
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxygen reduction on zirconium-stabilized-PdO surfaces: a first-principles study
Xiaofeng Yanga,
Zongbao Lib,
Xinyu Lia,
Ao Wanga,
Lichao Jia *a,
Bo Chia,
Jian Pua and
Jian Lia
*a,
Bo Chia,
Jian Pua and
Jian Lia
aCenter for Fuel Cell Innovation, School of Materials Science and Engineering, State Key Lab of Material Processing and Die & Mould Technology, Huazhong University of Science and Technology, Wuhan, 430074, China. E-mail: jialc@hust.edu.cn
bSchool of Material and Chemical Engineering, Tongren University, Guizhou 554300, China
First published on 22nd August 2017
Abstract
The oxygen reduction reaction properties on PdO and Zr-doped PdO surfaces, and the relative stability of the concerned surfaces, have been studied by first-principles calculations. The results demonstrate that the doped Zr element plays an important role in the stabilization of the PdO surfaces and the enhancement of the oxygen reduction activities on these surfaces. The Zr atoms prefer to replace the cus-Pd atoms on the PdO (101) surface, which is more stable than the Zr doped PdO (001) surface. The interaction between Pd and Zr could strengthen the hybridization between the adsorbed O 2p and Pd 4d states, thus lower the adsorption energy, improving the O2 adsorption performance. The adsorbed O2 molecules tend to be dissociated on the PdO (101) surface. It is also discovered that the increase of Zr doping concentration can also enhance the O2 adsorption properties and facilitate the subsequent dissociation.
1. Introduction
Solid oxide fuel cells (SOFCs) are a type of electrochemical device that convert chemical energy stored in hydrogen, carbon monoxide and hydrocarbon fuels into electrical power, in an efficient and environmentally friendly manner. In order to meet the requirements of commercialization in terms of cost and stability, the operating temperature should be below 850 °C. For intermediate and low temperature SOFCs, the performance of the cathode material is a critical bottle-neck issue, which restricts the development of SOFC technology.Sr-doped LaMnO3/yttria-stabilized zirconia (LSM/YSZ) based cermets are the most common cathodes in high-temperature SOFCs because of their high electrical conductivity, structural stability and high electrocatalytic activity for the oxygen reduction reaction (ORR).1–4 However, due to the negligibly low oxygen ion conductivity of LSM, its catalytic activity reduces significantly in the intermediate temperature range.5,6 Many efforts have been made to improve the electrochemical activity of the LSM cathode, such as compositing with mixed ionic–electronic conductors and modifying by precious metals.3,7
It has been proved that PdO is a promising modifier to enhance the electrochemical performance of the LSM-based cathodes by improving the oxygen adsorption and diffusion for intermediate temperature SOFCs (IT-SOFCs) in previous studies.8–11 Nevertheless, a significant growth and agglomeration of the infiltrated nano-sized PdO particles was observed under the conditions of high operating temperature and long-term cathodic current polarization,9,12 which probably due to the decomposition of PdO oxide to Pd metal.13,14 This poor stability of the nano-sized PdO particles during the polarization limits its application in IT-SOFCs. In previous study,15 it has been proved that adding 20 mol% of Zr element into PdO can significantly enhance its thermal stability, which finally shows no particles growth under the cathodic condition. In the meanwhile, our experimental results show that the catalytic activity of Zr-doped-PdO (PZO) is promoted. However, the enhancement mechanism of the adding Zr element remains to be studied. With the development of computer and computational theory, the first-principles calculations have been widely used in many different fields of materials science, especially in the study of SOFC electrodes.16,17 In the present work, we investigate the mechanism of the enhanced stability and catalytic activity of PZO based on the density functional theory (DFT) calculation.18
2. Computational methods
The first-principles calculations reported herein are implemented on the basis of spin polarized DFT and were carried out in the generalized gradient approximation (GGA-PBE),19,20 as performed in the Vienna Ab initio Simulation Package (VASP)21,22 and the projector augmented wave (PAW)23,24 approach to the core–valence interactions. The valence configurations used in this calculation are: 4d25s2 for Zr, 4d95s1 for Pd, and 2s22p4 for O. The optimization is carried out under the convergence criterion that a maximum residual force on any atom should be less than 0.02 eV Å−1. An energy cutoff of 400 eV is applied for the surfacewave expansion. Accurate Brillouin zone sampling is carried out in the Monkhorst–Pack lattice of 4 × 4 × 1.PdO has a tetragonal structure of P42/mmc.25 The calculated lattice constant for bulk PdO is a = 3.07 Å and c = 5.41 Å, which is substantially identical to previous theoretical study (a = 3.04, b = 5.34)25 and also in accordance with the experimental value of a = 3.05 Å and c = 5.50 Å.26 It has been theoretically and experimentally proved that the (101) surface of PdO is the most stable and optimal orientation surface,26 and adding 20 mol% Zr into PdO does not change its crystal structure.15 It is reasonable to select the PdO (101) surface as the optimal orientation surface for the ORR simulation. Furthermore, it is well known from the XRD standard card that the peaks of (101) and (001) surface of PdO are almost coincident. Thus, in this study, the (001) surface is also considered as well as the (101) surface.
