
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
SAPO-34 with a low acidity outer layer by epitaxial growth and its improved MTO performance
Chuiyan Kong,
Jiang Zhu,
Senyuan Liu and
Yao Wang *
*
Beijing Key Laboratory of Green Chemical Reaction Engineering and Technology, Department of Chemical Engineering, Tsinghua University, Beijing, 100084, China. E-mail: wang_yao@tsinghua.edu.cn
First published on 16th August 2017
Abstract
Although it is known that a low-silica SAPO-34 zeolite gives good MTO performance, the direct synthesis of a SAPO-34 zeolite with a low acid site density suffers from the problem of low yield and impure crystallinity. In this work, SAPO-34 zeolite samples with an appropriate low acid site density were synthesized as a low acid site outer layer on a high acid site core by epitaxial growth using a two-step hydrothermal synthesis. The samples were characterized by SEM, TEM, EDS-lining, XRD, ICP, XPS, TPD-NH3, TGA-Py and Ar adsorption. EDS-lining confirmed a silica distribution of a high-silica core and low-silica outer layer in the two-step synthesized SAPO-34 particles, and determined the thickness of low-silica outer layer to be 200 nm. The two-step synthesized samples have the CHA structure and gave good molecular sieve selectivity. The second-step growth used a low concentration of silica in the source to decrease the Si/Al ratio of the bulk solid phase, leading to the reduction of acid site density. Catalytic activity evaluation in a microreactor revealed that the two-step synthesized SAPO-34 zeolite gave both better MTO performance and hydrothermal stability compared to a zeolite produced by the one-step synthesis. The low acid site outer layer of the two-step synthesized samples had reduced coking and diffusion limitation, resulting in increased lifetime. This work provides a practical catalyst synthesis strategy of SAPO-34 catalysts for the MTO industry, by showing that a small high-silica core coated by a low-silica outer layer of appropriate thickness is a good structure for improved MTO performance.
1. Introduction
Ethene and propene are among the most important raw materials in the chemical industry. They are usually produced by the petroleum route. In view of the predicted petroleum shortage, the methanol-to-olefins (MTO) and methanol-to-propene (MTP) processes are expected to be important routes to produce light olefins from coal, natural gas and biomass instead of petroleum in the coming decades.1–3 The MTO reaction mechanism has been investigated for more than 30 years.2 The hydrocarbon pool mechanism proposed by Dahl and Kolboe4–6 is now been widely accepted. This has been refined by the dual cycle concept, which is comprised of the aromatic cycle and olefin cycle.7,8The MTO or MTP performance of a zeolite catalyst is determined by the zeolite framework topology, diffusivity, and acidity. On account of their unique porous structure and appropriate acid strength, the silicoaluminophosphate zeolite SAPO-34 (CHA) and aluminosilicate zeolite ZSM-5 (MFI) are the most successful catalysts for the MTO and MTP process.9–15 Compared to the medium-pore ZSM-5, the small-pore SAPO-34 can give much higher (>90%) light olefin selectivity and almost 100% methanol conversion, which are due to its cage structure and the small windows of the cages,16 while the medium-pore ZSM-5 has a higher selectivity to propene and some larger molecules. However, the small-pore SAPO-34 more rapidly loses activity because of more coke deposition than in the medium-pore ZSM-5.17–19 Conventional catalyst regeneration techniques, such as the burning of coke by oxidation in a regenerator, can be used to recover the activity. Since the SAPO-34 catalyst requires more frequent regeneration, a fluidized bed reactor and a fluidized bed regenerator are adopted to have the continuous reaction and regeneration of the SAPO-34 catalyst. Therefore, a SAPO-34 catalyst with good hydrothermal stability is required for stable performance in multiple reaction–regeneration cycles. Several SAPO-34 catalysts have been commercialized for the MTO process by UOP, SINOPEC, and Dalian Institute of Chemical Physics, and a modified ZSM-5 catalyst by Lurgi is used in their industrial MTP process.
In order to slow down the deactivation rate and prolong the MTO lifetime, many methods have been tried to reduce the diffusional resistance directly, such as decreasing the crystal size20–25 and synthesizing a hierarchical structure.13,26–35
In addition to the diffusional distance, the acid site density also significantly affects the SAPO-34 zeolite's MTO performance.38–40 It has been shown that shell deactivation usually occurs in the SAPO-34 catalyst during the MTO reaction and this prevents the internal acid sites from being fully utilized.41 Obviously, a high acid site density will lead to a more serious outer layer deactivation, resulting in the rapid deactivation and low products selectivity. Thus the low acid site density was achieved to improve the MTO performance by the synthesis of low-silica SAPO-34.19,38–40
However, it is worth noting that the direct synthesis of low-silica SAPO-34 may face the problem of the low yield and growth of impurity crystal. As silica is one of the SAPO-34 framework element, SAPO-34 crystals always grow slowly and gets low yield in the low-silica precursor solution. It has been proved that it is difficult to synthesize AlPO-34 zeolites containing no silica without help of the fluorine.42 The low-silica precursor solution tends to form SAPO-18 with the AEI structure.43–46 Even SAPO-5 may be formed in the low enough Si/Al ratio precursor solution. So it has to face the challenge for the one-step synthesis to lower the acid site density.
To get more SAPO-34 zeolite yield and avoid forming SAPO-18 and SAPO-5, adding the SAPO-34 crystal seeds into the precursor solution is an effective way.47 Norikazu et al.48–51 applied a two-step hydrothermal synthesis method to get a silicalite-1 outer layer to grow epitaxially on HZSM-5, and showed that this gave a high catalytic activity and excellent para-xylene selectivity, which was a result of removing the surface acid sites and protecting the HZSM-5 framework from damage at the same time. There have been few reports on epitaxial growth of SAPO-34 and its influence on the MTO performance. An epitaxial growth of SAPO-34 that modifies the Al, P and Si elements by the two-step hydrothermal synthesis is more complicated than the one of ZSM-5 zeolite only containing the Al and Si.
Meanwhile, if the high-silica SAPO-34 can grow epitaxially in the low-silica precursor solution, the SAPO-34 may form the unusual silica site distribution of a low-silica outer layer and high-silica core, which is different from the common SAPO-34 particles. It has been shown that the SAPO-34 zeolite synthesized by the common method have more silica on the outer surface than inside the crystal particle.47,52,53 The acid sites on the outer surface have no shape selectivity and can cause coking on the surface that blocks pores, resulting in shortening the lifetime and lowering product selectivity. Therefore, it is necessary to optimize the acid site distribution and decrease the surface acid density to achieve better MTO catalytic performance. Liu et al.52 showed that it was helpful for improving the catalyst lifetime and ethene selectivity to decrease the external acid site density with an oxalic acid treatment. Yang et al.47 developed a post-synthesis milling and recrystallization method to prepare small SAPO-34 crystals, and they also showed that reducing the Si enrichment on the external surface of SAPO-34 enhanced its catalytic performance in the MTO reaction.
