DOI:
10.1039/C7RA07017A
(Paper)
RSC Adv., 2017,
7, 39899-39905
Molecular design of organic small molecules based on diindole–diimide with fused aromatic heterocycles as donors for organic solar cells†
Received
24th June 2017
, Accepted 9th August 2017
First published on 15th August 2017
Abstract
A series of diindole–diimide based small molecules with different aromatic end groups have been designed and investigated theoretically as donor materials for organic solar cells (OSCs). Their optical, electronic, and charge transport properties have been investigated by density functional theory (DFT) and time-dependent density functional theory (TD-DFT). The calculation results showed that the designed molecules can lower the band gap and extend the absorption spectrum towards longer wavelengths. The optical, electronic, and charge transport properties of the designed molecules are affected by the introduction of different aromatic end groups. We have also predicted the mobility of molecules with benzo[d]thiazole and benzo[c]thiophene end groups. Their hole mobility values are 0.157 and 6.57 × 10−2 cm2 V−1 s−1, respectively. Our results reveal that the designed molecules are promising hole transport materials and donors with intense broad absorption spectra for OSCs.
Introduction
Organic solar cells (OSCs) have been regarded as potential energy resources due to their promising features, including lightweight, mechanical flexibility, low manufacturing cost, and so on.1 Unfortunately, the low power conversion efficiencies (PCEs) of OSCs remain the primary obstacle to their commercial application. To address the above issue, great efforts have been intensively devoted to improving the PCEs of OSCs.2 PCEs of bulk heterojunction (BHJ) polymer OSCs close to 10% have been reported.3 In recent years, as another branch of OSCs, OSCs based on small molecules have also received much attention.4 The PCE of small-molecule OSCs has recently reached 8%.5 Although the PCEs of small-molecule OSCs are still lower than those of their polymer counterparts, small molecules possess prominent advantages over their polymer counterparts, such as well-defined structures, higher charge carrier mobility, easier purification and synthesis, less batch to batch variation, and intrinsic monodispersity.6 These advantages make small molecules are more suited to mass production than polymer-based ones. Especially, compared with polymer molecule, the structure of small molecule can be tuned more easily, which is beneficial for effective sunlight absorption.7 However, the overall performance of OSCs based on the small molecules still lagged behind that of their polymer counterparts. Therefore, a perspective means how to improve the PCEs of small-molecule OSCs is to design and synthesize high-performance and desirable donor novel small molecules. In order to achieve high-performance of small molecules based OSCs, a low band gap (Eg) and a low highest occupied molecular orbital (HOMO) energy level are the two most important factors. In addition, the material should possess high carrier mobility. Nowadays, push–pull structures comprising electron donors and acceptors are regarded as one of the most promising materials in OSCs research.8 This push–pull structure can enhance intramolecular charge transfer (ICT) and lower the Eg of material, which extend the absorption spectrum towards longer wavelengths.9 At the same time, the HOMO and the lowest unoccupied molecular orbital (LUMO) energy and Eg levels can be tuned effectively by adjusting the push and pull units.10 Among the various push–pull organic compounds, the indole-containing molecules have been proved to be a promising building block for small-molecule OSCs owing to their strong light-capturing, excellent charge carrier mobility, and good chemical and environmental stability properties.11 Furthermore, the introduction of the electron-deficient aromatic imide group into this electron-rich π-system may lower the energy level of the π-conjugated semiconductors.12 Recently, indole-containing small-molecules for OSCs have been reported, they showed good charge transport, film morphology, and optical properties.13
Herein, we investigated the relationship between topologic structures and optical as well as charge transporting properties for diindole–diimide based small molecules. Furthermore, with the aim to extend the UV-vis in NIR region, the fused aromatic heterocycles as the electron donor were introduced to the π system of molecules. A series of novel diindole–diimide-based small molecules have been designed (Scheme 1). The optical and electronic properties of designed molecules have been investigated. We also select two compounds of the designed compounds as representation to predict their mobility.
 |
| | Scheme 1 Molecular structures of the designed compounds. | |
Computational methods
Ground neutral and charged state geometries of the designed compounds were optimized by using density functional theory (DFT) at the B3LYP/6-31G(d,p) level.14 The frontier molecular orbitals (FMOs) of the designed compounds were then calculated at this level of theory based on the optimized geometries. The HOMOs and LUMOs contributions of the individual fragments of the designed compounds to the respective FMO were also calculated. Additionally, the absorption spectra of the designed compounds were predicted using the time dependant density functional theory (TD-DFT) at the TD-B3LYP/6-31G(d,p) level of theory based on the optimized geometries.15 The molecule 1 was taken as an example to testify to the validity of the selected approach. The geometry optimizations of molecule 1 were carried out by the DFT method using the 6-31G(d,p) basis set. The absorptions were predicted using TD-DFT with the 6-31G(d,p) basis set based on the optimized geometries. The various functionals for all DFT and TD-DFT computations include B3LYP, PBE0, CAM-B3LYP, wB97XD, M062X, and LCWPBE. The longest λabs of molecule 1 are listed in ESI Table SI.† The results displayed in Table SI† showed that the TD-B3LYP/6-31G(d,p) method provided a better agreement with the reported experimental observations13 than those obtained with other methods, with the deviation being only 2 nm. In addition, our designed molecules are used as OSCs materials in the solid film. Therefore, the solvent effect for investigated system is negligible in this work. Thus, geometry optimizations, band gaps Eg, and absorption properties of designed molecules were carried out by the B3LYP/6-31G(d,p) method.