The PdO (101) surface is characterized by a 7-atomic layer slab of the (101) plane (Fig. 1(a)), the slabs are separate by a 15 Å thick vacuum layer to prevent the interaction between the two surfaces. Supercell with periodicity (1 × 3) is employed to simulate the O2 adsorption on the surface. One Pd atom of each plane of were replaced by a Zr atom in two different ways, which are indicated by Pd5/6Zr1/6O (101) A and Pd5/6Zr1/6O (101) B, respectively. For the PdO (001) surface, the supercell slab that consists of a 3 × 3 surface unit cell with 6 atomic layers, each of which contains 9 atoms, is constructed. A vacuum layer of 20 Å is also introduced into this structure (see Fig. 1(d)). One or two Pd atoms of each plane of such surfaces are replaced by the Zr atoms to modelt Pd8/9Zr1/9O (001) and Pd7/9Zr2/9O (001) surfaces. During the geometry optimization, the bottom two layers were fixed at corresponding bulk positions, the upper layers and the absorbate are fully relaxed.
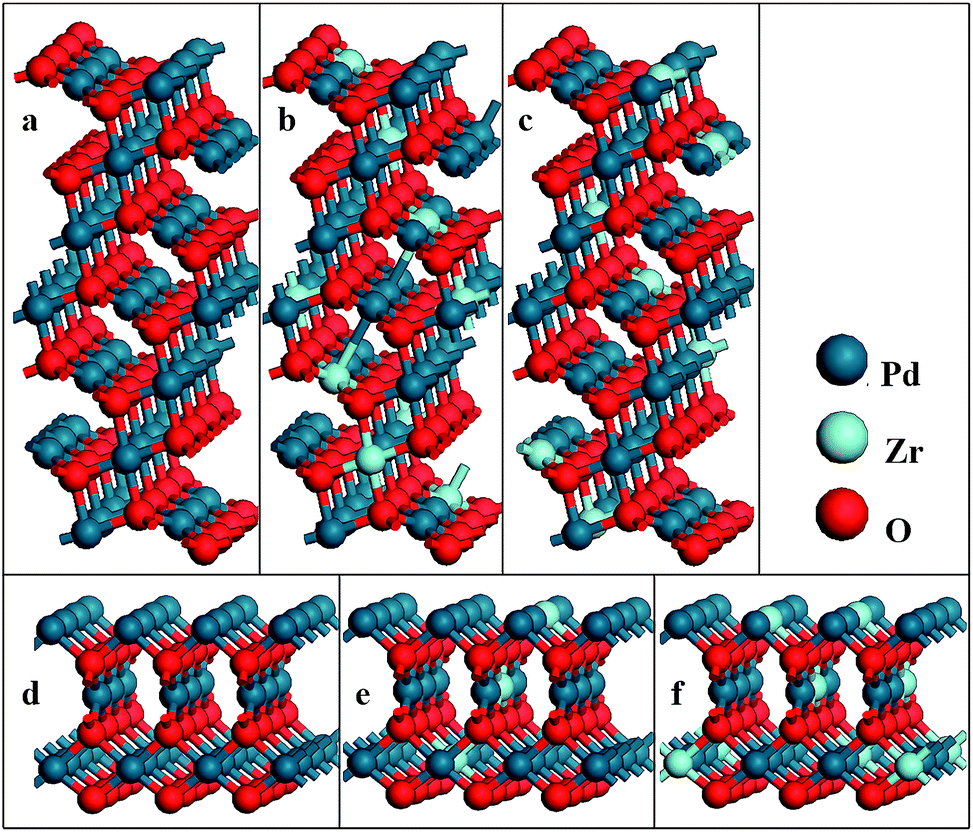 | ||
| Fig. 1 Models of (a) PdO (101); (b) Pd5/6Zr1/6O (101) A; (c) Pd5/6Zr1/6O (101) B; (d) PdO (001); (e) Pd8/9Zr1/9O (001) and (f) Pd7/9Zr2/9O (001) surfaces. | ||
The oxygen adsorption on the cathode surface is the first step and also a critical step during the cathode reaction process.27 The adsorption energy (Eads) of O2 is a criterion to determine the difficulty and stability of the adsorption, which is defined by
| Eads = Etotal − Eslab − EO2, | (1) |
3. Results and discussion
3.1 Adsorption of oxygen molecule on PdO (101) and PZO (101) surfaces
The stoichiometric PdO (101) surface consists of alternating lines of threefold-coordinated (cus) unsaturated and fourfold-coordinated (4f) saturated Pd and O atoms28 as shown in Fig. 2 In this study, we simulate the adsorption of oxygen molecule on these 4 kinds of adsorption sites. According to the results of structural relaxation, Fig. 3 shows the configuration of the PdO after adsorbing a O2 on these adsorption sites, respectively. The result shows that oxygen adsorption can be achieved only on the 4f O and cus Pd site. However, the adsorbed oxygen on the two adsorption site is ultimately bonded with the cus Pd atom. The detailed structural parameters of oxygen adsorption achieved on the 4f O and cus Pd two adsorption sites has been compared in the Fig. 4, it is obvious that the two structures are approximately identical. Consequently, it is found that the oxygen molecule can only be adsorbed on the