In this work, the epitaxial growth on the SAPO-34 particles by a two-step hydrothermal method was studied. And the effect of epitaxial growth on the silica distribution in the particles was carefully confirmed. Meanwhile, the influence on the catalytic performance and hydrothermal stability caused by the epitaxial growth of SAPO-34 was discussed. From the results, a two-step synthesis strategy to improve the MTO catalytic performance for SAPO-34 was proposed.
2. Experimental
2.1 Synthesis of the materials
SAPO-34 zeolite samples were synthesized by the traditional hydrothermal method using the method of Hendrik et al.54 Aluminium iso-propoxide (Al(OPri)3), phosphoric acid (H3PO4, 85 wt%), colloidal silica (SiO2, 20 wt%) and tetraethylammonium hydroxide solution (TEAOH, 25 wt%) were used as the aluminum source, phosphorus source, silicon source and structure directing agent (SDA).The two-step hydrothermal synthesized process of the samples is shown in Scheme 1. The precursor solution of the first-step hydrothermal synthesis was prepared with the following molar compositions: 1.0Al2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4.0P2O5:0.60SiO2:8.0TEAOH:212H2O. Aluminium iso-propoxide, TEAOH solution and colloidal silica were mixed and stirred at room temperature for one hour. Then phosphoric acid was added dropwise into the clear precursor solution. The mixture was stirred for one hour before it was placed in a Teflon-lined stainless steel autoclave for hydrothermal growth. The precursor solution was heated and stirred at 180 °C for 72 h. The solid product was separated by centrifugation, washed with deionized water 3 times and dried at 90 °C overnight. The SDA was removed by calcination in air at 600 °C for 4 h. The sample obtained from the first-step hydrothermal synthesis with 0.30 Si/Al ratio in the precursor solution was named 1st0.30.
:4.0P2O5:0.60SiO2:8.0TEAOH:212H2O. Aluminium iso-propoxide, TEAOH solution and colloidal silica were mixed and stirred at room temperature for one hour. Then phosphoric acid was added dropwise into the clear precursor solution. The mixture was stirred for one hour before it was placed in a Teflon-lined stainless steel autoclave for hydrothermal growth. The precursor solution was heated and stirred at 180 °C for 72 h. The solid product was separated by centrifugation, washed with deionized water 3 times and dried at 90 °C overnight. The SDA was removed by calcination in air at 600 °C for 4 h. The sample obtained from the first-step hydrothermal synthesis with 0.30 Si/Al ratio in the precursor solution was named 1st0.30.
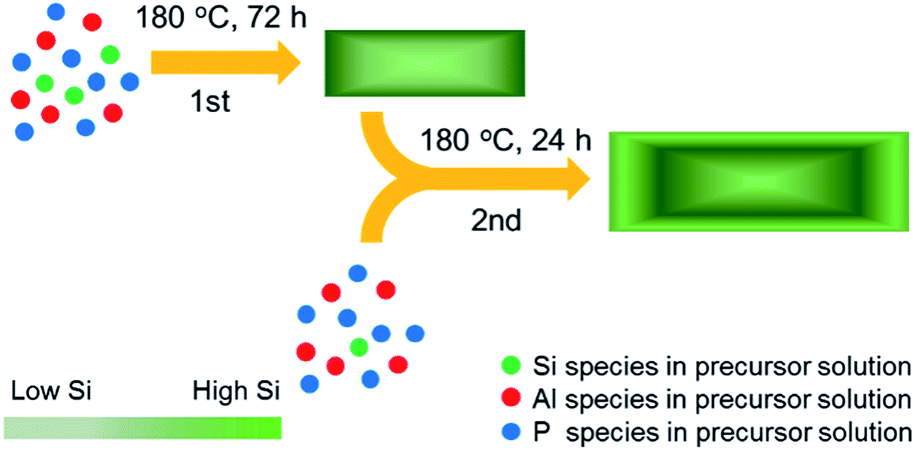 | ||
| Scheme 1 Schematic of the epitaxial growth of SAPO-34 zeolite with a low-acid outer layer and high-acid core by the two-step hydrothermal synthesis. | ||
The precursor solution of the second-step hydrothermal synthesis was prepared with the following molar compositions: 1.0Al2O3:4.0P2O5:ySiO2:8.0TEAOH:218H2O, where y was 0.30, 0.20 and 0.10. The 1st0.30 sample was first dried and calcined before it was added to the precursor solution in the weight ratio of 1.0 SAPO-34:74.0 precursor solution. After this, the second-step hydrothermal process was the same as the first step except that the crystallization time was 24 h. We named the samples from the second-step hydrothermal synthesis 2nd0.15, 2nd0.10 and 2nd0.05 according to the Si/Al ratio of the second-step precursor solution.
A hydrothermal treatment (HT) was carried out to test the hydrothermal stability of the catalysts. The samples named with the HT were treated with saturated steam at 600 °C for 3 hours.
2.2 Characterization
A scanning electron microscope (SEM), Model JEOL JSM-7401, was used to observe the morphology of the samples. The Si, Al and P distributions in the SAPO-34 particles were characterized by the energy dispersive spectrometer lining (EDS lining) of a transmission electron microscope (TEM), model JEM-2010. The framework type of the samples was characterized by X-ray diffraction (XRD) with a Bruker D8/Advance instrument (transmission mode, Cu Kα radiation, 5° min−1, 5–45°). The total acid site density was measured by the temperature programmed desorption of ammonia (TPD-NH3), which was performed on a Quantachrome ChemBET Pulsa. The BET total surface area and microscopic volume were determined by Ar adsorption and desorption using a Quantachrom Autosorb iQ2 instrument. The chemical composition was analyzed by inductively coupled plasma atomic emission spectrometry (ICP-AES) using a SPECTRO ARCOS instrument. The surface chemical composition was characterized by X-ray photoelectron spectroscopy (XPS) using a Thermo Fisher 250XI instrument.The surface acid density was characterized by the TGA-Py method. The pyridine is too big to enter the pores of SAPO-34 zeolite, so it is only absorbed by the acid sites on the surface. The sample was heated to 500 °C and kept for 1 h in order to drive off water and other impurity gases adsorbed by the samples. After the samples were cooled to 50 °C, pyridine was injected and adsorbed for 2 h. To remove the pyridine absorbed physically, the temperature was increased to 150 °C and held for 1 h. Then the temperature was increased from 150 °C to 500 °C and kept at 500 °C for 1 h. The mass loss after 150 °C was taken as the amount of pyridine absorbed by the surface acid sites chemically.
