
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ti(Phen)(OC2H5)2Cl2: a highly efficient pre-catalyst for selective oxidation of organic sulfides to sulfoxides by hydrogen peroxide†
Rong-Hui Wuab,
Jing Wua,
Ming-Xin Yua and
Long-Guan Zhu *a
*a
aDepartment of Chemistry, Zhejiang University, Hangzhou 310027, People's Republic of China. E-mail: chezlg@zju.edu.cn
bKey Laboratory of Chemical Utilization of Forestry Biomass of Zhejiang Province, Zhejiang A & F University, Lin'an, Zhejiang Province 311300, China
First published on 14th September 2017
Abstract
A newly developed and structurally characterized titanium complex [Ti(Phen)(OC2H5)2Cl2] (Phen = 1,10-phenanthroline) is reported as a homogeneous pre-catalyst for the selective oxidation of organic sulfides to sulfoxides with a stoichiometric amount of 30% aq. H2O2 in CH3OH at room temperature. This catalytic system showed high yield (95–100%) and good selectivity (92–100%) during the catalytic reaction in a short period of time.
Introduction
Catalytic oxygenation of organic sulfides has attracted great interest during the past decade for its final products, sulfoxides and sulfones, which are important intermediates for the synthesis of chemically and biologically significant molecules1 and also in the process of desulfurization of fuels.2 Among different sulfide oxidation reactions, controlling the reaction of organic sulfides to sulfoxides over the fully oxidized sulfones is an important challenge for their potential applications as chiral auxiliaries in organic synthesis and as important functionalities in many biologically active molecules.3 As a consequence, new methods for the formation of sulfoxides have been developed for chemoselective oxidation of sulfides.For the oxidation of sulfides to sulfones, a very wide variety of oxidizing reagents and catalysts have been investigated. Oxidants such as organic peracids, inorganic oxides, oxygen and hydrogen peroxide have been used for the oxidation of sulfides to sulfoxides.4 Among these oxidants, hydrogen peroxide has been widely used since it is readily available, non-toxic and cheap, and forms water as the sole by-product.5 But in the absence of catalyst, the oxidation of sulfides to sulfoxides employing aqueous hydrogen peroxide is very slow, especially at room temperature. Accordingly, aqueous hydrogen peroxide as the terminal oxidant and a Lewis acid as an activator have been developed to promote this transformation.
Over the past few decades, some transition-metal catalysts such as vanadium,6 iron,7 manganese8 and titanium have been diffusely used to catalyze the selective oxidation of sulfides to sulfoxides in the presence of hydrogen peroxide. Among these catalytic systems, titanium complexes have proved themselves as very successful catalysts to activate hydrogen peroxide in sulfoxide syntheses, due to their cheapness and low toxicity, inertness to redox processes, as well as high activity and selectivity even using equimolecular amounts of H2O2.9 Thus, titanium complexes with different ligands have been used as catalysts to promote oxidation reaction of organic sulphides. Kagan10 and Modena11 have initially reported highly enantioselective sulfoxidation by using titanium–tartrate complexes modified with H2O and tartrate as the catalyst. Then, various titanium complexes with different types of ligands such as diol,12 binol,13 salen14 have been used for this type of oxidation. It is noticeable that many of the reported examples of titanium mediated sulfoxidation reactions were performed under “Ligand Assisted Catalysis (LAC)” conditions in situ generation of the catalyst in the reaction mixture. Consequently, a few of structurally characterized titanium complexes15 have been developed for the sulfoxidation reaction in recent years.
Herein, we present a mononuclear titanium(IV) complex Ti(Phen)(OC2H5)2Cl2 (1), and describe its synthesis, characterization, and as an effective homogeneous pre-catalyst for the selective oxidation of sulfides at ambient temperature in a short period of time.
Results and discussion
The preparation of complex 1 was directly finished in the mixed solution. The reaction of titanium(IV) chloride with 1,10-phenanthroline in ethanol led to the clear solution, and then about two days later, colourless block-shaped single crystals were isolated at room temperature. The single crystal X-ray diffraction of complex 1 proved that it is a hexa-coordinated mononuclear titanium(IV) species with space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The ORTEP diagram of the pre-catalyst shows distorted octahedral geometry (Fig. 1) with two chlorine groups occupying the apical positions and the 1,10-phenanthroline N atoms and two ethoxy group O atoms in the equatorial sites.