cus-Pd site of PdO (101) surface. The calculated adsorption energies and structural parameters for PdO (101) surface are listed in Table 1. The negative values here imply that the adsorption process is an exothermic reaction. Therefore, the more negative the Eads, the more stable the adsorbed structure. Bader charge analysis indicates that the configurations on the Pd5/6Zr1/6O (101) B surface correspond to a superoxide (O2−) state.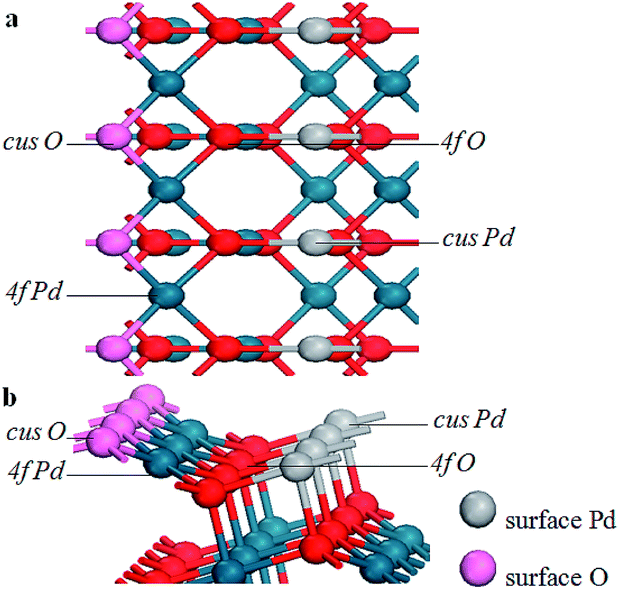 | ||
| Fig. 2 The PdO (101) surface comprised of cus-Pd, cus-O, 4f-Pd, and 4f-O atoms. (a) Top view and (b) side view. | ||
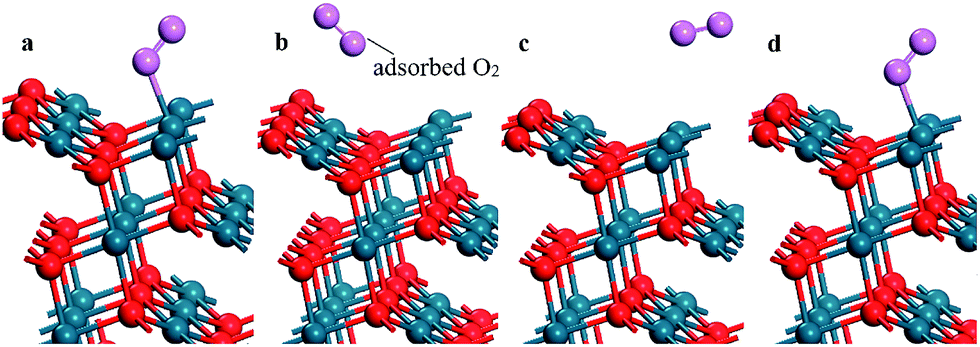 | ||
| Fig. 3 The configuration of the PdO after adsorbing a O2 on the (a) 4f O; (b) 4f Pd; (c) cus O and (d) cus Pd site, respectively. | ||
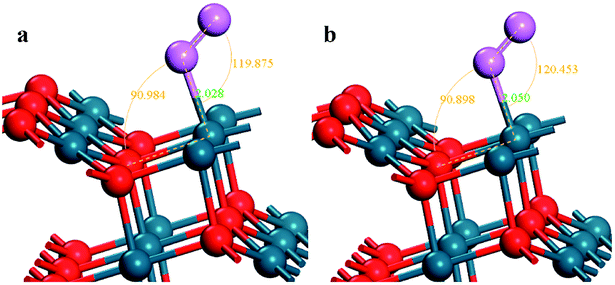 | ||
| Fig. 4 The detailed structural parameters of the O2 adsorption achieved on the (a) 4f O and (b) cus Pd site, respectively. | ||
| Site | Eads (eV) | dO–Pd (Å) | dO–Zr (Å) | dO–O (Å) | Charges (e) | |
|---|---|---|---|---|---|---|
| PdO (101) | cus-Pd | −0.552 | 2.050 | — | 1.195 | 0.197 |
| Pd5/6Zr1/6O (101) A | cus-Pd | −0.523 | 2.145 | — | 1.189 | 0.165 |
| Pd5/6Zr1/6O (101) B | cus-Pd | −1.003 | 2.011 | — | 1.279 | 0.627 |
| cus-Zr | −0.812 | 2.159 | 2.166 | 1.313 | 0.345 |
Since only the cus-Pd site is the stable adsorption site on the PdO (101) surface, we construct two different structures to model the Pd5/6Zr1/6O (101). In the structure of Pd5/6Zr1/6O (101) A, a 4f-Pd atom of each layer is substituted by a Zr atom, while in Pd5/6Zr1/6O (101) B a cus-Pd atom is replaced. Therefore, there are only cus-Pd sites as the possible adsorption sites on the Pd5/6Zr1/6O (101) A surface and cus-Pd and cus-Zr sites on the Pd5/6Zr1/6O (101) B surface. The adsorption energies and structural parameters of ORR on PdO and Zr-doped-PdO (101) surfaces are summarized in Table 1. In the structure of Pd5/6Zr1/6O (101) A, the cus-Pd and the 4f-Zr atoms are too far from each other to make them interact weakly, so that the adsorption of oxygen releases less energy than the adsorption occurred on PdO (101) surface. On the contrary, on the Pd5/6Zr1/6O (101) B surface, the cus-Pd and the cus-Zr atoms are very closed which will affect the electronic structures of the surface atoms, leading to strong adsorptions of oxygen. Furthermore, the longer O–O bond indicates that the Pd5/6Zr1/6O (101) B surface is better for the subsequent dissociation of the adsorbed oxygen molecule.