Generally, the Marcus theory has been applied to estimate the charge transfer rate:16,17
| |
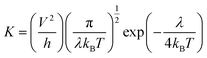 | (1) |
where
T and
kB are the temperature and the Boltzmann constant, correspondingly,
λ and
V the reorganization energy and transfer integral, respectively. Clearly, the two key parameters
λ and
V determine the charge transfer rate. For the
λ, it is composed of two parts: external reorganization energy (
λext) and internal reorganization energy. The
λext is the effect of the surrounding media in bulk materials, while the
λint is the measure of relaxation in molecular geometry.
18 In this work, we focus only on the
λint and neglect the
λext because the
λext values are much smaller than their
λint counterparts in pure organic condensed phases.
19 The
λint can be expressed as:
| | |
λ = λ1 + λ2 = (E±0 − E±±) + (E0± − E00)
| (2) |
where
E00 and
E±0 represent the energy of the neutral and cation/anion species with the optimized structure of the neutral species, respectively, while
E0± and
E±± are the energies of the neutral and cation/anion species with the optimized cation/anion structure, respectively. The
λint for electron (
λe) and hole (
λh) were calculated at the B3LYP/6-31G(d,p) level.
20
The V can be calculated by:21,22
| |
Vij = 〈ϕ01|![[F with combining circumflex]](https://www.rsc.org/images/entities/i_char_0046_0302.gif) 0|ϕ02〉 0|ϕ02〉
| (3) |
ϕ01 and
ϕ02 mean the HOMOs or LUMOs of the two monomers in the dimer.
0 is the Kohn–Sham Hamiltonian of the dimer. The molecular orbitals and density matrix of the two individual monomers are applied to calculate the
0. In this work, the
V were calculated at the pw91pw91/6-31G(d) level.
23 The module polymorph of software package Materials Studio
24 was employed to predicte the molecular crystal structure. The polymorph predictor calculations are restricted to the ten most popular space groups,
P2
1/
c,
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif)
,
P2
12
12
1,
C2/
c,
P2
1,
Pbca,
Pna2
1,
Cc,
Pbcn, and
C2. The compass force field was used for the prediction. The geometry of the cluster models was taken from B3LYP/6-31G(d,p) level. van der Waals and Coulomb interactions were evaluated by using the Ewald summation method with a cutoff of 6 Å, and the Ewald accuracy tolerance was set to 0.0001 kcal mol
−1.
The carrier mobility μ can be evaluated by Einstein equation:
| |
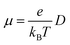 | (4) |
The diffusion constant D can be obtained by equation:25
| |
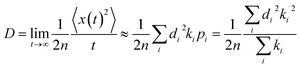 | (5) |
here
d is the intermolecular center-to-center distance, while
k is the hopping rate due to charge transfer to neighbour.
n = 3 means the spatial dimension of the crystal
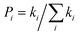
represents the probability of the specific hopping route. This means that it is a three-dimension averaged diffusion process. All the calculations are carried out using the Gaussian 09 program.
26
Results and discussion
Frontier molecular orbital
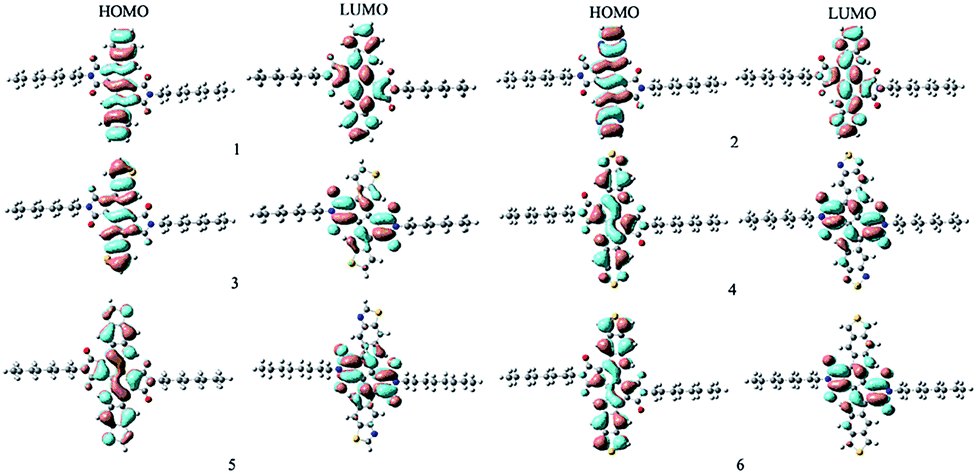
With the aim to characterize the optical, electronic, and charge transport properties, the distribution of electron density in HOMOs and LUMOs for the designed compounds are plotted in Fig. 1. Table 1 presents the EHOMO, ELUMO, Eg and the HOMOs and LUMOs contributions of the 3,8-dihydroindolo[7,6-g]indole-diimide fragments (IID) and aromatic end groups (Ar) fragments (in %) to the FMO. As visualized in Fig. 1, both the HOMOs and LUMOs of the designed compounds exhibited π characteristics. Furthermore, the distribution patterns of HOMOs and LUMOs delocalized across the whole molecular backbone because of the planar and rigid conjugated molecular geometry. The results displayed in Table 1 reveal that the HOMOs of the designed compounds are mainly localized on the IID and Ar fragments. On the other hand, the LUMOs mainly reside at the IID fragments with minimal density on Ar fragments. The contributions of IID fragments of LUMOs are larger than 78.8%, while the contributions of Ar fragments are within 8% except the corresponding contribution of Ar fragment is 21.2% for 5. As expect, the designed compounds exhibited obvious the ICT character of the vertical S0 → S1 transition due to the planar and rigid molecular structure. Analysis of the FMOs indicates that the HOMOs → LUMOs excitations results in transfer of electron density from the Ar fragments to IID fragments for the designed compounds. The percentages of charge transfer from Ar to IID fragments for 1–6 are 23, 7.3, 60.8, 35.5, 26.4, and 56.6%, respectively. It suggests that the Ar fragments serve as donors and IID fragments serve as acceptors for the designed compounds.
