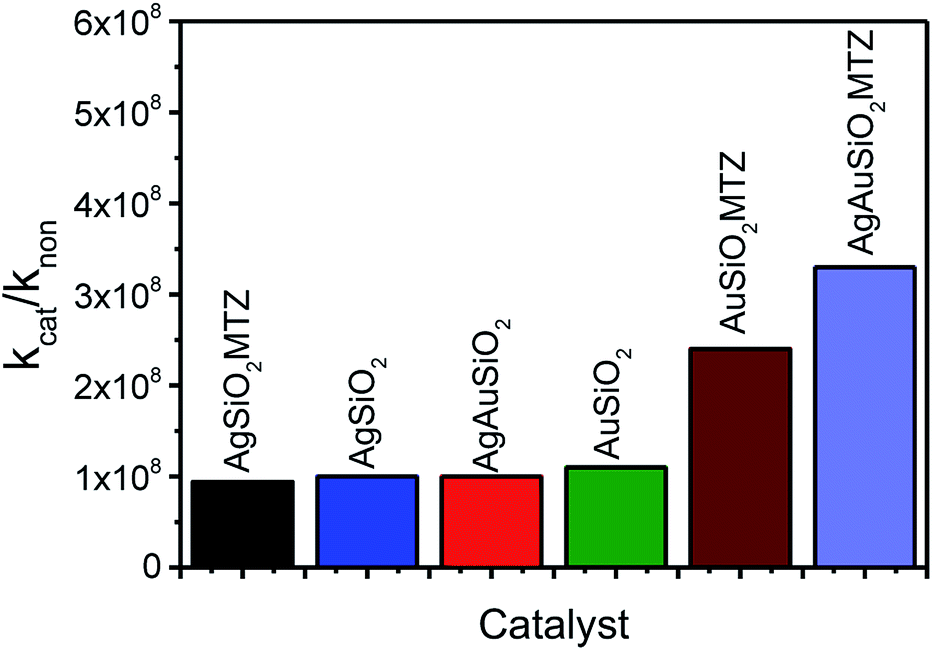 | ||
| Fig. 8 Rate enhancements of the nanocatalysts with Paraoxon (kcat in g−1 h−1) compared to the reaction with water (kH2O in g−1 h−1).45 | ||
Conclusions
Herein, we functionalized Ag, Au and bimetallic AgAu NPs (supported on SiO2) with MTZ, in order to obtain nanocatalysts comprising freely available imidazole groups. Results evidence that the NPs were successfully coated and that MTZ interacts preferably via sulfur atom over Au NPs and via the imidazole nitrogen over Ag NPs. The nanocatalysts were highly effective towards OP cleavage. For example, Fig. 9 illustrates the efficiency of detoxification of the toxic OP Paraoxon with the nanocatalysts, that is given as the time in days taken to degrade 50% of the OP. Normally in water, the lifetime of Paraoxon is extremely high and the estimated time to degrade 50% is over 600 years.45 In the presence of the nanocatalysts, this time is reduced to 7 days with the most reactive AgAuSiO2–MTZ.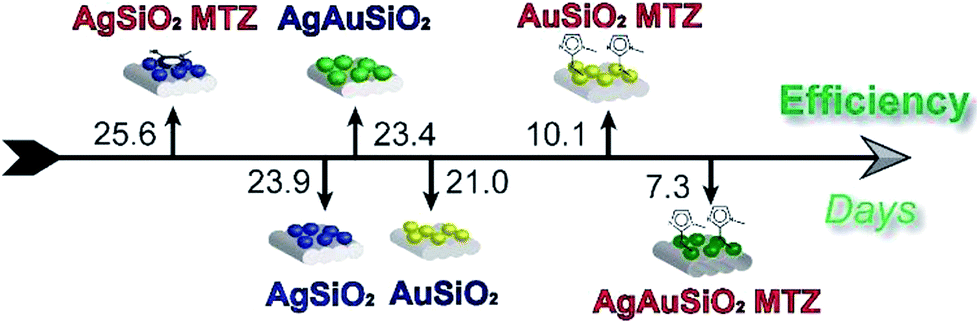 | ||
| Fig. 9 Efficiency for the detoxification of Paraoxon by the nanocatalysts, given as the time in days required to degrade 50%. | ||
The higher activity of Au-derived nanocatalysts was attributed to the imidazole group that is free to react with the OP, since in this case Au–S interactions is preferable. When Ag–N interactions takes place, the reactivity of imidazole is reduced.