In order to examine the relative difficulty for Zr element to incorporate into the surface structure, the defect formation energy (ΔEi) of these two kinds of structures are calculated by the equation:
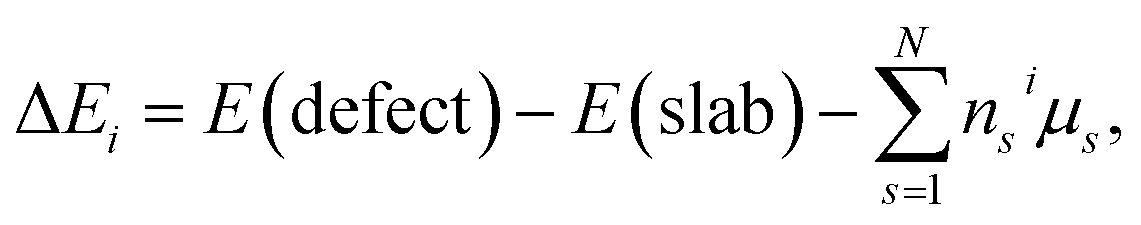 | (2) |
3.2 Adsorption of oxygen molecule on PdO (001) and PZO (001) surfaces
For the PdO (001) surface, four possible adsorption sites are considered in this study. Fig. 5(a) shows different O2 adsorption configurations, which are the single-coordinated atop Pd site (labeled as T), the twofold-coordinated “bridge” site which is above a lattice oxygen (OB), the twofold-coordinated “bridge” site below which is empty (EB), and the fourfold-coordinated “hollow” site (H).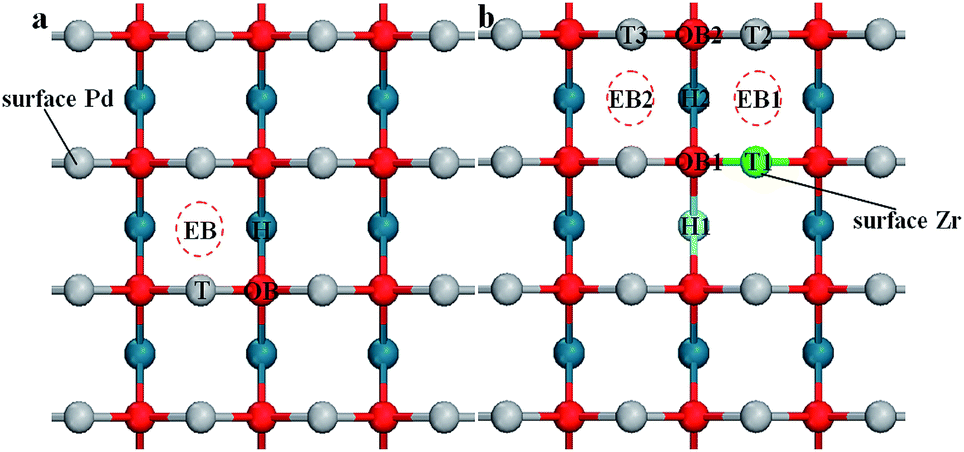 | ||
| Fig. 5 Top view of the adsorption sites of (a) PdO (001) surface and (b) Pd8/9Zr1/9O (001) surface. | ||
The adsorption energies can also be derived from eqn (1). Table 2 summarizes the predicted adsorption energies and structural parameters for these four adsorbed configurations. As with the adsorptions on the PdO (101) surface, adsorption of O2 on the PdO (001) surface is also exothermic. The Pd–O bond length of the T site is found to be shorter than those of the others, which means that the Pd–O bond in the “T” configuration is the strongest one. However, when the O2 molecule is adsorbed on the T site, it forms only one Pd–O bond to obtain a Pd–O–O structure instead of the Pd–O–Pd structure. While on the other adsorption sites, the Pd–O–Pd structure make the O2 molecule be adsorbed strongly on the surface, leading to lower adsorption energies. The adsorption on the hollow site shows lowest adsorption energy, indicating the most stable adsorption site, which can be attributed to its high-symmetry. After being adsorbed on the H site, the O2 molecule tends to be disassociated, the O–O bond length increases to 1.609 Å.