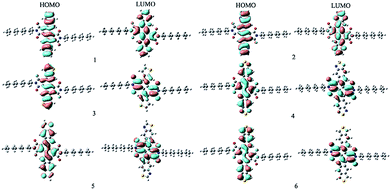 |
| | Fig. 1 The FMOs of the designed compounds at the B3LYP/6-31G(d,p) level. | |
Table 1 The FMOs energy and band gaps Eg, and FMOs molecular orbital contribution (%) from individual fragments (in %) to the FMOs of the designed compounds at the B3LYP/6-31G(d,p) level
| Species |
HOMO |
LUMO |
Eg |
| EHOMO |
Ara |
IIDb |
ELUMO |
Ar |
IID |
| Ar: aromatic groups. IID: 3,8-dihydroindolo[7,6-g]indole-diimide fragments. |
| 1 |
−5.494 |
27.8 |
72.2 |
−2.764 |
4.8 |
95.2 |
2.730 |
| 2 |
−6.106 |
12.1 |
87.9 |
−2.973 |
4.8 |
95.2 |
3.133 |
| 3 |
−5.520 |
65.3 |
34.7 |
−2.808 |
4.5 |
95.5 |
2.712 |
| 4 |
−5.571 |
42.7 |
57.3 |
−3.098 |
7.2 |
92.8 |
2.473 |
| 5 |
−5.516 |
47.6 |
52.4 |
−2.900 |
21.2 |
78.8 |
2.616 |
| 6 |
−4.811 |
62.4 |
37.6 |
−2.724 |
5.8 |
94.2 |
2.087 |
To gain a deeper insight into the influence of the optical and electronic properties, the EHOMO, ELUMO, and Eg were analyzed. From Table 1, one can find that both the EHOMO and EHOMO values of 2–5 decrease compared with those of parent compound 1, respectively. Interestingly, the EHOMO and EHOMO values of 6 increases compared with those of parent compound 1, respectively. The trend of the EHOMO is 6 > 1 > 5 > 3 > 4 > 2 while ELUMO is 6 > 1 > 3 > 5 > 2 > 4. On the other hand, the Eg is in the order of 2 > 1 > 3 > 5 > 4 > 6. These results indicate that molecule with benzo[c]thiophene (6) end group possesses lower both EHOMO and ELUMO, while molecules with pyrazine (2), thiophene (3), benzo[c][1,2,5]thiadiazole (4), benzo[d]thiazole (5) end groups have higher EHOMO and ELUMO compared with molecule with benzene end group. The Eg value of molecule with pyrazine end group are larger, while the corresponding values of molecules with thiophene, benzo[c][1,2,5]thiadiazole, benzo[d]thiazole, benzo[c]thiophene end groups are smaller than that of molecules with benzene end group. These results suggest that the different end groups have effects on the EHOMO, ELUMO, and Eg for the compounds under investigation.
Absorption spectra
Table 2 presents the longest wavelength of absorption λabs, the oscillator strength f, main configurations, and the absorption region R of the designed molecules. R corresponds to the difference of the longest and shortest wavelength values with oscillator strength larger than 0.01 considering the first fifteen excited states. The corresponding λabs and f of the first fifteen excited states for the designed compounds are listed in Table SII in ESI.† We also present the wavelength with the largest f of absorption in Table 2. From the data in Table 2 and Table SII in ESI,† one can find that the 1 has three main absorption peaks at about 542 (f = 0.127), 339 (f = 0.321), and 318 (f = 0.283) nm. These results are in good agreement with the experimental results namely, absorption peaks at about 540, 340, and 320 nm, which are all observed in experiments.13 Furthermore, the designed molecules have two main absorption peaks at about 350 and 500–700 nm. We aim to obtain a broad and intense absorption region which can match the sunlight spectrum by the introduction of aromatic end groups. Therefore, we mainly focus on the UV-vis regions absorption of the designed molecules. The major assignments of the lowest S0 → S1 electronic transitions for 1–6 are mainly as HOMOs → LUMOs. It can be noted that the λabs of 3–6 exhibit bathochromic shifts, while the corresponding λabs of 2 has hypsochromic shift compared with that of the parent compound 1. The λabs values are in the order of 6 > 4 > 5 > 3 > 1 > 2, which is in excellent agreement with the corresponding reverse order of Eg values displayed in Table 1. It indicates that the introduction of pyrazine (2) end group decrease, while the introduction of thiophene (3), benzo[c][1,2,5]thiadiazole (4), benzo[d]thiazole (5), benzo[c]thiophene (6) end groups increase the λabs values compared with parent molecules with benzene end group. The sequence of R is 6 > 3 > 5 > 4 > 1 > 2. It suggests that the introduction of pyrazine end group end results in smaller R value, while the introduction of thiophene, benzo[c][1,2,5]thiadiazole, benzo[d]thiazole, benzo[c]thiophene end groups lead to the increase of R values compared with parent molecules with benzene end group. In general, larger oscillator strength means larger experimental absorption coefficient. The order of the predicted f is in the decreasing order of 6 > 5 > 4 > 1 > 3 > 2. It indicates that the introduction of benzo[c][1,2,5]thiadiazole, benzo[d]thiazole, benzo[c]thiophene end groups increases, while the introduction of pyrazine and thiophene end groups decreases slightly the f values compared with benzene end group for the designed molecules. Furthermore, from Table 2 one can find that the λabs and R values of molecules with benzo[c][1,2,5]thiadiazole, benzo[d]thiazole, benzo[c]thiophene end groups are larger than those of other molecules. It suggests that molecules with benzo[c][1,2,5]thiadiazole, benzo[d]thiazole, benzo[c]thiophene end groups can lower the material band gap and extend the absorption spectrum towards longer wavelengths. The λabs and R values of molecules with pyrazine and thiophene end groups are smaller slightly than those of parent molecule 1. Therefore, the designed compounds possess the large λabs, f, and R values. The designed molecules could be used as donors with intense broad absorption spectra for solar cell material, particularly for 4–6.