2.3 Catalytic experiments
The MTO reaction was tested in a quartz tubular fixed bed reactor at 400 °C and atmospheric pressure. 20 mg of the sample mixed with 60 mg quartz (120–180 mesh) was activated at 400 °C in an Ar gas flow of 20 ml min−1 for 1 h before the MTO reaction. The methanol was fed in by a carrier gas (Ar) using a saturator containing methanol at room temperature. The weight hourly space velocity (WHSV) was 4.0 h−1. Another flow line of Ar gas was used to keep the methanol feed mole fraction at 4.2%. An online gas chromatograph (Aglient 7890A) was used to analyze the composition of the gas products. The conversion of methanol and product selectivity were calculated on a carbon basis. Both methanol and dimethylether (DME) were regarded as reactants.3. Results and discussion
3.1 Morphology, framework type and elemental distribution
The SEM images of the samples are shown in Fig. 1. The 1st0.30 sample exhibited 200 nm thick sheet-like morphology (Fig. 1a). The samples grew much bigger but still remained sheet-like after the second-step hydrothermal synthesis (Fig. 1b–d). Meanwhile, the surface of these samples was much rougher compared to the 1st0.30 sample. It seems that the 1st0.30 sample acted as the crystal core which continued to grow after it was added into the low-silica hydrothermal environment. | ||
| Fig. 1 SEM images of the samples: (a) 1st0.30; (b) 2nd0.15; (c) 2nd0.10; (d) 2nd0.05. | ||
As shown in Fig. 2, the elemental distribution was characterized by TEM EDS-lining. The EDS-lining method55–58 not only showed the element distribution in the zeolite, but also measured approximately the thickness of the outer layer grown on the samples epitaxially. The P and Al distribution are kept highly consistent and indicated the edge and thickness of a particle. When the P and Al signals begin to increase, it means the light spot of the characterization has moved onto the edge of the particle. Conversely, the P and Al signals will be reduced when the light spot for the characterization moves out of the edge of the particle. The P and Al intensities show the thickness of the particle in the scanning area of EDS-lining.
 | ||
| Fig. 2 EDS-lining and TEM images of samples: (a) and (b) 1st0.30 before and after HT; (c) and (d) 2nd0.15 before and after HT; (e) and (f) 2nd0.10 before and after HT; (g) and (h) 2nd0.05 before and after HT. In the EDS-lining results, H and L means the high and low silica content. The diameter of EDS-lining spot was 10 nm. | ||
The Si signal obviously increased on the edge of the particle of 1st0.30 sample, showing the Si enrichment of the external surface of the SAPO-34 samples (Fig. 2a and b). This is in agreement with the results in other reports.47,52,53 The P, Al and Si signals of the 1st0.30 sample increased and decreased synchronously. However, after the 2nd-step hydrothermal synthesis, the Si signal changed in a wider range than the P and Al signals (Fig. 2c–h). This could be explained by that the SAPO-34 crystals were coated by a low-silica outer layer with a thickness of about 200 nm. It indicates that a low-silica outer layer has grown epitaxially onto the 1st-step synthesized SAPO-34 in the 2nd-step hydrothermal synthesis. The P, Al and Si distribution in the SAPO-34 crystals showed few changes after the HT (Fig. 2d, f and h), which means the low-silica outer layer could remain stable in the 600 °C saturated steam for 3 hours.
The XRD images of samples and that of reference SAPO-34 are shown in Fig. 3. The XRD pattern of the 1st0.30 sample was in agreement with CHA-type framework, and it remained unchanged after the second-step hydrothermal synthesis. It suggested that the 2nd-step hydrothermal growth tended to be epitaxial growth on the 1st-step synthesized sample and it kept the same framework, as opposed to new nucleation and growth independently of the already-present zeolite. That is, the 1st0.30 sample acted with the same function as “crystal seeds”. Combined with the XRD and EDS-lining results (Fig. 2 and 3), it proved that the epitaxial growth on the SAPO-34 was achieved by the two-step hydrothermal synthesis, and formed an unusual silica distribution with a low-silica outer layer and a high-silica core. There are very small peaks at 5–6 2θ degree in Fig. 3, which indicate there were impurity crystals of SAPO-5 in the SAPO-34 samples. However, since these peaks were very small and SAPO-5 crystals were not seen by SEM, as shown in Fig. 1, it was believed that the amount of impurity crystals was limited and did not influence the results.
 | ||
| Fig. 3 XRD images of samples and standard SAPO-34 peaks. | ||
3.2 Composition, acid site density and textural property
The Si/Al mole ratios are shown in Table 1, together with the yield and mass increase in the 2nd-step hydrothermal growth. After the 2nd hydrothermal growth, the mass of two-step synthesized samples increased by at least 180%. And when the silica content in the 2nd precursor solution became higher, the higher increasing mass and yield were achieved (Table 1), indicating that the high silica environment was conductive to the growth of SAPO-34 zeolite.| Samples no. | Si/Al ratio | ΔMd/% | Yielde/% | ||
|---|---|---|---|---|---|
| Rprecursora | Rbulkb | Rsurfacec | |||
| a Rprecursor is the Si/Al ratio of the precursor solution.b Rbulk is the Si/Al ratio of the bulk phase of the sample measured by ICP.c Rsurface is the Si/Al ratio of the surface of the sample measured by XPS, which can penetrate at most 10 nm thickness into the surface.d ΔM is the mass increase in the 2nd-step growth.e Yield is the ratio of the actual product mass divided by the highest mass that can be synthesized. | |||||
| 1st0.30 | 0.30 | 0.24 | 0.52 | — | 62 |
| 2nd0.15 | 0.15 | 0.16 | 0.32 | 210 | 63 |
| 2nd0.10 | 0.10 | 0.15 | 0.20 | 185 | 60 |
| 2nd0.05 | 0.05 | 0.13 | 0.13 | 180 | 55 |
In agreement with the EDS-lining results (Fig. 2a), the surface Si/Al ratio of 1st0.30 sample was higher than the average value of its bulk phase, showing the surface enrichment of silica (Table 1). About 2 times mass increment of two-step synthesized samples led to a decrease of both surface and bulk phase's silica content (Table 1). The 2nd-step growth could result in less enrichment of silica on the surface, and even no difference in the silica content between the surface and bulk phase when the 2nd-step precursor solution had a low enough Si/Al ratio, such as the 2nd0.05 sample.
The acid site density and strength measured by TPD-NH3 and TGA-Py are shown in Fig. 4 and Table 2. The total acid site density was calculated from the peak in the high-temperature area of TPD-NH3 measurement, which was due to Brönsted acid sites,59–61 acting as the catalytic sites in the MTO process. The temperature of the peak center in the high temperature area represents the acid strength. There was a positive correlation between the total acid site density from TPD-NH3 and the Si/Al ratio of their bulk phase (Tables 1 and 2). The bulk Si/Al ratio of the 1st0.30 sample in Table 1 implied that if all Si atoms were isolated as Si4Al, the acid site density would be 1.98 mmol g−1. The actual acid site density measured by TPD-NH3 was 1.02 mmol g−1 before the HT, as shown in Table 2. This implies that there were silicon islands in the 1st 0.30 sample and nearly half of the Si atoms were not acidic as defects or atoms inside silicon islands. For the two-step synthesized samples, the low silica content in the 2nd-step precursor solution reduced both the Si/Al ratio and the total acid site density of the samples.