| Eads (eV) | dO–Pd (Å) | dO–O (Å) | Charges (e) | |
|---|---|---|---|---|
| T | −0.055 | 2.067 | 1.191 | 0.367 |
| OB | −1.540 | 2.118 | 1.211 | 0.479 |
| EB | −1.283 | 2.092 | 1.227 | 0.378 |
| H | −1.677 | 2.249 | 1.609 | 0.836 |
The doping element Zr will change the surface chemical environment of PdO, subsequently introducing more possible adsorption sites on the (001) surface of Zr-doped-PdO. As Fig. 5 shows, there are 3 kinds of T adsorption sites, 2 OB sites, 2 EB sites and 2H sites on the Pd8/9Zr1/9O (001) surface. The adsorption energies and structural parameters for O2 adsorption on different sites are summarized in Table 3. Here we focus on two factors: the adsorption energy determines the difficulty of the O2 adsorption, while the O–O bond length represents the ability for the following O2 dissociation. Comparing with Table 2, although the adsorption energies of adsorption on OB and EB sites of the Pd8/9Zr1/9O (001) surface are higher than those on the PdO (001) surface, the bond lengths become longer. Furthermore, the adsorptions on the H sites provide the lowest adsorption energies and the adsorption O2 molecules are completely dissociated. Thus we conjecture that the introduction of Zr element will promote the O2 adsorption (especially on the H sites) and dissociation. In addition, the Bader charge analysis reveals that the adsorbed oxygen molecule are O2− on the H site of PdO (001) surface, while they are dissociated O− on the H site of Pd8/9Zr1/9O (001) surface.
| Eads (eV) | dO–Pd (Å) | dO–Zr (Å) | dO–O (Å) | Charges (e) | |
|---|---|---|---|---|---|
| T1 | −0.893 | — | 2.047 | 1.216 | 0.544 |
| T2 | −0.809 | 2.023 | — | 1.204 | 0.303 |
| T3 | −0.072 | 2.084 | — | 1.192 | 0.318 |
| OB1 | −1.157 | 2.025 | 2.185 | 1.243 | 0.612 |
| OB2 | −1.057 | 2.082 | — | 1.251 | 0.543 |
| EB1 | −0.940 | 2.121 | 2.140 | 1.262 | 0.694 |
| EB2 | −0.710 | 2.104 | — | 1.228 | 0.422 |
| H1 | −2.376 | 2.101 | 2.050 | 3.455 | 2.100 |
| H2 | −6.554 | 2.070 | 2.064 | 3.247 | 1.888 |
In order to verify our inference, more Zr atoms are introduced into the PdO (001) surface to construct a Pd7/9Zr2/9O (001) surface structure, on which the O2 adsorptions are also simulated. Only the typical adsorption sites (T, OB, EB and H) are considered here, and the corresponding adsorption energies, structural parameters are listed in Table 4. The same conclusion can be reached since the adsorption energies are lower on the OB, EB, and H sites, and the adsorbed O2 molecules are ready to be dissociated. It is worth mentioning that the oxygen adsorbed on the H site of PZO surface is completely dissociated, with greatly enlarging of O–O distance from gaseous value of 1.209 to 3.455, 3.247 and 2.699 Å (Fig. 6). It is for this reason that oxygen adsorption on the H site can achieve such a low adsorption energy. These results can be proved by Bader charge analysis, which indicate that 1.980e charges have been transferred to the adsorbed dissociated oxygen ions on the OB and H sites of on the Pd7/9Zr2/9O (001) surface.