Table 2 Predicted λabs, corresponding f and R, and main configurations of 1–6 at the TD-B3LYP/6-31G(d,p)//B3LYP/6-31G(d,p) level
| Species |
λabs |
F |
Main assignment |
R |
| Experimental data for 1 from ref. 13. Data in the parentheses are the wavelength with the largest f values. Data in the parentheses are the f values. |
| 1 |
542 (339)b |
0.127 (0.321)c |
H → L (0.70) |
236 |
| 2 |
473 (371) |
0.084 (0.210) |
H → L (0.69) |
140 |
| 3 |
559 (308) |
0.088 (0.550) |
H → L (0.70) |
259 |
| 4 |
595 (353) |
0.175 (1.265) |
H → L (0.70) |
242 |
| 5 |
565 (334) |
0.188 (1.001) |
H → L (0.70) |
244 |
| 6 |
708 (346) |
0.195 (1.251) |
H → L (0.70) |
362 |
| Expa |
540 (340) |
|
|
|
Reorganization energy
In order to design good candidates for OSCs devices, one key factor is to understand the relationship between molecular structure and charge transport property of material. As mentioned above, the reorganization energy is one major parameter determining the charge transfer rate.16,17 The calculated reorganization energies for hole and electron are listed in Table 3. From Table 3, one can find that the λe of 1–6 are larger than that of the typical electron transport material (λe = 0.276 eV), tris(8-hydroxyquinolinato)aluminum(III) (Alq3).27 On the contrary, the λh values of 1–6 are smaller than that of the typical hole transport material (λh = 0.290 eV), N,N′-diphenyl-N,N′-bis(3-methlphenyl)-(1,1′-biphenyl)-4,4′-diamine (TPD).28 This indicates that the hole transfer rates of 1–6 might be higher than that of TPD. Furthermore, λh values of 2 and 3 are larger, while the corresponding values of 4–6 are smaller than that of 1. It suggests that the introduction of pyrazine and thiophene end groups decrease, while the introduction of benzo[c][1,2,5]thiadiazole, benzo[d]thiazole, benzo[c]thiophene end groups increase the and hole transfer rates compared with parent molecule 1. It implies that the designed molecules can be used as promising hole transport materials from the stand point of the smaller reorganization energy.
Table 3 Calculated λe and λh (both in eV) of 1–6 at the B3LYP/6-31G(d,p) level
| Species |
λh |
λe |
| 1 |
0.173 |
0.329 |
| 2 |
0.261 |
0.355 |
| 3 |
0.266 |
0.340 |
| 4 |
0.104 |
0.280 |
| 5 |
0.129 |
0.322 |
| 6 |
0.068 |
0.288 |
Charge-carrier mobility
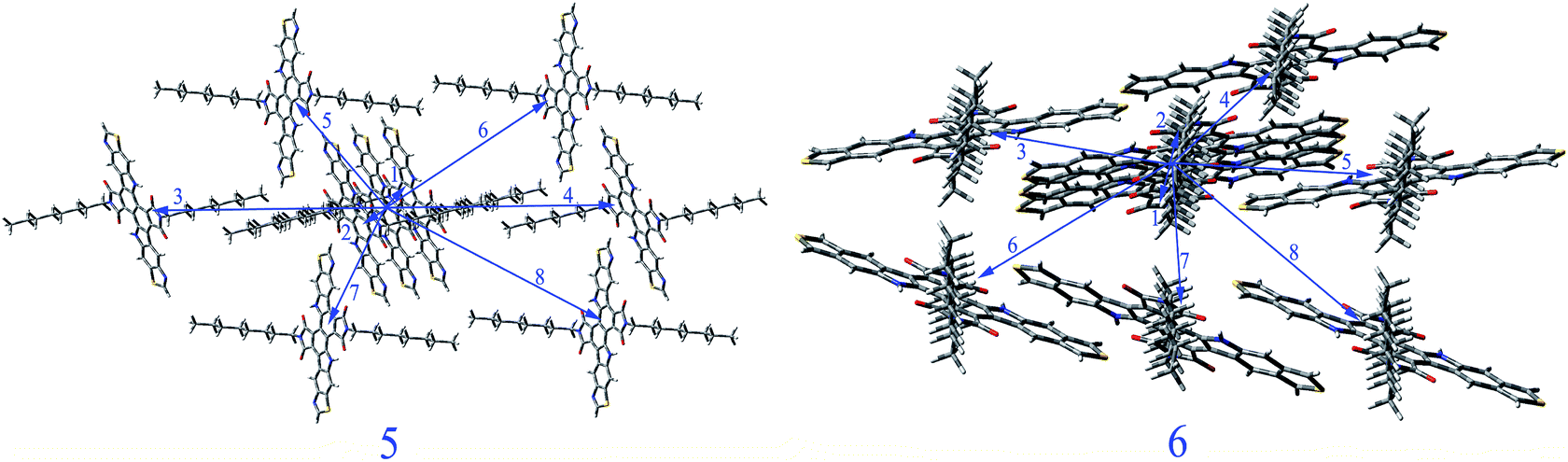
We select 5 and 6 as representation to study their charge transport property. The total energies and lattice constants of the predicted crystal structures for 5 and 6 in different space groups are summarized in Tables SIII and SIV in ESI,† respectively. The crystal structures of 5 and 6 with the lowest total energies belong to space groups P21 and Pbcn respectively and shown in Fig. SI in ESI.† Thus, we predict the mobility of 5 and 6 in these two space groups, respectively. The main carrier hoping pathways of 5 and 6 are selected based on their optimized crystal structures, respectively. The selected pathways (dimers) of 5 and 6 in space groups P21 and Pbcn respectively are depicted in Fig. 2. The calculated transfer integrals and mobility of 5 and 6 for holes and electrons in space groups P21 and Pbcn are listed in Table 4. The hole or electron coupling means the orbital coupling of the neighboring molecules. The FMO