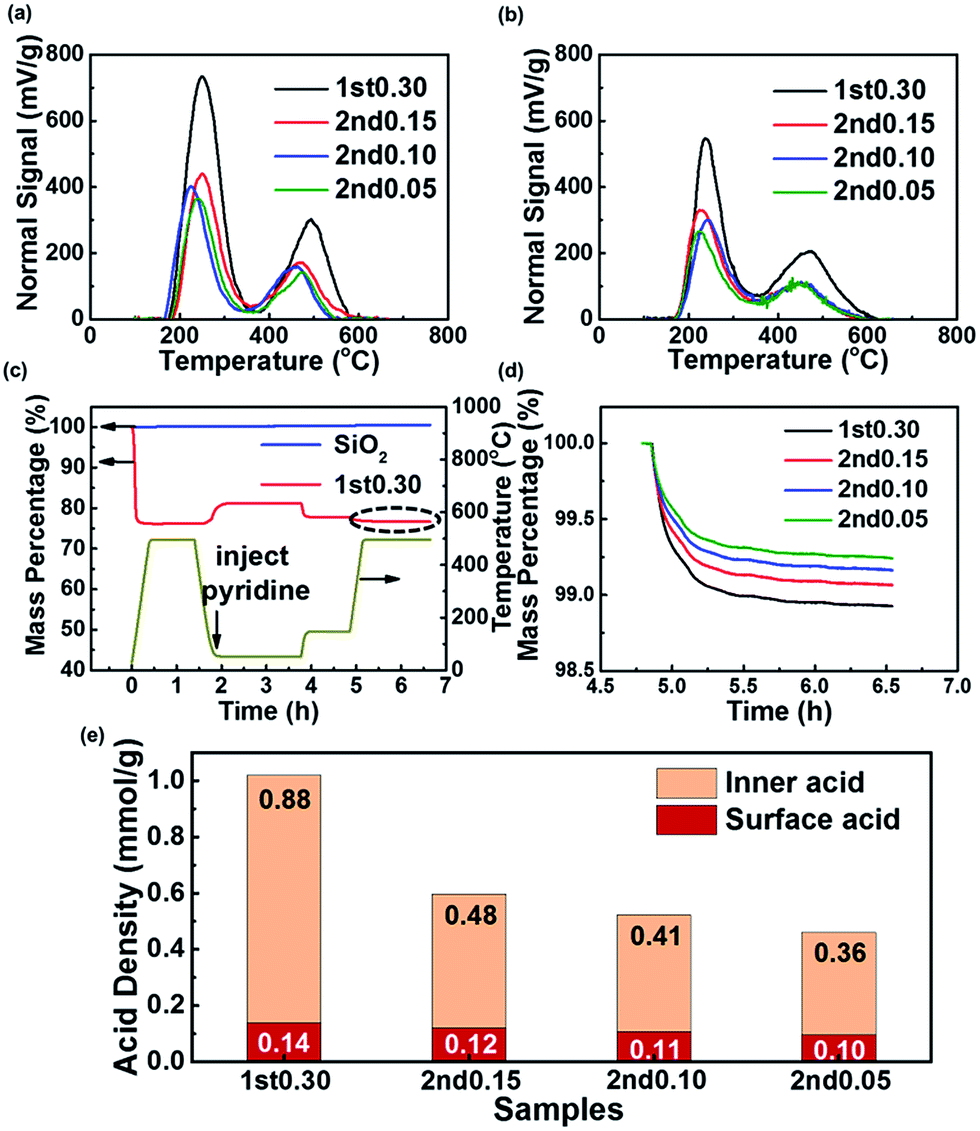 | ||
| Fig. 4 (a) TPD-NH3 results from the samples before the HT; (b) TPD-NH3 results of samples after the HT; (c) pyridine temperature programmed adsorption and desorption TGA results of sample 1st0.30 and quartz sand and temperature programed TGA; (d) mass loss of the samples when the temperature increased from 150 °C to 500 °C and kept at 500 °C for 1 h, taking the mass percentage as 100% when the temperature began to rise from 150 °C. This compares the sample's dash line area in (c); (e) inner and surface acid site density of the samples. | ||
| Sample no. | Atotala (mmol g−1) | Tstrongb (°C) | ||||
|---|---|---|---|---|---|---|
| Before HT | After HT | +/− | Before HT | After HT | +/− | |
| a Atotal is the total acid density calculated from high-temperature area of TPD-NH3 curve.b Tstrong is the peak temperature in the high-temperature area. | ||||||
| 1st0.30 | 1.02 | 0.74 | −27% | 494 | 473 | −4% |
| 2nd0.15 | 0.60 | 0.49 | −19% | 470 | 453 | −4% |
| 2nd0.10 | 0.52 | 0.45 | −13% | 461 | 452 | −2% |
| 2nd0.05 | 0.46 | 0.40 | −12% | 474 | 442 | −7% |
The TGA-Py method was used to measure the surface acid site density, using the mass loss after 150 °C as due to pyridine absorbed chemically by the surface acid sites. The mass of the quartz sand did not show any change because it had no acidity and did not absorb any pyridine chemically, which was different from the SAPO-34 samples (Fig. 4c). The low-silica outer layer caused the decrease of the surface acid site density (Fig. 4d). So the two-step hydrothermal synthesis was an effective method to reduce the surface acid density of the SAPO-34.
Results in Fig. 4e compared the acid site density and distribution of the samples furtherly. A similar method has been used for a ZSM-5 zeolite.62,63 The interior acid density was obtained by subtracting the surface acid density from the total acid density. Although the 2nd-step hydrothermal synthesis caused both the interior and surface acid site density to decrease, the interior acid site density decreased more (Fig. 4e). This suggested that the two-step synthesis had more impact on the interior acid site than the surface acid, which was due to the surface enrichment of silica and much mass increase after the 2nd-step growth. The low-silica outer layer synthesized by the two-step method had a large influence on the acid site density and site distribution.
A HT could decrease both the acid site density and acid strength of the SAPO-34 zeolite (Table 2). In the high-temperature saturated steam atmosphere, the acid sites in the framework were hydrolyzed, leading to the decrease of the total acid site density.11,37,64–66 After the HT, the acid site density of 1st0.30 sample decreased to 0.76 mmol g−1, which means more Si atoms had been hydrolyzed into defects or collected into islands during the HT process. Although all the samples showed a total acid site density decrease after the HT, it was interesting to note that the two-step synthesized samples showed a smaller reduction than the 1st0.30 sample (Table 2 and Fig. 4a and b). And the 2nd0.05 sample with the lowest acid site density lost the fewest acid site density after the HT (Table 2). It seemed that the SAPO-34 with low acid site density was more stable during the HT with respect to the acid site density.
The acid strength of all the samples also decreased after the HT to a comparable degree (Table 2). As the silica species on the borders of the silica island show the strongest acidity and they were always firstly hydrolyzed,67,68 the average acid strength of SAPO-34 showed a decrease after the HT. In conclusion, thanks to the low acid site density, the two-step synthesized samples showed higher hydrothermal stability of the acid sites compared with the one-step synthesized samples.