. The ORTEP diagram of the pre-catalyst shows distorted octahedral geometry (Fig. 1) with two chlorine groups occupying the apical positions and the 1,10-phenanthroline N atoms and two ethoxy group O atoms in the equatorial sites.
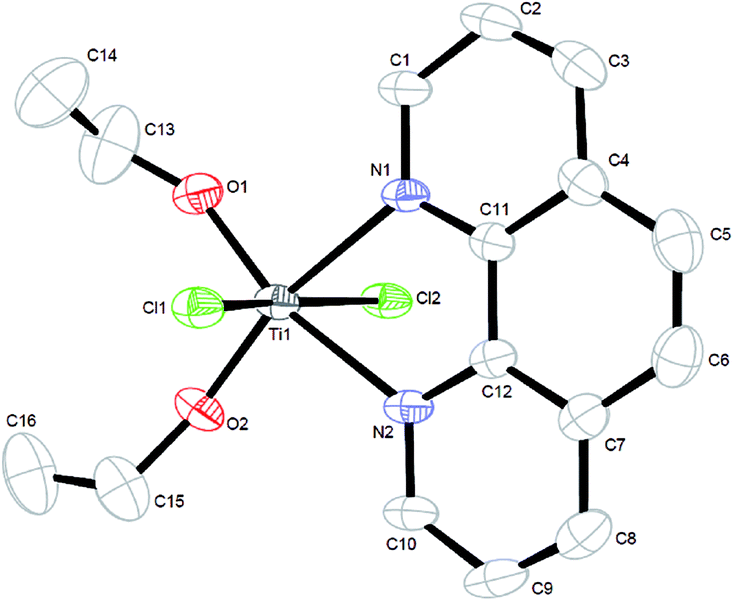 | ||
| Fig. 1 ORTEP drawing of Ti(Phen)(OC2H5)2Cl2. The H atoms are omitted for clarity. | ||
To further study the purity and repeatability of the crystals, PXRD experiments were carried out for complex 1. The PXRD experimental and computer simulated patterns of the corresponding complex are shown in ESI data (Fig. S1 and S2, ESI†). They showed that the synthesized bulk materials and the measured single crystals are same. The thermal gravimetric analyses (TGA) of complex 1 were carried out at the rate of 10 °C min−1 under an inert (N2) atmosphere (Fig. 2). The TGA curve of complex 1 shows a gradual weight-loss step of 22.96% in the temperature range of 25–155 °C, corresponding to the escape of two oxyethyl groups from the framework (calcd 23.14%). By further increasing the temperature, a successive decomposition step occurs and the framework of complex 1 was destroyed. Other characterization data and spectra can be found in ESI data.†
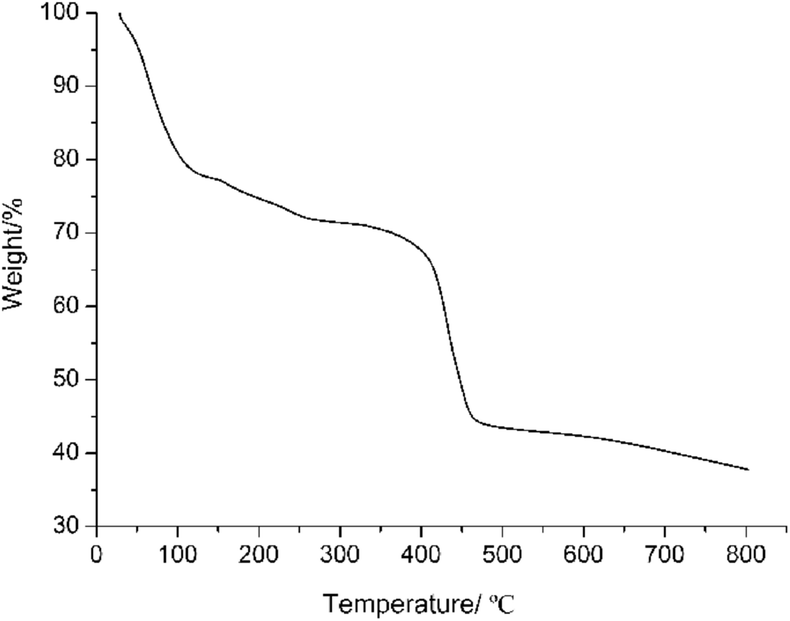 | ||
| Fig. 2 TGA curve of compound 1. | ||
Our interest in oxidation inspired us to use this complex as a catalyst for sulfide oxidation. In order to achieve optimum conditions, we performed a set of preliminary experiments on methyl phenyl sulfide (MPS) as a model substrate using 30% aqueous hydrogen peroxide in the presence of a catalytic amount of complex 1 at room temperature. The reaction was initially carried out in several solvents (Table 1, entries 1–7) using 0.0025 mmol of pre-catalyst. Interestingly, both methanol and ethanol are found to be highly effective as the solvents, affording high selectivity and conversion in the presence of the pre-catalyst (Table 1, entries 2–3). We also performed the oxidation of several other sulfides in methanol and ethanol maintaining the same conditions (Table S1, ESI†). The results showed methanol was better than ethanol in the oxidation of the other sulfides and therefore, methanol was chosen as solvent in the following experiments. The attention was then turned on to the stoichiometric ratios for the oxidizing agent. 1 eq. of H2O2 was found most useful for maximum conversion of the substrates. A higher amount of an oxidizing agent led to sulfone formation (Fig. S3, ESI†). Furthermore, we carried out the oxidation with different amount of the pre-catalyst under the same conditions (Table 1, entries 2 and 8–11). As the pre-catalyst amount increased from 1 × 10−4 mmol to 2.5 × 10−3 mmol, the conversion and selectivity of the probe reaction increased from 6% to 99% and 54% to 98%, respectively. A further increase in the pre-catalyst amount led to an insignificant effect on the yield of the desired product. Moreover, in a control experiment conducted in the absence of the pre-catalyst, very little conversion of methyl phenyl sulfide to sulfoxide or sulfone was observed under analogous conditions (Table 1, entry 12). Further screening with 0.0025 mmol of Ti(OPi)4 was also carried out, but none of them provided the product in a yield as high as that obtained in the presence of complex 1 (Table 1, entries 13–14). Lastly, the stability and reuse of the pre-catalyst was investigated by adding to the reaction mixture new charges of substrate and oxidant in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Interestingly, the pre-catalyst was reused for up to 8 cycles without significant loss in its activity and selectivity (Fig. S4, ESI†). Thus, complex 1 plays a crucial role in facilitating the partial oxidation reactions.