distributions and relative position of the interacting molecules are two major parameters that determine the electronic coupling.29 From Table 4, it can be noted that 5 possesses the largest absolute electron and hole coupling values in pathways 1 and 2 in space group P21. For 6, the largest absolute electron and hole coupling are located in pathways 4 and 7 in space group Pbcn. The orientation of the interacting molecules is the key factor of hole or electron coupling for 5 and 6. The values of hole mobility of 5 and 6 for P21 and Pbcn space groups (0.157 and 6.57 × 10−3 cm2 V−1 s−1) are larger than that of TPD (1.0 × 10−3 cm2 V−1 s−1),30 respectively. The values of electron mobility of 5 and 6 for P21 and Pbcn space groups are 1.15 × 10−2 and 2.46 × 10−4 cm2 V−1 s−1, respectively. Furthermore, the values of both hole and electron mobility of 5 are larger than those of 6, which is in agreement with the corresponding reverse order of reorganization energies for hole and electron values displayed in Table 3, respectively. It indicates shows that the stacking structure is the most important factor for molecular mobility property.
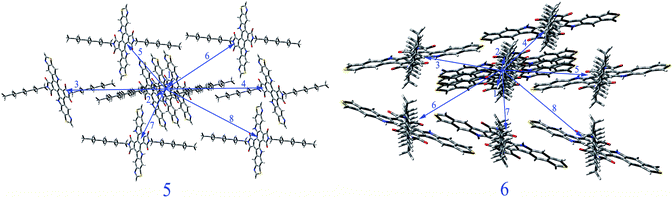 |
| | Fig. 2 Crystal structures and hopping routes of 5 in P21 space group and 6 in Pbcn space group. | |
Table 4 The hole and electron coupling (in eV) in all of the nearest neighbor pathways and the distance (in Å) for 5 and 6 in P21 and Pbcn space groups respectively
| 5 |
Pathway |
Distance |
Electron coupling |
Hole coupling |
| P21 |
1 |
4.809 |
−7.70 × 10−3 |
−2.80 × 10−2 |
| 2 |
4.809 |
−7.70 × 10−3 |
−2.80 × 10−2 |
| 3 |
31.560 |
6.30 × 10−12 |
−9.14 × 10−10 |
| 4 |
31.560 |
6.30 × 10−12 |
−9.14 × 10−10 |
| 5 |
14.189 |
−3.20 × 10−5 |
−3.85 × 10−5 |
| 6 |
27.553 |
3.10 × 10−14 |
1.23 × 10−12 |
| 7 |
14.189 |
−3.20 × 10−5 |
−3.85 × 10−5 |
| 8 |
27.553 |
3.10 × 10−14 |
1.23 × 10−12 |
| Drift mobility |
|
|
1.15 × 10−3 |
0.157 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| 6 |
| Pbcn |
1 |
30.038 |
1.47 × 10−9 |
−3.67 × 10−8 |
| 2 |
30.038 |
1.47 × 10−9 |
−3.67 × 10−8 |
| 3 |
18.369 |
−1.21 × 10−7 |
2.37 × 10−6 |
| 4 |
7.972 |
1.90 × 10−3 |
−1.80 × 10−3 |
| 5 |
18.369 |
−1.21 × 10−7 |
2.37 × 10−6 |
| 6 |
20.643 |
3.46 × 10−10 |
1.75 × 10−10 |
| 7 |
7.883 |
−8.16 × 10−4 |
−7.20 × 10−3 |
| 8 |
20.643 |
3.46 × 10−10 |
1.75 × 10−10 |
| Drift mobility |
|
|
2.46 × 10−4 |
6.57 × 10−2 |
Considering the reorganization energy and charge-carrier mobility, the designed molecules can be made as hole transfer materials high hole mobility. It is well established that the mobility of charge carriers play a dominant role in the efficiency of OSCs. The high hole mobility is a basic requirement for effective photovoltaic active donors, to ensure effective charge carrier transport to the electrode and reduce the photocurrent loss in OSCs.31 Additionally, the high hole mobility for the donors as a hole transport layer is also crucial for the carrier transport to enhancement in both short-circuit current density (Jsc) and fill factor (FF) of the devices, and consequently the overall PCE of OSCs.32 The calculation results suggest that the designed molecules can be made as donors material with intense broad absorption spectra and hole transfer materials using for OSCs.