The textural properties of the samples before and after the HT are shown in Table 3. The micropore volume depends on the amount of zeolite material in the samples, so it represents the growth completeness of the SAPO-34 zeolites. The reduction of non-framework material can increase the micropore volume. After the 2nd-step growth, the textural properties of two-step synthesized samples remained almost unchanged, compared with the 1st0.30 sample. It implied that crystal epitaxial growth on the SAPO-34 particles in two-step method had little effect on the textural properties, but great effect on the acid site density and distribution.
| Sample no. | Stotala (m2 g−1) | Vmicrob (cm3 g−1) | ||||
|---|---|---|---|---|---|---|
| Before HT | After HT | +/− | Before HT | After HT | +/− | |
| a Stotal is the total surface area calculated from the BET equation.b Vmicro is the micropore volume calculated by the Ar isothermal adsorption and desorption curve with the NLDFT equilibrium model. | ||||||
| 1st0.30 | 521 | 539 | +3% | 0.27 | 0.29 | +7% |
| 2nd0.15 | 526 | 603 | +15% | 0.28 | 0.33 | +18% |
| 2nd0.10 | 530 | 574 | +8% | 0.28 | 0.31 | +11% |
| 2nd0.05 | 516 | 525 | +2% | 0.27 | 0.30 | +11% |
After the HT, all the samples had micropore volume increases to varying degrees (Table 3). On the one hand, the high-temperature saturated steam drives some non-framework atom into the framework and these start to recrystallize during the HT, resulting in increasing the micropore volume.37,64,69 On the other hand, the hydrolysis of hot steam can damage the framework of SAPO-34 zeolites, which cause the decrease of micropore volume.11 In the HT process, the samples were more influenced by the recrystallization than the damage, so the framework of the samples were furtherly improved. The obvious increase of the 2nd0.10 sample's total surface area and micropore volume has been noted, which implied a good MTO performance after the HT.
3.3 Catalytic performance and coking distribution
The MTO performance of the fresh samples are shown in Fig. 5. All the samples gave an initial conversion close to 100%, but the 1st0.30 sample was rapidly deactivated. Although the 2nd0.15 sample's particle size was bigger than that of the 1st0.30 sample, the 2nd0.15 sample had a lower acid site density, which could offset the negative effect of the increase in diffusion resistance. So the 1st0.30 and 2nd0.15 samples showed the similar catalytic performance (Fig. 1, 2 and 5a). Compared to the 2nd0.15 sample, the outer layer with the lower silica content of the 2nd0.10 and 2nd0.05 samples improved their catalytic lifetime (Fig. 5a). It implied that the low-silica outer layer inhibited coking on the outer layer of the crystals and kept the interior acid sites more available for methanol.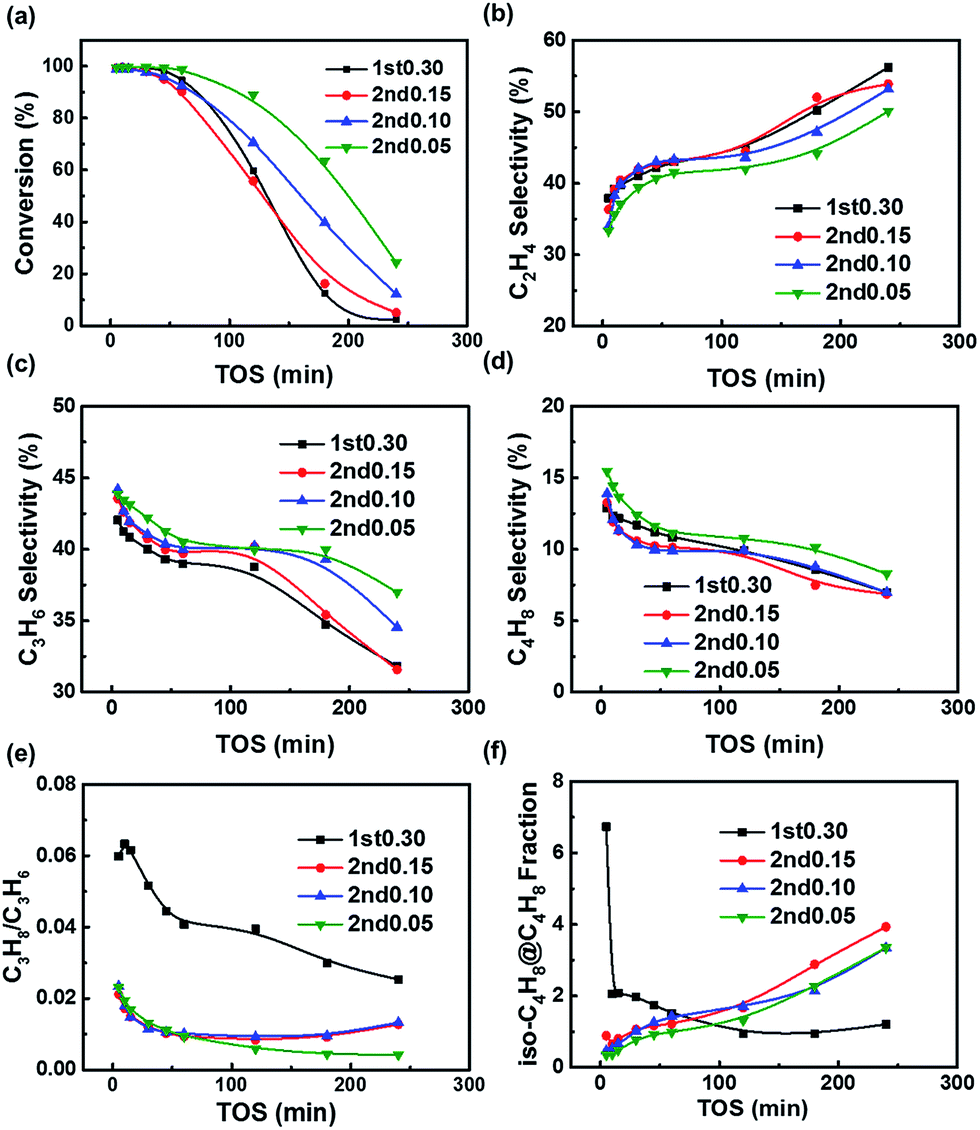 | ||
| Fig. 5 MTO results of the fresh samples: (a) conversion of methanol; (b) C2H4 selectivity; (c) C3H6 selectivity; (d) C4H8 selectivity; (e) C3H8/C3H6; (f) iso-C4H8 fraction referenced to all C4H8. Experimental conditions: T = 400 °C, WHSV = 4 h−1, methanol partial pressure = 4260 Pa. | ||
All the samples showed a C2/C3 ratio that increased continuously with time on stream (Fig. 5b and c). This could be explained by that coke formation reduced the void volume of the cavities, resulting in enhanced transition shape selectivity and favoring the formation of smaller molecules.70,71 The two-step synthesized samples tended to synthesize propene and butene, while the 1st0.30 sample had a higher ethene selectivity (Fig. 5b–d). It suggested that the low-silica outer layer inhibited the diffusional resistance caused by the coking, in favor of the diffusion of propene and butene.