:1. Interestingly, the pre-catalyst was reused for up to 8 cycles without significant loss in its activity and selectivity (Fig. S4, ESI†). Thus, complex 1 plays a crucial role in facilitating the partial oxidation reactions.
| Entry | Solvent | Catalyst (amount of catalyst) | Conv.b (%) | Sele.c (%) | |
|---|---|---|---|---|---|
| Sulfoxide | Sulfone | ||||
| a Reaction conditions: 1.00 mmol of methyl phenyl sulfide, 1.00 mmol of 30% H2O2, 1 × 10−2 to 1 × 10−4 mmol Ti catalyst, in the special solvent (5 mL) at 25 °C (20 min). Products were quantified by GC analysis and characterized by the standard substance.b The GC yields (%) are measured relative to the starting sulphide.c Selectivity to sulfoxide = (sulfoxide/(sulfoxide + sulfone)) × 100. | |||||
| 1 | Water | Complex 1 (2.5 × 10−3 mmol) | 100 | 80 | 20 |
| 2 | Methanol | 99 | 98 | 2 | |
| 3 | Ethanol | 99 | 98 | 2 | |
| 4 | Acetonitrile | 23 | 89 | 11 | |
| 5 | Acetone | 14 | 82 | 18 | |
| 6 | Tetrahydrofuran | 28 | 82 | 18 | |
| 7 | Dichloromethane | 11 | 95 | 5 | |
| 8 | Methanol | Complex 1 (1 × 10−4 mmol) | 6 | 54 | 46 |
| 9 | Methanol | Complex 1 (1 × 10−3 mmol) | 53 | 96 | 4 |
| 10 | Methanol | Complex 1 (5 × 10−3 mmol) | 99 | 97 | 3 |
| 11 | Methanol | Complex 1 (1 × 10−2 mmol) | 99 | 96 | 4 |
| 12 | Methanol | — | 8 | 81 | 19 |
| 13 | Methanol | Ti(OPi)4 (2.5 × 10−3 mmol) | 33 | 88 | 12 |
| 14 | Methanol | Ti(OPi)4 (2.5 × 10−3 mmol + 2.5 × 10−3 mmol phen) | 37 | 87 | 13 |
To study the scope of this procedure, a series of structurally diverse sulfides with different electronic and steric effects were then examined under the optimized reaction conditions. The reactions went on well affording high yields (95–99%) and excellent sulfoxide selectivity (92–100%) under the optimum conditions. The results are presented in Table 2. As it can be seen aliphatic sulfides undergo oxidation reactions more easily than aromatic substrates. It is notable that sulfides were chemoselectively oxidized in the presence of some oxidation prone functional groups such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and –OH (Table 2, entries 3 and 5). However, the oxidation of aryl sulfides containing electron-withdrawing groups (Table 2, entries 5–8) was sluggish producing the corresponding sulfoxides, which may be due to the presence of electron-withdrawing groups reduces the electron density on the sulfur atom thereby decreasing its nucleophilicity. Regarding sulfoxidation of benzyl phenyl sulfide, diphenylsulfide and dibenzothiophene, due to their large steric hindrance, a drastic decrease of the catalytic activity is observed with excellent selectivity to the sulfoxides (Table 2, entries 9–11). Quite significantly, catalytic activity of diphenylsulfide is greatly inferior to that of benzyl phenyl sulfide, which is probably due to the influence of conjugation effect.