Conclusions
In summary, a series of diindole–diimide-based small molecules with different aromatic end groups have been systematically investigated for OSCs applications. Analysis of the FMOs indicates that the vertical S0 → S1 transition results in transfer of electron density from the Ar fragments to IID fragments for the designed compounds. The introduction of different aromatic end groups affects the optical, electronic, and charge transport properties of the designed compounds. The molecules can lower the material band gap and extend the absorption spectrum towards longer wavelengths. Our results show that the molecules under investigation own the longest wavelength of absorption spectra, oscillator strength, and absorption region values. In addition, the mobility values of 5 and 6 as representation are also investigated. The values of hole mobility of 5 and 6 are 0.157 and 6.57 × 10−2 cm2 V−1 s−1, respectively, which are larger than that of TPD (1.0 × 10−3 cm2 V−1 s−1). On the basis of the investigated results, the designed molecules are expected to be promising candidates for donors with intense broad absorption spectra and hole transport materials for OSCs.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial supports from the NSFC (No. 21563002), the Natural Science Foundation of Inner Mongolia Autonomous Region (No. 2015MS0201), and the Research Program of Sciences at Universities of Inner Mongolia Autonomous Region (No. NJZZ235) are gratefully acknowledged.
Notes and references
-
(a) R. F. Service, Science, 2011, 332, 293 CrossRef PubMed
 ;
(b) H. X. Shang, H. J. Fan, Y. Liu, W. P. Hu, Y. F. Li and X. W. Zhan, Adv. Mater., 2011, 23, 1554–1557 CrossRef CAS PubMed ;
(c) Y. Zou, A. Najari, P. Berrouard, S. Beaupré, R. B. Aich, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2010, 132, 5330–5331 CrossRef CAS PubMed ;
(d) Z. He, C. Zhong, X. Huang, W. Wong, H. Wu, L. Chen, S. Su and Y. Cao, Adv. Mater., 2011, 23, 4636–4643 CrossRef CAS PubMed ;
(e) Y. Lin, Y. F. Li and X. W. Zhan, Chem. Soc. Rev., 2012, 41, 4245–4272 RSC ;
(f) B. Walker, C. Kim and T. Q. Nguyen, Chem. Mater., 2011, 23, 470–482 CrossRef CAS .
;
(b) H. X. Shang, H. J. Fan, Y. Liu, W. P. Hu, Y. F. Li and X. W. Zhan, Adv. Mater., 2011, 23, 1554–1557 CrossRef CAS PubMed ;
(c) Y. Zou, A. Najari, P. Berrouard, S. Beaupré, R. B. Aich, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2010, 132, 5330–5331 CrossRef CAS PubMed ;
(d) Z. He, C. Zhong, X. Huang, W. Wong, H. Wu, L. Chen, S. Su and Y. Cao, Adv. Mater., 2011, 23, 4636–4643 CrossRef CAS PubMed ;
(e) Y. Lin, Y. F. Li and X. W. Zhan, Chem. Soc. Rev., 2012, 41, 4245–4272 RSC ;
(f) B. Walker, C. Kim and T. Q. Nguyen, Chem. Mater., 2011, 23, 470–482 CrossRef CAS . -
(a) Y. Liang and L. Yu, Polym. Rev., 2010, 50, 454–473 CrossRef CAS ;
(b) N. F. Montcada, B. Pelado, A. Viterisi, J. Albero, J. Coro, P. D. L. Cruz, F. Langa and E. Palomares, Org. Electron., 2013, 14, 2826–2832 CrossRef CAS .
-
(a) J. You, L. Dou, K. Yoshimura, T. Kato, K. Ohya, T. Moriarty, K. Emery, C. C. Chen, J. Gao, G. Li and Y. Yang, Nat. Commun., 2013, 4, 1446–1456 CrossRef PubMed ;
(b) G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS .
-
(a) B. Walker, C. Kim and T. Q. Nguyen, Chem. Mater., 2011, 23, 470–482 CrossRef CAS ;
(b) D. Demeter, T. Rousseau, P. Leriche, T. Cauchy, R. Po and J. Roncali, Adv. Funct. Mater., 2011, 21, 4379–4387 CrossRef CAS .
-
(a) A. K. K. Kyaw, D. H. Wang, V. Gupta, J. Zhang, S. Chand, G. C. Bazan and A. J. Heeger, Adv. Mater., 2013, 25, 2397–2402 CrossRef CAS PubMed ;
(b) A. K. K. Kyaw, D. H. Wang, V. Gupta, W. L. Leong, L. Ke, G. C. Bazan and A. J. Heeger, ACS Nano, 2013, 7, 4569–4577 CrossRef CAS PubMed ;
(c) J. Y. Zhou, Y. Zuo, X. J. Wan, G. K. Long, Q. Zhang, N. Wang, Y. S. Liu, Z. Li, G. R. He, C. X. Li, B. Kan, M. Li and Y. S. Chen, J. Am. Chem. Soc., 2013, 135, 8484–8575 CrossRef CAS PubMed .
-
(a) Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245–4272 RSC ;
(b) A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed ;
(c) Z. He, C. Zhong, X. Huang, W. Wong, H. Wu, L. Chen, S. Su and Y. Cao, Adv. Mater., 2011, 23, 4636–4643 CrossRef CAS PubMed .
- T. Zhang, M. Liu, Q. Zeng, Z. Wu, L. Piao and S. Zhao, RSC Adv., 2013, 3, 13259–13264 RSC .
-
(a) X. Li, W. C. H. Choy, L. Huo, F. Xie, W. E. I. Sha, B. Ding, X. Guo, Y. Li, J. Hou, J. You and Y. Yang, Adv. Mater., 2012, 24, 3046–3052 CrossRef CAS PubMed ;
(b) X. Hu, M. Shi, J. Chen, L. Zuo, L. Fu, Y. Liu and H. Chen, Macromol. Rapid Commun., 2011, 32, 506–511 CAS ;
(c) Y. Huan, X. Guo, F. Liu, L. Huo, Y. Chen, T. P. Russell, C. C. Han, Y. Li and J. Hou, Adv. Mater., 2012, 24, 3383–3389 CrossRef PubMed ;
(d) J. Roncali, Acc. Chem. Res., 2009, 42, 1719–1730 CrossRef CAS PubMed .