For the C3 products, the hydrogen transfer of propene is an important side reaction because it can not only decrease the propene selectivity, but also tend to produce coke.36,72 Propane/propene is called hydrogen transfer index, representing the degree of hydrogen transfer reaction of propene to propane in SAPO-34 zeolite. All the two-step synthesized samples exhibited a lower hydrogen transfer index than the 1st0.30 sample (Fig. 5e). On the one hand, all the two-step synthesized samples could suppress coking on the shell and decrease the diffusion resistance, thanks to the acid site distribution of a low-acid outer layer and high-acid core formed by the two-step hydrothermal method. Decreasing the diffusion resistance could speed up the mass transfer of propene and lower the propene concentration in the particles, resulting in inhibiting hydrogen transfer. On the other hand, it was helpful for inhibiting the hydrogen transfer to decrease the acid site density. The low acid site density could decrease the contacting chance between the acid sites and propene, preventing the hydrogen transfer. It indicated that the low acid site density and diffusional resistance helped suppress the hydrogen transfer of propene and improved the product selectivity. When taking both the lifetime and product selectivity into consideration, the two-step hydrothermal method could be deduced to give a good acid site density and distribution for the SAPO-34 zeolite to improve the MTO performance.
The fraction of iso-butene in butene is mainly affected by the surface acid site density of SAPO-34 molecular sieves. As one of the four isomers of butene, the fraction of iso-butene in butene is 46% under the thermodynamic equilibrium of 400 °C. However, the fraction of iso-butene produced by SAPO-34 zeolite during the MTO reaction is less than 10% (Fig. 5f), because the molecular volume of iso-butene is relatively large, and the diffusion of iso-butene is obviously limited under the influence of SAPO-34 shape selectivity, which makes iso-butene fraction lower than the thermodynamic equilibrium value. Iso-butene is too large to exit from the small windows of the SAPO-34 zeolite, so it is mainly produced by the isomerization on the surface. Obviously, the fraction of iso-butene in the total butenes can be regarded as a probe of surface acid site density.
The fraction of iso-butene in butene is determined by the relative activity of the internal acid sites and the external acid sites. When the activity of the internal acid sites is relatively high, the fraction of iso-butene in butene is reduced under the shape selectivity of SAPO zeolite. Otherwise, more iso-butene will be produced by the isomerization reaction catalyzed by the surface acid sites, giving rise to increasing iso-butene fraction in the butene.
In the initial stage of the reaction, the fresh 1st0.30 sample had the most surface acid sites according to the TGA-Py results (Fig. 4e), which meant that its isomerization of butene on the surface of catalysts was the highest, so the fraction of iso-butene in the 1st0.30 sample of the initial stage was much higher than that of other samples (Fig. 5f). But as coking usually occurred on the outer surface firstly and the isomerization reaction was inhibited at the same time, the 1st0.30 sample's iso-butene fraction decreased rapidly. With the progress of the reaction, the SAPO-34 particles were coked and the formation of butene also slowed down. Relatively, the rate of isomerization did not decrease obviously and the fraction of iso-butene began to rise for the two-step synthesized samples, while the fraction of iso-butene of the 1st0.30 sample remained at a lower level compared with the two-step synthesized samples (Fig. 5f). The 1st0.30 sample had the most interior acid site density, so the formation of butene was still dominant and the iso-butene fraction didn't become more even when the SAPO-34 sample was severely deactivated.
The C, Al, P and Si elemental EDS-lining images of completely deactivated SAPO-34 particles are shown in Fig. 6. The 1st0.30 and 2nd0.15 samples had Al, P and Si distributions that remained unchanged after coking (Fig. 2a and c and 6). There was still Si enrichment of the surface on the 1st0.30 particle. The 2nd0.15 sample's low-silica outer layer was also shown to be stable after coking.
 | ||
| Fig. 6 C, Al, Si and P elemental EDS-lining images of completely deactivated samples after the MTO reaction: (a) and (b) 1st0.30; (c) and (d) 2nd0.15. | ||
The 1st0.30 sample had a coking distribution after the MTO reaction that followed the silica distribution, which gathered on the outer layer of the particles (Fig. 6a and b). It could be explained by Si surface enrichment of SAPO-34 particles and diffusional limitation. Although diffusional limitation still existed, the coking was evenly distributed in the two-step synthesized particles due to the low-silica outer layer (Fig. 6c and d). The absence of a lot of coke on the outer layer of the SAPO-34 particles helped to inhibit pores blocking and weaken diffusional limitation to some extent, and allowed more interior acid sites to be fully utilized. In addition, it could help reactants and products diffuse more easily, leading to improving catalytic efficiency and inhibit hydrogen transfer. This was why optimizing the acid site distribution could prolong the catalytic lifetime and reduce byproducts selectivity (Fig. 5a and e).
The comparison of the methanol conversion of the samples before and after the HT is shown in Fig. 7. The two-step synthesized samples showed better hydrothermal stability than the one-step synthesized sample (Fig. 7). With the high-temperature saturated steam, all samples started to recrystallize, especially for the two-step synthesized samples. Compared to the one-step synthesized samples, the micropore volume of two-step synthesized samples increased more, meaning that more cages could be used (Table 3). Meanwhile, the recrystallization of high-temperature saturated steam made two-step synthesized SAPO-34 samples loss a lower proportion of acid sites during the hydrothermal process (Table 2). The less loss of acid site density and the more increase in micropore volume ensure the better hydrothermal stability of the two-step synthesized samples relative to the one-step synthesized samples.
 | ||
| Fig. 7 Methanol conversion of the samples before and after the HT. | ||
It was interesting to note that the 2nd0.15 sample had its lifetime prolonged after the HT, which was different from the other samples. Its micropore volume increased 18% during the hydrothermal process (Table 2). The obvious increase of its micropore volume was helpful for catalysis, despite the modest reduction of its acid site density. Thus, the two-step hydrothermal synthesis showed good hydrothermal stability, which was important for the MTO industrial application.
As a model study, this work not only proves the feasibility of epitaxial growth of a low-silica shell on the high-silica SAPO-34, but also shows the better MTO catalytic performance and hydrothermal stability, compared to the common one-step synthesized sample. The two-step synthesized samples achieve high molecular sieves yield in the synthesis, and remain the CHA structure, preventing forming other impurity crystals. It will be a great advantage if one-step synthesized low-silica sample is taken into comparison. Meanwhile, the two-step synthesized samples have an unusual acid distribution of high-acid core and low-acid outer layer. The low-acid outer layer inhibits coking on the outer layer and weakens diffusional limitation, which can cause better utilization of interior acid sites and prolong the catalytic lifetime. In addition, a proper acid site density and distribution formed by the two-step synthesized method can help suppress the hydrogen transfer of propene and improves the product selectivity.