Overall, we were able to detoxify OP efficiently which is promising destroying toxic stocks and also to design monitoring devices for these compounds. These perspectives are aligned with the importance of the issue as evidenced by the Peace Nobel Prize in 2013 awarded by the Organization for the Prohibition of Chemical Weapons “for its extensive efforts to eliminate chemical weapons”.46
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge the financial support from UFPR, CNPq, CAPES, Fundação Araucária, L'Oréal-UNESCO-ABC National, Institute of Science and Technology of Carbon Nanomaterials (INCT-Nanocarbon).References
- S. C. L. Kamerlin, P. K. Sharma, R. B. Prasad and A. Warshel, Q. Rev. Biophys., 2013, 46, 1–132 CrossRef CAS PubMed.
- F. H. Westheimer, Science, 1987, 235, 1173–1178 CAS.
- A. J. Kirby, B. S. Souza and F. Nome, Can. J. Chem., 2015, 93, 422–427 CrossRef CAS.
- S. C. Kamerlin, P. K. Sharma, R. B. Prasad and A. Warshel, Q. Rev. Biophys., 2013, 46, 1 CrossRef CAS PubMed.
- K. Kim, O. G. Tsay, D. A. Atwood and D. G. Churchill, Chem. Rev., 2011, 111, 5345–5403 CrossRef CAS PubMed.
- Y. C. Yang, J. A. Baker and J. R. Ward, Chem. Rev., 1992, 92, 1729–1743 CrossRef CAS.
- R. T. Delfino, T. S. Ribeiro and J. D. Figueroa-Villar, J. Braz. Chem. Soc., 2009, 20, 407–428 CrossRef CAS.
- A. M. Manfredi, W. Demos, E. H. Wanderlind, B. V. Silva, A. C. Pinto, B. S. Souza and F. Nome, J. Phys. Org. Chem., 2016, 29, 600–603 CrossRef CAS.
- E. S. Orth, J. E. S. Fonsaca, T. G. Almeida, S. H. Domingues, J. G. L. Ferreira and A. J. G. Zarbin, Chem. Commun., 2014, 50, 9891–9894 RSC.
- J. Fonsaca, L. Hostert, E. S. Orth and A. J. Zarbin, J. Mater. Chem. A, 2017, 5, 9591–9603 CAS.
- P. V. Attwood, M. J. Piggott, X. L. Zu and P. G. Besant, Amino Acids, 2007, 32, 145–156 CrossRef CAS PubMed.
- K. Hofmann, Imidazole and its derivatives, Interscience Publishers, New York, 1953 Search PubMed.
- J. G. Ferreira and E. S. Orth, J. Braz. Chem. Soc., 2017, 28, 1760–1767 Search PubMed.
- P. Xu, S. Guo, H. Yu and X. Li, Small, 2014, 10, 2404–2412 CrossRef CAS PubMed.
- M. H. Kuchma, C. B. Komanski, J. Colon, A. Teblum, A. E. Masunov, B. Alvarado, S. Babu, S. Seal, J. Summy and C. H. Baker, Nanomedicine, 2010, 6, 738–744 CrossRef CAS PubMed.
- L. Bromberg, L. Chen, E. P. Chang, S. Wang and T. A. Hatton, Chem. Mater., 2010, 22, 5383–5391 CrossRef CAS.
- L. Bromberg and T. A. Hatton, Ind. Eng. Chem. Res., 2007, 46, 3296–3303 CrossRef CAS.
- Y. Zheng, C. Duanmu and Y. Gao, Org. Lett., 2006, 8, 3215–3217 CrossRef CAS PubMed.
- L. Y. Kuo, A. K. Bentley, Y. A. Shari'ati and C. P. Smith, Organometallics, 2012, 31, 5294–5301 CrossRef CAS.
- L. Pasquato, F. Rancan, P. Scrimin, F. Mancin and C. Frigeri, Chem. Commun., 2000, 2253–2254 RSC.
- L. Rassaei, F. Marken, M. Sillanpää, M. Amiri, C. M. Cirtiu and M. Sillanpää, TrAC, Trends Anal. Chem., 2011, 30, 1704–1715 CrossRef CAS.
- J. Zeng, Q. Zhang, J. Chen and Y. Xia, Nano Lett., 2009, 10, 30–35 CrossRef PubMed.