C and –OH (Table 2, entries 3 and 5). However, the oxidation of aryl sulfides containing electron-withdrawing groups (Table 2, entries 5–8) was sluggish producing the corresponding sulfoxides, which may be due to the presence of electron-withdrawing groups reduces the electron density on the sulfur atom thereby decreasing its nucleophilicity. Regarding sulfoxidation of benzyl phenyl sulfide, diphenylsulfide and dibenzothiophene, due to their large steric hindrance, a drastic decrease of the catalytic activity is observed with excellent selectivity to the sulfoxides (Table 2, entries 9–11). Quite significantly, catalytic activity of diphenylsulfide is greatly inferior to that of benzyl phenyl sulfide, which is probably due to the influence of conjugation effect.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Time | Conv.b (%) | Sele.b,c (%) | TOFd | |
| Sulfoxide | Sulfone | |||||
| a Reaction conditions: sulfide:30% H2O2:catalyst = 1:1:0.0025 mmol; methanol: 5 mL, 25 °C.b Conversion and selectivity were determined by GC.c Products were characterized by GC-MS.d TOF (product mol × catalyst mol−1 × h−1). |
||||||
| 1 |  |
15 min | 97 | 99 | 1 | 1544 |
| 2 |  |
15 min | 100 | 99 | 1 | 1598 |
| 3 | 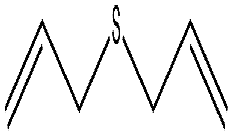 |
15 min | 98 | 98 | 2 | 1574 |
| 4 | 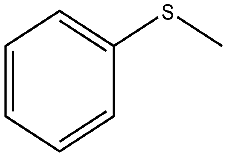 |
20 min | 99 | 98 | 2 | 1182 |
| 5 | 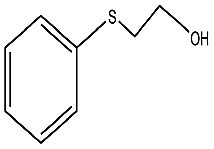 |
50 min | 96 | 99 | 1 | 459 |
| 6 | 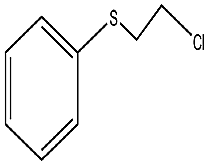 |
65 min | 99 | 97 | 3 | 364 |
| 7 | 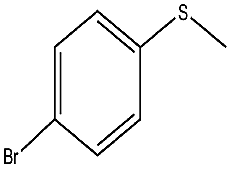 |
30 min | 98 | 98 | 2 | 787 |
| 8 | 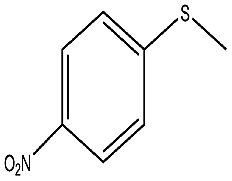 |
145 min | 98 | 95 | 5 | 163 |
| 9 | 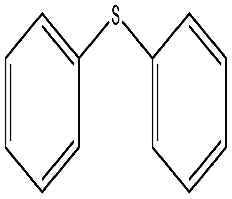 |
360 min | 98 | 92 | 8 | 65 |
| 10 | 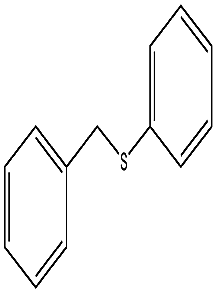 |
45 min | 98 | 96 | 4 | 524 |
| 11 |  |
24 h | 95 | 100 | 0 | 12 |
In order to obtain more insight on the nature of titanium-based oxidant species, the thianthrene 5-oxide was used as a mechanistic probe for the sulfoxidation reactions (Table 3, entries 1–2). The results show that thianthrene 5-oxide was predominantly oxidized at the sulfide for complex 1/H2O2 catalytic system. Meanwhile, complex 1/H2O2 shows higher activity than Ti(OPi)4/H2O2 under same conditions. We also performed the competitive experiment by putting together p-chlorophenyl methyl sulfide and methyl phenyl sulfoxide one against the other to compete for the titanium complex (Table 3, entries 3–4). A much higher oxidative reactivity of p-ClC6H4SCH3 was observed for complex 1/H2O2 catalytic system, which is in accordance with the competition experiment on thianthrene 5-oxide.