4. Conclusions
A low-silica SAPO-34 zeolite, that is, a low acid site density SAPO-34 zeolite, shows better MTO performance, but it suffers the problem of low yield and impure crystal when synthesized by a one-step hydrothermal method. This work demonstrated a practical two-step hydrothermal synthesis strategy that gave SAPO-34 zeolite with the acid site distribution of a high-silica core and a low-silica outer layer. EDS-lining confirmed that a low-silica outer layer was epitaxially grown on a high-silica core, and measured the thickness of the low-silica outer layer as 200 nm. During the second-step hydrothermal synthesis in a low-silica precursor solution, the high-silica core acted as crystal seed for the growth of the low-silica outer layer and also inhibited the formation of impure crystal.Diffusion resistance has a significant impact on the activity and product selectivity in the MTO reaction. The core–shell SAPO-34 zeolite here showed a longer catalyst lifetime, because it benefited from that most of the catalytic reaction occurred in the low acid site outer layer. Unlike the one-step synthesized high-silica sample, the low acid site outer layer of the two-step synthesized samples showed inhibited coking and reduced diffusion limitation, resulting in increased lifetime. After a hydrothermal treatment, the two-step synthesized samples showed less reduction of acid site density and a larger increase in the micropore volume than the one-step synthesized sample, that is, they showed better hydrothermal stability than the one-step synthesized sample.
To study the epitaxial growth by EDS-lining, relatively large particles were formed in the first-step growth and used as core seeds in the second-step growth. It was deduced that for industrial use, by using a smaller high-silica SAPO-34 zeolite as “crystal seed” and with the thickness of the low-silica outer layer optimized, even better MTO performance will be achieved.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the National Natural Science Foundation of China (21173125) and the National High Technology Research and Development Program of China (863 Program, 2012AA051001) for their financial support for this study.References
- M. Stocker and J. Weitkamp, Microporous Mesoporous Mater., 1999, 29, 1 CrossRef CAS.
- M. Stocker, Microporous Mesoporous Mater., 1999, 29, 3–48 CrossRef CAS.
- U. Olsbye, S. Svelle, M. Bjorgen, P. Beato, T. V. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810–5831 CrossRef CAS PubMed.
- I. M. Dahl and S. Kolboe, J. Catal., 1996, 161, 304–309 CrossRef CAS.
- I. M. Dahl and S. Kolboe, J. Catal., 1994, 149, 458–464 CrossRef CAS.
- I. M. Dahl and S. Kolboe, Catal. Lett., 1993, 20, 329–336 CrossRef CAS.
- M. Bjorgen, S. Svelle, F. Joensen, J. Nerlov, S. Kolboe, F. Bonino, L. Palumbo, S. Bordiga and U. Olsbye, J. Catal., 2007, 249, 195–207 CrossRef CAS.
- S. Svelle, F. Joensen, J. Nerlov, U. Olsbye, K.-P. Lillerud, S. Kolboe and M. Bjorgen, J. Am. Chem. Soc., 2006, 128, 14770–14771 CrossRef CAS PubMed.
- H. Yamazaki, H. Shima, H. Imai, T. Yokoi, T. Tatsumi and J. N. Kondo, J. Phys. Chem. C, 2012, 116, 24091–24097 CAS.
- S. Ilias and A. Bhan, J. Catal., 2012, 290, 186–192 CrossRef CAS.
- Z. Li, J. Martinez-Triguero, P. Concepcion, J. Yu and A. Corma, Phys. Chem. Chem. Phys., 2013, 15, 14670–14680 RSC.
- W. Dai, G. Wu, L. Li, N. Guan and M. Hunger, ACS Catal., 2013, 3, 588–596 CrossRef CAS.
- X. Chen, D. Xi, Q. Sun, N. Wang, Z. Dai, D. Fan, V. Valtchev and J. Yu, Microporous Mesoporous Mater., 2016, 234, 401–408 CrossRef CAS.
- Q. Sun, N. Wang, R. Bai, X. Chen and J. Yu, J. Mater. Chem. A, 2016, 4, 14978–14982 CAS.
- B. Gao, M. Yang, Y. Qiao, J. Li, X. Xiang, P. Wu, Y. Wei, S. Xu, P. Tian and Z. Liu, Catal. Sci. Technol., 2016, 6, 7569–7578 CAS.
- J. F. Haw and D. M. Marcus, Top. Catal., 2005, 34, 41–48 CrossRef CAS.
- S.-G. Lee, H.-S. Kim, Y.-H. Kim, E.-J. Kang, D.-H. Lee and C.-S. Park, J. Ind. Eng. Chem., 2014, 20, 61–67 CrossRef CAS.
- J. Liang, H. Y. Li, S. Zhao, W. G. Guo, R. H. Wang and M. L. Ying, Appl. Catal., 1990, 64, 31–40 CrossRef CAS.
- A. Izadbakhsh, F. Farhadi, F. Khorasheh, S. Sahebdelfar, M. Asadi and Y. Z. Feng, Appl. Catal., A, 2009, 364, 48–56 CrossRef CAS.
- D. Chen, K. Moljord, T. Fuglerud and A. Holmen, Microporous Mesoporous Mater., 1999, 29, 191–203 CrossRef CAS.
- N. Nishiyama, M. Kawaguchi, Y. Hirota, D. Van Vu, Y. Egashira and K. Ueyama, Appl. Catal., A, 2009, 362, 193–199 CrossRef CAS.
- S. Lin, J. Li, R. P. Sharma, J. Yu and R. Xu, Top. Catal., 2010, 53, 1304–1310 CrossRef CAS.
- Q. Sun, Y. Ma, N. Wang, X. Li, D. Xi, J. Xu, F. Deng, K. B. Yoon, P. Oleynikov, O. Terasaki and J. Yu, J. Mater. Chem. A, 2014, 2, 17828–17839 CAS.
- G. Yang, Y. Wei, S. Xu, J. Chen, J. Li, Z. Li, J. Yu and R. Xu, J. Phys. Chem. C, 2013, 117, 8214–8222 CAS.
- K. Y. Lee, H.-J. Chae, S.-Y. Jeong and G. Seo, Appl. Catal., A, 2009, 369, 60–66 CrossRef CAS.
- D. Xi, Q. Sun, J. Xu, M. Cho, H. S. Cho, S. Asahina, Y. Li, F. Deng, O. Terasaki and J. Yu, J. Mater. Chem. A, 2014, 2, 17994–18004 CAS.
- J. Zhu, Y. Cui, Y. Wang and F. Wei, Chem. Commun., 2009, 3282–3284 RSC.
- H. Yang, Z. Liu, H. Gao and Z. Xie, J. Mater. Chem., 2010, 20, 3227–3231 RSC.
- Y. Li, Y. Huang, J. Guo, M. Zhang, D. Wang, F. Wei and Y. Wang, Catal. Today, 2014, 233, 2–7 CrossRef CAS.
- F. Schmidt, S. Paasch, E. Brunner and S. Kaskel, Microporous Mesoporous Mater., 2012, 164, 214–221 CrossRef CAS.
- A. Z. Varzaneh, J. Towfighi, S. Sahebdelfar and H. Bahrami, J. Anal. Appl. Pyrolysis, 2016, 121, 11–23 CrossRef CAS.
- A. Z. Varzaneh, J. Towfighi and S. Sahebdelfar, Microporous Mesoporous Mater., 2016, 236, 1–12 CrossRef CAS.
- Q. Sun, N. Wang, G. Guo, X. Chen and J. Yu, J. Mater. Chem. A, 2015, 3, 19783–19789 CAS.