| Eads (eV) | dO–Pd (Å) | dO–Zr (Å) | dO–O (Å) | Charges (e) | |
|---|---|---|---|---|---|
| T | −0.900 | — | 2.081 | 1.221 | 0.575 |
| OB | −6.607 | 2.004 | 2.065 | 3.300 | 1.98 |
| EB | −1.347 | 2.080 | 2.103 | 1.299 | 0.742 |
| H | −5.437 | 2.044 | 2.135 | 2.699 | 1.852 |
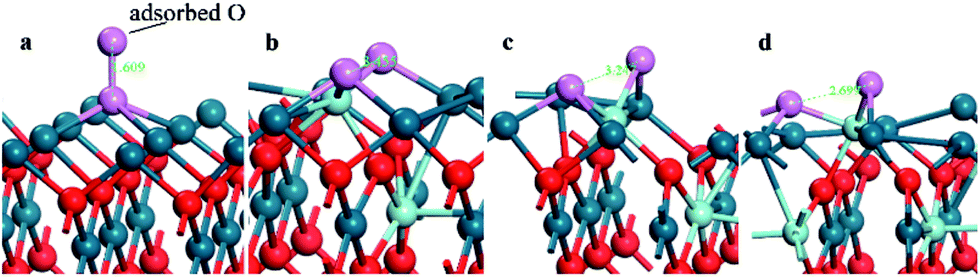 | ||
| Fig. 6 The configuration of the PdO after adsorbing a O2 on the (a) H site of PdO (001) surface; (b) H1 and (c) H2 site of Pd8/9Zr1/9O (001) surface and (d) H site of Pd7/9Zr2/9O (001). | ||
3.3 Relative stability of PdO and PZO surfaces
As with the adsorption and dissociation properties, the stability also is an important evaluation criterion of the cathode materials for SOFCs. In order to estimate the stability of the PdO and PZO surfaces considered in this study, the formation energies of the surface structures are calculated and compared in Table 5. Since the different structures have different atomic number, the formation energies per O atom (Eform) is employed to keep the comparability. The value of the Eform is defined by:| Eform = (Esubstrate − NPd × EPd-bulk − NZr × EZr-bulk − NO × EO-bulk)/NO, | (3) |
| Slab | Eform (kJ mol−1) |
|---|---|
| PdO (101) | −93.470 |
| Pd5/6Zr1/6O (101) A | −156.771 |
| Pd5/6Zr1/6O (101) B | −185.474 |
| PdO (001) | −8.075 |
| Pd8/9Zr1/9O (001) | −88.938 |
| Pd7/9Zr2/9O (001) | −164.388 |
As in shown in Table 5, for the (101) surfaces, the stability is in the order of Pd5/6Zr1/6O (101) B > Pd5/6Zr1/6O (101) A > PdO (101); and for the (001) surfaces, the order is Pd7/9Zr2/9O (001) > Pd8/9Zr1/9O (001) > PdO (001). Thus, a conclusion that the addition of Zr element will increase the stability of the PdO surfaces. Moreover, compared the results of (101) surfaces to those of (001) surfaces, it is found that the (101) surface is much more stable than (001) surface. Such corollary is in consistent with the previous result that PdO is in the form of PdO (101) surface under the working condition.
3.4 Electronic structures of PdO and PZO (101) surfaces
In order to explore the interaction between the adsorbed O2 and the surfaces, and the deep mechanism of the Zr improved ORR performance, partial densities of states (PDOS) calculations are carried out on the O2 molecular adsorbed PdO and PZO surfaces. Considering that the (101) surface is the stable surface under the operating condition and the O2 molecule could only be adsorbed on the cus-Pd (Zr) sites of the surface, PDOS of top-one-layer atoms and adsorbed oxygen atoms on PdO (101) and Pd5/6Zr1/6O (101) B surface are calculated and plotted in Fig. 7. As shown in Fig. 7(a), when the O2 molecule is adsorbed on the cus-Pd site of PdO (101) surface, hybridization between Pd 4d and adsorbed O 2p states can be found around −7.6 eV, indicating the bonding states. The overlapping means that there will be electronic exchange between the two atoms, and they are bonded by chemical bond. Thus, the bond length is smaller than the sum of the atomic radius. However, the anti-bonding states can also be found ranging from −1 to 0.5 eV, which are partly occupied by electrons. The occupied anti-bonding states can neutralize some bonding states, weakening the adsorption. For the Pd5/6Zr1/6O (101) B surface, as described previously, there are two kinds of adsorption sites: the cus-Pd and cus-Zr sites. When the adsorption occurs on the cus-Zr site (Fig. 7(b)), there also exist the bonding and anti-bonding states. The difference is that the Zr 4d states mainly locate in the conducting bands and the anti-bonding states are unoccupied, resulting in a lower adsorption energy compared to that on the PdO (101) surface. Due to the interactions between Pd and Zr, the Pd 4d states move towards to higher states. After adsorption, as shown in Fig. 7(c), the hybridization between O 2p states and Pd 4d states are around the energy level of 1.5 eV, and −7 to −5 eV. The splitting and broadening of peaks indicate a strong interaction between Pd and the adsorbed O atom, the charge transfer between Pd and the adsorbed O becomes easier, which facilitate the adsorption, leading to the lowest adsorption energy. The overlap center moves to high energy level, thus the Pd–O bond length is longer than that on the PdO (101) surface.