| Entry | Substrate | Catalyst | Conversion (%) | Product distribution (%) | |||
|---|---|---|---|---|---|---|---|
| (i) | (ii) | (1) | (2) | (3) | |||
a Reaction conditions: sulfides or sulfoxides:30% H2O2:catalyst = 1:1:0.0025 mmol; methanol: 5 mL, 25 °C.b Products was determined by HPLC and reaction equation is described as follows. 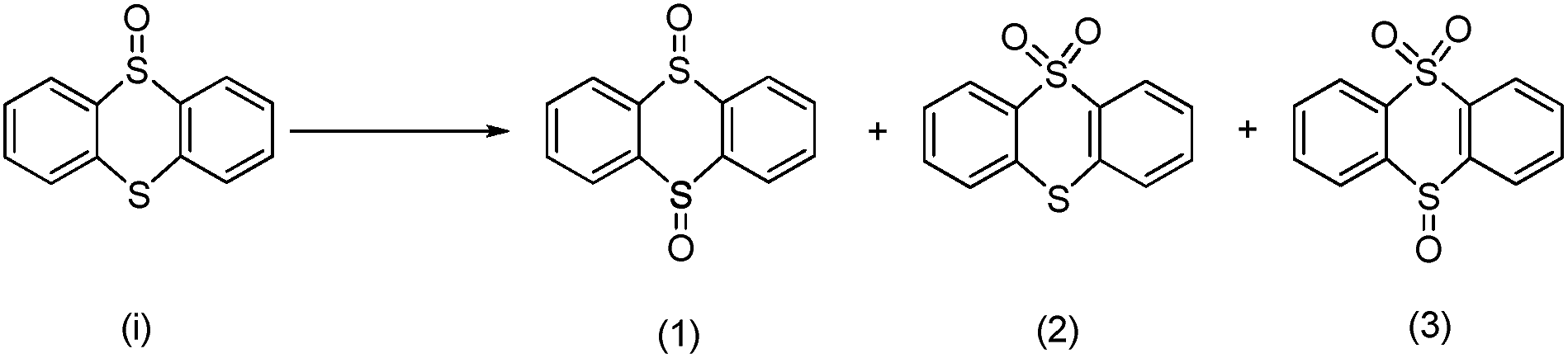 c Products was determined by GC and reaction equation is described as follows. c Products was determined by GC and reaction equation is described as follows. 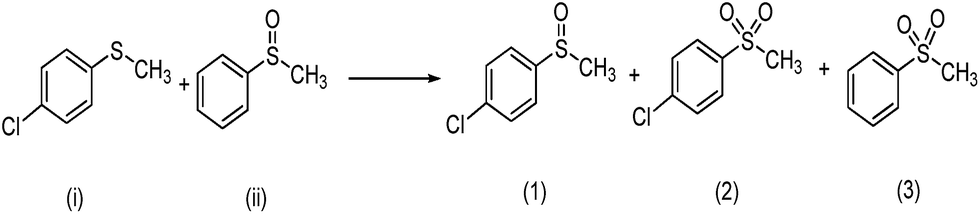 |
|||||||
| 1b | 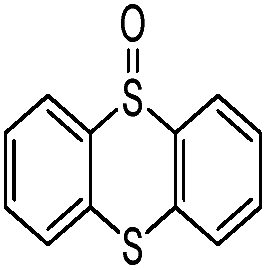 |
Ti(OPi)4 | 7 | 90 | 10 | — | |
| 2 | Complex 1 | 11 | 94 | 6 | — | ||
| 3c | 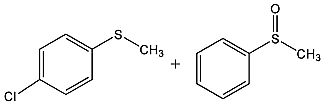 |
Ti(OPi)4 | 47 | 7 | 79 | 7 | 14 |
| 4 | Complex 1 | 92 | 4 | 93 | 3 | 4 | |
The active species in this system is still unknown. However, the generally accepted mechanism for titanium-based oxidation pre-catalysts involves the formation of titanium-based active intermediate species, Ti-hydroperoxo or Ti-peroxo species in the oxygen transfer process.16 It is largely accepted that in the H2O2 activation by titanium complexes, the active species originates from exchange of the alkoxy group by the oxidant.15a,b,17 According to these above results and analysis, a plausible mechanism for the oxidation of sulfides over the pre-catalyst is proposed in Scheme 1.