- Q. Sun, N. Wang, D. Xi, M. Yang and J. Yu, Chem. Commun., 2014, 50, 6502–6505 RSC.
- X. Chen, A. Vicente, Z. Qin, V. Ruaux, J.-P. Gilson and V. Valtchev, Chem. Commun., 2016, 52, 3512–3515 RSC.
- F. D. P. Mees, P. Van Der Voort, P. Cool, L. R. M. Martens, M. J. G. Janssen, A. A. Verberckmoes, G. J. Kennedy, R. B. Hall, K. Wang and E. F. Vansant, J. Phys. Chem. B, 2003, 107, 3161–3167 CrossRef CAS.
- Z. Li, J. Martinez-Triguero, J. Yu and A. Corma, J. Catal., 2015, 329, 379–388 CrossRef CAS.
- S. Wilson and P. Barger, Microporous Mesoporous Mater., 1999, 29, 117–126 CrossRef CAS.
- A. Izadbakhsh, F. Farhadi, F. Khorasheh, S. Sahebdelfar, M. Asadi and Z. F. Yan, Microporous Mesoporous Mater., 2009, 126, 1–7 CrossRef CAS.
- W. Dai, X. Wang, G. Wu, N. Guan, M. Hunger and L. Li, ACS Catal., 2011, 1, 292–299 CrossRef CAS.
- D. Mores, E. Stavitski, M. H. F. Kox, J. Kornatowski, U. Olsbye and B. M. Weckhuysen, Chem.–Eur. J., 2008, 14, 11320–11327 CrossRef CAS PubMed.
- L. Marchese, A. Frache, E. Gianotti, G. Martra, M. Causa and S. Coluccia, Microporous Mesoporous Mater., 1999, 30, 145–153 CrossRef CAS.
- J. Chen, J. Li, C. Yuan, S. Xu, Y. Wei, Q. Wang, Y. Zhou, J. Wang, M. Zhang, Y. He, S. Xu and Z. Liu, Catal. Sci. Technol., 2014, 4, 3268–3277 CAS.
- Y. Wang, S.-L. Chen, Y.-J. Jiang, Y.-Q. Cao, F. Chen, W.-K. Chang and Y.-L. Gao, RSC Adv., 2016, 6, 104985–104994 RSC.
- T. Alvaro-Munoz, C. Marquez-Alvarez and E. Sastre, Top. Catal., 2016, 59, 278–291 CrossRef CAS.
- J. Chen, J. Li, Y. Wei, C. Yuan, B. Li, S. Xu, Y. Zhou, J. Wang, M. Zhang and Z. Liu, Catal. Commun., 2014, 46, 36–40 CrossRef CAS.
- M. Yang, P. Tian, C. Wang, Y. Yuan, Y. Yang, S. Xu, Y. He and Z. Liu, Chem. Commun., 2014, 50, 1845–1847 RSC.
- D. Van Vu, M. Miyamoto, N. Nishiyama, Y. Egashira and K. Ueyama, J. Catal., 2006, 243, 389–394 CrossRef.
- D. Van Vu, M. Miyamoto, N. Nishiyama, S. Ichikawa, Y. Egashira and K. Ueyama, Microporous Mesoporous Mater., 2008, 115, 106–112 CrossRef.
- D. V. Vu, M. Miyamoto, N. Nishiyama, Y. Egashira and K. Ueyama, Catal. Lett., 2009, 127, 233–238 CrossRef.
- M. Miyamoto, T. Kamei, N. Nishiyama, Y. Egashira and K. Ueyama, Adv. Mater., 2005, 17, 1985–1991 CrossRef CAS.
- G. Liu, P. Tian, Q. Xia and Z. Liu, J. Nat. Gas Chem., 2012, 21, 431–434 CrossRef CAS.
- G. Liu, P. Tian, Y. Zhang, J. Li, L. Xu, S. Meng and Z. Liu, Microporous Mesoporous Mater., 2008, 114, 416–423 CrossRef CAS.
- H. van Heyden, S. Mintova and T. Bein, Chem. Mater., 2008, 20, 2956–2963 CrossRef CAS.
- A. Lombard, A. Simon-Masseron, L. Rouleau, A. Cabiac and J. Patarin, Microporous Mesoporous Mater., 2010, 129, 220–227 CrossRef CAS.
- M. Okamoto and Y. Osafune, Microporous Mesoporous Mater., 2011, 143, 413–418 CrossRef CAS.
- G. Yang, N. Tsubaki, J. Shamoto, Y. Yoneyama and Y. Zhang, J. Am. Chem. Soc., 2010, 132, 8129–8136 CrossRef CAS PubMed.
- J. Bao, J. He, Y. Zhang, Y. Yoneyama and N. Tsubaki, Angew. Chem., Int. Ed., 2008, 47, 353–356 CrossRef CAS PubMed.
- R. Barthos, F. Lonyi, G. Onyestyak and J. Valyon, J. Phys. Chem. B, 2000, 104, 7311–7319 CrossRef CAS.
- F. Lonyi and J. Valyon, Microporous Mesoporous Mater., 2001, 47, 293–301 CrossRef CAS.
- N. Katada, H. Igi, J. H. Kim and M. Niwa, J. Phys. Chem. B, 1997, 101, 5969–5977 CrossRef CAS.
- J. Zhang, W. Qian, C. Kong and F. Wei, ACS Catal., 2015, 5, 2982–2988 CrossRef CAS.
- Z. Zhu, Z. Xie, Q. Chen, D. Kong, W. Li, W. Yang and C. Li, Microporous Mesoporous Mater., 2007, 101, 169–175 CrossRef CAS.
- Y. Ouyang, Y. Luo and X. Shu, Pet. Process. Petrochem., 2009, 40, 22–25 CAS.
- M. Briend, R. Vomscheid, M. J. Peltre, P. P. Man and D. Barthomeuf, J. Phys. Chem., 1995, 99, 8270–8276 CrossRef CAS.
- A. Buchholz, W. Wang, A. Arnold, M. Xu and M. Hunger, Microporous Mesoporous Mater., 2003, 57, 157–168 CrossRef CAS.
- G. A. V. Martins, G. Berlier, S. Coluccia, H. O. Pastore, G. B. Superti, G. Gatti and L. Marchese, J. Phys. Chem. C, 2007, 111, 330–339 CAS.
- G. Sastre, D. W. Lewis and C. R. A. Catlow, J. Phys. Chem. B, 1997, 101, 5249–5262 CrossRef CAS.
- G. Liu, P. Tian, J. Li, D. Zhang, F. Zhou and Z. Liu, Microporous Mesoporous Mater., 2008, 111, 143–149 CrossRef CAS.
- D. Chen, D. Yang, Q. Wang and Z. Y. Jiang, Ind. Eng. Chem. Res., 2006, 45, 4110–4116 CrossRef CAS.
- D. Chen, K. Moljord and A. Holmen, Microporous Mesoporous Mater., 2012, 164, 239–250 CrossRef CAS.
- Y. Cui, Y. Wang and F. Wei, CIESC J., 2015, 66, 2982–2989 CAS.
| This journal is © The Royal Society of Chemistry 2017 |