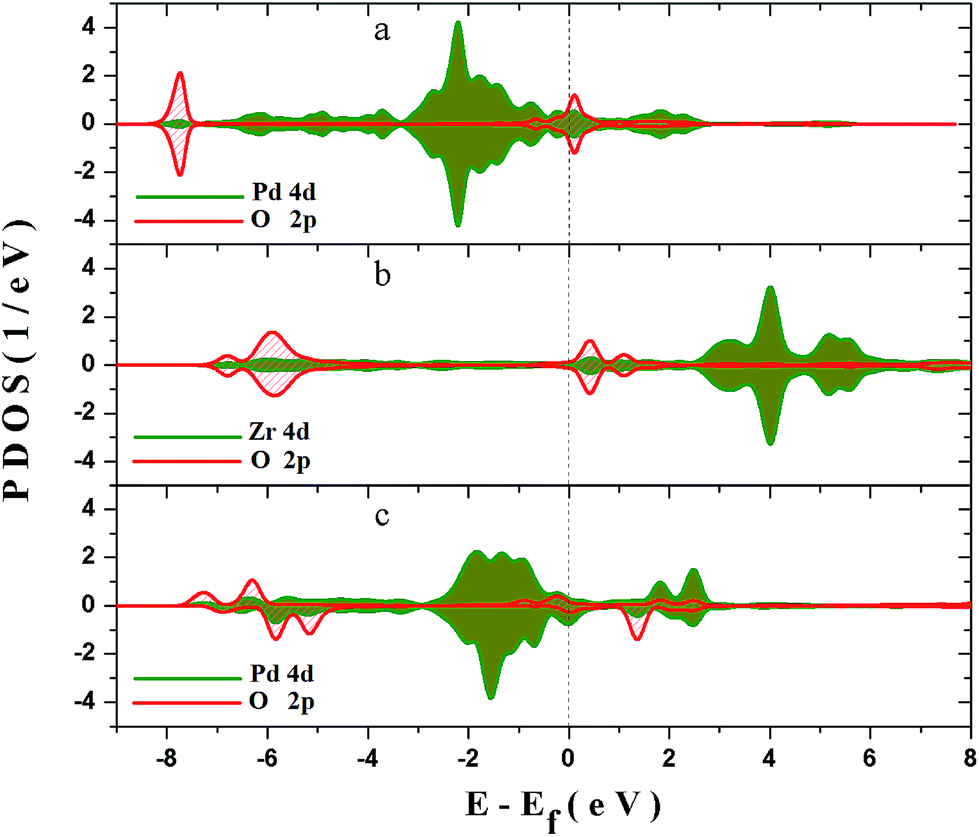 | ||
| Fig. 7 PDOS for different adsorption configurations: (a) O2 adsorbed on cus-Pd site of PdO (101) surface; (b) O2 adsorbed on cus-Zr site of Pd5/6Zr1/6O (101) B surface and (c) O2 adsorbed on cus-Pd site of Pd5/6Zr1/6O (101) B surface. | ||
The charge distribution and transfer are of great importance to understanding the interactions between atoms. Therefore, the difference electron density of the clean and O2 adsorbed Pd5/6Zr1/6O (101) B surfaces are calculated and shown in Fig. 8, those of the top two layers and the adsorbed O2 molecule are the main concerns in this study. Before adsorption, as represented in Fig. 8(a), it can be seen that there is a low electron-density region around the Zr atom. The electron density increases when the O2 molecule is adsorbed on the Zr atom, as is shown in Fig. 8(b), the electrons go into the antibonding orbital of the O2 molecule, which promotes the cleavage of the O2 molecule. Fig. 8(c) shows a high electron-density region between adsorbed O and surface Pd atom, which shows that there is a stronger orbital hybridization between the two atoms. The hybridized electronic orbitals act as a bridge to make the transmission of electrons easier. From the comparison of Fig. 8(b) and (c), it is found that the O atom adsorbed on the cus-Pd site receives more electrons than that on the cus-Zr site, indicating that the interaction between O and Pd is stronger than the Zr case. This is a good explanation for the predicted adsorption results.
 | ||
| Fig. 8 The section patterns for difference electron density of (a) Pd5/6Zr1/6O (101) B surface; (b) Pd5/6Zr1/6O (101) B surface with adsorbed O2 on cus-Zr and (c) Pd5/6Zr1/6O (101) B surface with adsorbed O2 on cus-Pd. | ||
4. Conclusions
In summary, first-principles calculations have been performed to investigate the stability of the Zr doped PdO surfaces and the O2 adsorption properties on which. The following conclusions can be made:(1) The incorporation of Zr can stabilize the PdO (101) and (001) surfaces, and the doped (101) surface is more stable than the (001) surface. Zr atom prefers to substitute for the cus-Pd atom on the (101) surface.
(2) The lowest O2 adsorption energies on the PdO (101) and the Zr-doped PdO (101) surfaces are determined to be −0.552 eV and −1.003 eV respectively. The lower adsorption energy indicates that the addition of Zr makes the adsorption on cus-Pd site easier and more stable than the pure surface, thereby improving the ORR performance.
(3) The adsorption results on the Zr-doped PdO (001) surfaces suggest that these surfaces are suitable for adsorbing oxygen, and oxygen molecules tend to dissociate in some sites. Besides, the increase of Zr doping concentration will enhance the O2 adsorption properties and facilitate the subsequent dissociation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is financially supported by the National Natural Science Foundation of China (51502103, 51472099), Guangdong Province (2013B090500051) and Shandong Province (2015ZDXX0602A02).References
- S. P. Jiang, Y. J. Leng, S. H. Chan and K. A. Khor, Electrochem. Solid-State Lett., 2003, 6, A67, DOI:/10.1149/1.1558351.