-
(a) J. Zhang, D. Den, C. He, M. Zhang, Z. Zhang, Z. Zhang and Y. Li, Chem. Mater., 2011, 23, 817–822 CrossRef CAS ;
(b) Y. Sun, G. C. Welch, W. L. Leong, C. J. Takacs, G. C. Bazan and A. J. Heeger, Nat. Mater., 2012, 11, 44–48 CrossRef CAS PubMed .
-
(a) B. Yin, L. Yang, Y. Liu, Y. Chen, Q. Qi, F. Zhang and S. Yin, Appl. Phys. Lett., 2010, 97, 023303 CrossRef ;
(b) Y. Liu, J. Zhou, X. Wan and Y. Chen, Tetrahedron, 2009, 65, 5209–5215 CrossRef CAS ;
(c) J. Zhang, D. Den, C. He, M. J. Zhang, Z. G. Zhang, Z. J. Zhang and Y. F. Li, Chem. Mater., 2010, 23, 817–822 CrossRef .
-
(a) P. F. Xia, X. J. Feng, J. Lu, S. W. Tsang, R. Movileanu, Y. Tao and M. S. Wong, Adv. Mater., 2008, 20, 4810–4815 CrossRef CAS ;
(b) J. Sim, K. Do, K. Song, A. Sharma, S. Biswas, G. D. Sharma and J. Ko, Org. Electron., 2016, 30, 122–130 CrossRef CAS ;
(c) D. Yang, Y. Jiao, Y. Huang, T. Zhuang, L. Yang, Z. Lu, X. Pu, H. Sasabe and J. Kido, Org. Electron., 2016, 32, 179–186 CrossRef CAS ;
(d) G. D. Sharma, M. A. Reddy, K. Ganesh, S. P. Singh and M. Chandrasekharam, RSC Adv., 2014, 4, 732–742 RSC ;
(e) F. Baert, C. Cabanetos, M. Allain, V. Silvestre, P. Leriche and P. Blanchard, Org. Lett., 2016, 18, 1582–1585 CrossRef CAS PubMed ;
(f) J. Sim, H. Lee, K. Song, S. Biswas, A. Sharma, G. D. Sharma and J. Ko, J. Mater. Chem. C, 2016, 4, 3508–3516 RSC ;
(g) B. Liu, Q. Liu, D. You, X. Li, N. Y. Aruta and W. Zhu, J. Mater. Chem., 2012, 22, 13348–13356 RSC .
-
(a) J. Gao, Y. Li and Z. Wang, Org. Lett., 2013, 15, 1366–1369 CrossRef CAS PubMed ;
(b) S. L. Suraru, U. Zschieschang, H. Klauk and F. Würthner, Chem. Commun., 2011, 47, 11504–11506 RSC ;
(c) Y. Dong and J. Xie, J. Mol. Sci., 2014, 30, 118–123 CAS .
- Z. Zhao, Z. Wang, X. Zhang, S. Gao, X. Yang, Z. Duan and X. Gao, ChemPlusChem, 2015, 80, 57–61 CrossRef CAS .
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS ;
(b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS ;
(c) B. J. Lynch, P. L. Fast, M. Harris and D. G. Truhlar, J. Phys. Chem. A, 2000, 104, 4811–4815 CrossRef CAS .
-
(a) C. R. Zhang, W. Z. Liang, H. S. Chen, Y. H. Chen, Z. Q. Wei and Y. Z. Wu, J. Mol. Struct., 2008, 862, 98–104 CrossRef CAS ;
(b) J. Sun, J. Song, Y. Zhao and W. Z. Liang, J. Chem. Phys., 2007, 127, 234107–234113 CrossRef PubMed .
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef CAS .
- R. A. Marcus, Annu. Rev. Phys. Chem., 1964, 15, 155–196 CrossRef CAS .
- V. Lemaur, M. Steel, D. Beljonne, J. L. Brédas and J. Cornil, J. Am. Chem. Soc., 2005, 127, 6077–6086 CrossRef CAS PubMed .
-
(a) D. L. Cheung and A. Troisi, J. Phys. Chem. C, 2010, 114, 20479–20488 CrossRef CAS ;
(b) D. P. McMahon and A. Trois, J. Phys. Chem. Lett., 2010, 1, 941–946 CrossRef CAS ;
(c) S. D. Motta, E. D. Donato, F. Negri, G. Orlandi, D. Fazzi and C. Castiglioni, J. Am. Chem. Soc., 2009, 131, 6591–6598 CrossRef PubMed .
- M. E. Köse, W. J. Mitchell, N. Kopidakis, C. H. Chang, S. E. Shaheen, K. Kim and G. Rumbles, J. Am. Chem. Soc., 2007, 129, 14257–14270 CrossRef PubMed .
- T. Fujita, H. Nakai and H. Nakatsuji, J. Chem. Phys., 1996, 104, 2410–2417 CrossRef CAS .
- A. Troisi and G. Orlandi, J. Phys. Chem. A, 2006, 110, 4065–4070 CrossRef CAS PubMed .
- B. Liu, X. Chen, Y. Zou, L. Xiao, X. Xu, Y. He, L. Li and Y. Li, Macromolecules, 2012, 45, 6898–6905 CrossRef CAS .