- C. Jin, J. Han, F. Chu, X. Wang and R. Guo, Langmuir, 2017, 33, 4520–4527 CrossRef CAS PubMed.
- T. S. Rodrigues, A. G. da Silva, A. B. de Moura, I. G. Freitas and P. H. Camargo, RSC Adv., 2016, 6, 62286–62290 RSC.
- M. V. Petri, R. A. Ando and P. H. Camargo, Chem. Phys. Lett., 2012, 531, 188–192 CrossRef CAS.
- C. M. Kisukuri, D. J. Palmeira, T. S. Rodrigues, P. H. Camargo and L. H. Andrade, ChemCatChem, 2016, 8, 171–179 CrossRef CAS.
- P. Pengo, S. Polizzi, L. Pasquato and P. Scrimin, J. Am. Chem. Soc., 2005, 127, 1616–1617 CrossRef CAS PubMed.
- T. S. Rodrigues, A. H. da Silva, A. G. da Silva, D. G. Ceara, J. F. Gomes, J. M. Assaf and P. H. Camargo, Catal. Sci. Technol., 2016, 6, 2162–2170 CAS.
- T. S. Rodrigues, A. G. da Silva, A. Macedo, B. W. Farini, R. d. S. Alves and P. H. Camargo, J. Mater. Sci., 2015, 50, 5620–5629 CrossRef CAS.
- A. G. da Silva, T. S. Rodrigues, A. Macedo, R. T. da Silva and P. H. Camargo, Quim. Nova, 2014, 37, 1716–1720 CAS.
- G. N. Bowers, R. B. McComb, R. Christensen and R. Schaffer, Clin. Chem., 1980, 26, 724–729 CAS.
- L. J. Daumann, L. Marty, G. Schenk and L. R. Gahan, Dalton Trans., 2013, 42, 9574–9584 RSC.
- K. TekaiaáElhsissen, J. Mater. Chem., 1996, 6, 573–577 RSC.
- Y. Sun, B. Mayers and Y. Xia, Adv. Mater., 2003, 15, 641–646 CrossRef CAS.
- M. Muniz-Miranda, F. Muniz-Miranda and A. Pedone, Phys. Chem. Chem. Phys., 2016, 18, 5974–5980 RSC.
- N. Biswas, S. Thomas, A. Sarkar, T. Mukherjee and S. Kapoor, J. Phys. Chem. C, 2009, 113, 7091–7100 CAS.
- D. De Faria, H. Gil and A. De Queiroz, J. Mol. Struct., 1999, 478, 93–98 CrossRef CAS.
- J. García-Aguilar, D. Cazorla-Amorós and Á. Berenguer-Murcia, Appl. Catal., A, 2017, 538, 139–147 CrossRef.
- T. A. Saleh, M. M. Al-Shalalfeh and A. A. Al-Saadi, Sci. Rep., 2016, 6, 32185 CrossRef CAS PubMed.
- B. Pergolese, M. Muniz-Miranda and A. Bigotto, J. Phys. Chem. B, 2004, 108, 5698–5702 CrossRef CAS.
- P. Galinetto, A. Taglietti, L. Pasotti, P. Pallavicini, G. Dacarro, E. Giulotto and M. Grandi, J. Appl. Spectrosc., 2016, 82, 1052–1059 CrossRef CAS.
- E. S. Orth, E. H. Wanderlind, M. Medeiros, P. S. M. Oliveira, B. G. Vaz, M. N. Eberlin, A. J. Kirby and F. Nome, J. Org. Chem., 2011, 76, 8003–8008 CrossRef CAS PubMed.
- E. H. Wanderlind, Cinética e mecanismo de hidrólise do triéster dietil 2,4-dinitrofenil fosfato, Master thesis, Federal University of Santa Catarina, 2014.
- A. Torrents and A. T. Stone, Environ. Sci. Technol., 1991, 25, 143–149 CrossRef CAS.
- E. S. Orth, T. G. Almeida, V. B. Silva, A. R. M. Oliveira, F. M. M. Ocampos and A. Barison, J. Mol. Catal. A: Chem., 2015, 403, 93–98 CrossRef CAS.
- https://www.nobelprize.org/nobel_prizes/peace/laureates/2013/opcw-facts.html, accessed in May, 5, 2017.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07059d |
| This journal is © The Royal Society of Chemistry 2017 |