- M. K. Jeon, C. H. Lee, G. I. Park and K. H. Kang, J. Power Sources, 2012, 216, 400, DOI:/10.1016/j.jpowsour.2012.05.107.
- P. Qiu, A. Wang, J. Li, Z. B. Li, L. C. Jia, B. Chi, J. Pu and J. Li, J. Power Sources, 2016, 327, 408, DOI:/10.1016/j.jpowsour.2016.07.074.
- L. C. Jia, K. Li, D. Yan, X. Wang, B. Chi, J. Pu, J. Li and S. L. Yuan, RSC Adv., 2015, 5, 7761, DOI:/10.1039/c4ra08705d.
- Y. Jiang, S. Z. Wang, Y. H. Zhang, J. W. Yan and W. Z. Li, Solid State Ionics, 1998, 110, 111, DOI:/10.1016/S0167-2738(98)00111-8.
- Y. Sakaki, Y. Takeda, A. Kato, N. Imanishi, O. Yamamoto, M. Hattori, M. Iio and Y. Esaki, Solid State Ionics, 1999, 118, 187, DOI:/10.1016/S0167-2738(98)00440-8.
- F. L. Liang, J. Chen, S. P. Jiang, B. Chi, J. Pu and J. Li, Electrochem. Commun., 2009, 11, 1048, DOI:/10.1016/j.elecom.2009.03.009.
- F. L. Liang, J. Chen, S. P. Jiang, B. Chi, J. Pu and J. Li, Electrochem. Solid-State Lett., 2008, 11, B213, DOI:/10.1149/1.2987693.
- N. Q. Duan, D. Yan, B. Chi, J. Pu and J. Li, Sci. Rep., 2015, 5, 8174, DOI:/10.1038/srep08174.
- V. A. C. Haanappel, A. Mai and J. Mertens, Solid State Ionics, 2006, 177, 2033, DOI:/10.1016/j.ssi.2005.12.038.
- J. Chen, F. L. Liang, L. N. Liu, S. P. Jiang, B. Chi, J. Pu and J. Li, J. Power Sources, 2008, 183, 586, DOI:/10.1016/j.jpowsour.2008.05.082.
- F. L. Liang, J. Chen, S. P. Jiang, F. Z. Wang, B. Chi, J. Pu and J. Li, Fuel Cells, 2009, 9, 636, DOI:/10.1002/fuce.200800078.
- F. Wang, A. Wang, J. Chen, B. Chi, J. Pu and J. Li, Int. J. Hydrogen Energy, 2014, 39, 14421, DOI:/10.1016/j.ijhydene.2014.02.051.
- F. Liang, J. Chen, B. Chi, J. Pu, S. P. Jiang and J. Li, J. Power Sources, 2011, 196, 153, DOI:/10.1016/j.jpowsour.2010.07.011.
- H. Zhang, J. Gromek, G. Fernando and H. L. Marcus, Mater. Sci. Eng., A, 2004, 366, 248, DOI:/10.1016/j.msea.2003.07.004.
- L. C. Jia, X. Wang, W. L. Li, K. Li, B. Chi, J. Pu, J. Li and S. L. Yuan, J. Power Sources, 2014, 253, 138, DOI:/10.1016/j.jpowsour.2013.10.146.
- M. L. Liu, M. E. Lynch, K. Blinn, F. M. Alamgir and Y. M. Choi, Mater. Today, 2011, 14, 534, DOI:/10.1016/S1369-7021(11)70279-6.
- D. S. Sholl and J. A. Steckel, Density Functional Theory: A Practical Introduction, John Wiley & Sons, Inc., Hoboken, NJ, 2009 Search PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhai, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671, DOI:/10.1103/PhysRevB.46.6671.
- J. P. Perdew, K. Burke and M. Emzerhof, Phys. Rev. Lett., 1996, 77, 3865, DOI:/10.1103/PhysRevLett.77.3865.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169, DOI:/10.1103/PhysRevB.54.11169.
- S. Stolbov, M. A. Ortigoza and T. S. Rahman, J. Phys.: Condens. Matter, 2009, 21, 474226, DOI:/10.1088/0953-8984/21/47/474226.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758, DOI:/10.1103/PhysRevB.59.1758.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953, DOI:/10.1103/PhysRevB.50.17953.
- D. B. Rogers, R. D. Shannon and J. L. Gillson, J. Solid State Chem., 1971, 3, 314, DOI:/10.1016/0022-4596(71)90045-4.
- J. Rogal, K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 075421, DOI:/10.1103/PhysRevB.69.075421.
- H. C. Ham, G. S. Hwang, J. Han, S. P. Yoon, S. W. Nam and T. H. Lim, Catal. Today, 2016, 263, 11, DOI:/10.1016/j.cattod.2015.07.054.
- A. Antony, A. Asthagiri and J. F. Weaver, J. Chem. Phys., 2013, 139, 2172, DOI:/10.1063/1.4819909.
| This journal is © The Royal Society of Chemistry 2017 |