- Materials Studio, Accelrys Inc., San Diego, CA, 2005 Search PubMed .
- W. Q. Deng and W. A. Goddard, J. Phys. Chem. B, 2004, 108, 8614–8621 CrossRef CAS .
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery Jr, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian 09, Gaussian, Inc, Wallingford, CT, 2009 Search PubMed .
- B. C. Lin, C. P. Cheng, Z. Q. You and C. P. Hsu, J. Am. Chem. Soc., 2005, 127, 66–67 CrossRef CAS PubMed .
- N. E. Gruhn, D. A. da Silva Filho, T. G. Bill, M. Malagoli, V. Coropceanu, A. Kahn and J. L. Brédas, J. Am. Chem. Soc., 2002, 124, 7918–7919 CrossRef CAS PubMed .
- C. Wang, F. Wang, X. Yang, Q. Li and Z. Shuai, Org. Electron., 2008, 9, 635–640 CrossRef CAS .
- K. Shizu, T. Sato, A. Ito, K. Tanaka and H. Kaji, J. Mater. Chem., 2011, 21, 6375–6382 RSC .
- G. D. Sharma, M. Anil Reddy, K. Ganesh, S. P. Singhb and M. Chandrasekharam, RSC Adv., 2014, 4, 732–742 RSC .
- Y. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS PubMed .
Footnote |
| † Electronic supplementary information (ESI) available: Calculated the longest wavelength of absorption λmax and corresponding oscillator strength f of molecule 1 by various methods with 6-31G(d,p) basis set; the λabs and corresponding oscillator strength f and absorption region R of the first fifteen excited states for 1–6 obtained by the TD-B3LYP/6-31G(d,p) level. The calculated total energies and crystal cell parameters of 5 and 6 in different space groups. Herringbone structures of 5 in P21 space group and 6 in Pbcn space group. See DOI: 10.1039/c7ra07017a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab and
Ahmad Irfancd
*ab and
Ahmad Irfancd
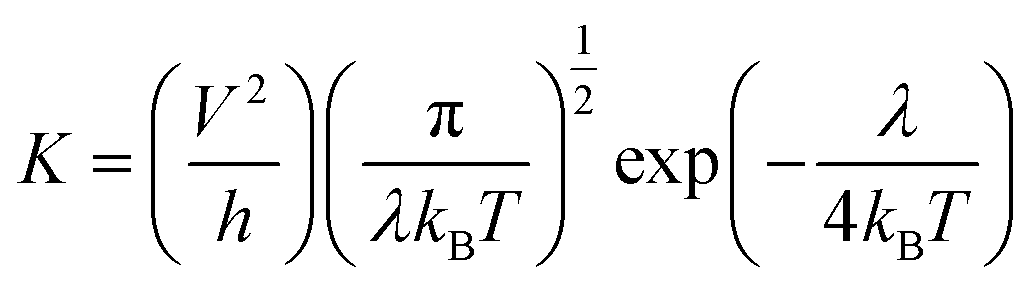
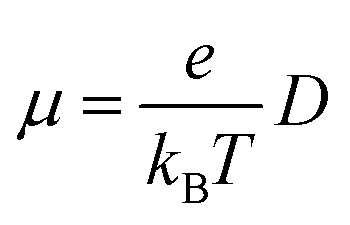
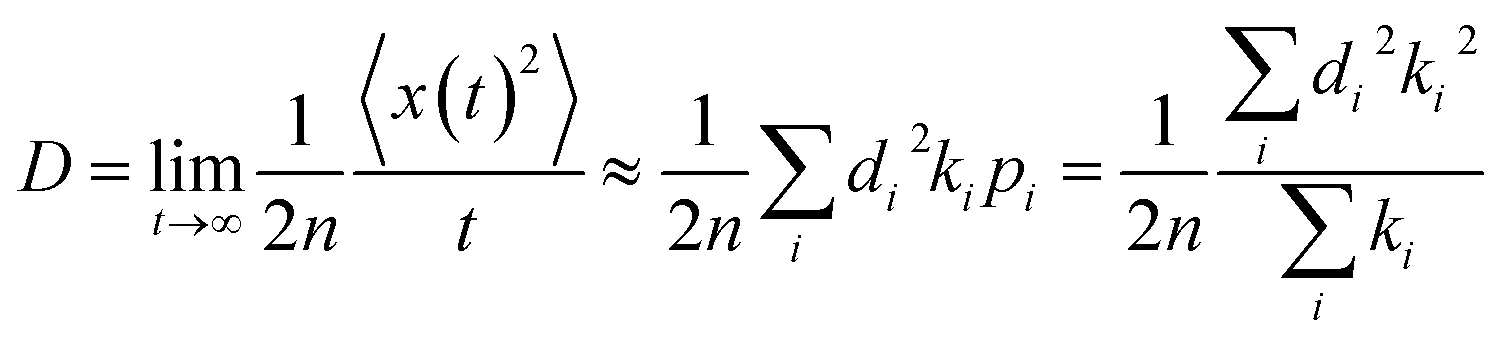
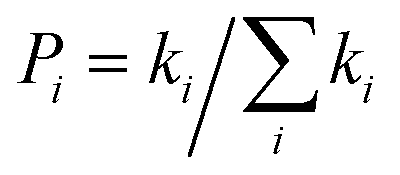 represents the probability of the specific hopping route. This means that it is a three-dimension averaged diffusion process. All the calculations are carried out using the Gaussian 09 program.26
represents the probability of the specific hopping route. This means that it is a three-dimension averaged diffusion process. All the calculations are carried out using the Gaussian 09 program.26