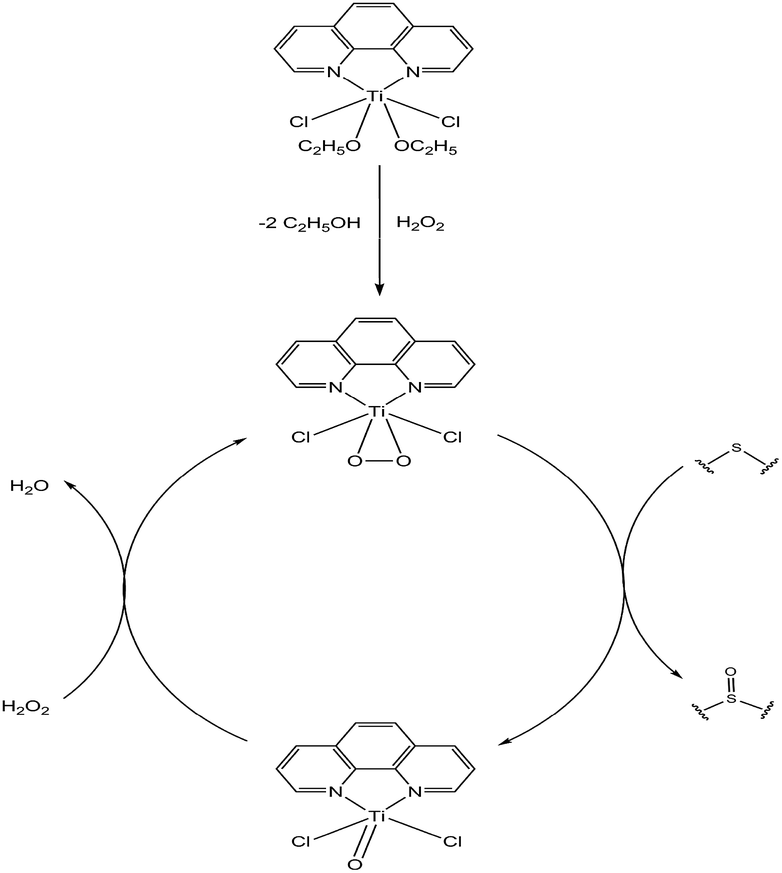 | ||
| Scheme 1 Plausible mechanism for the oxidation of thioethers over the complex 1. | ||
Conclusions
In conclusion a mononuclear titanium(IV) complex was prepared and fully characterized. The results reported inhere clearly indicate that it is possible to obtain a mild and chemoselective synthesis of sulfoxides from both aromatic and aliphatic sulfides, using as low as 2.5 × 10−3 mmol of complex 1 as a homogeneous pre-catalyst and the stoichiometric amount of H2O2 as an oxidant in CH3OH at room temperature. It is worth noting that complex 1 showed remarkable stability in the oxidation medium, being reused for up to 8 cycles without appreciable loss in its activity. Additionally the experimental results and related mechanism study suggests that titanium Ti-peroxo species are believed to catalyze the oxidation of sulfides into sulfoxides and sulfones.Experimental section
Materials and instruments
Thianthrene 5-oxide was synthesised according to the method described in the literature.18 All other reagents were commercially available and used without further purification. Elemental analyses for C, H, N were performed on a Perkin-Elmer analyzer model 1110. Infrared spectrum of the sample prepared in the form of KBr pellet was recorded on a Nicolet Nexus 470 infrared spectrophotometer in the range between 400 and 4000 cm−1. The UV-Vis spectra were obtained on a Varian Bio 2550 UV-Visible spectrophotometer in CH3OH at room temperature. Thermogravimetric analyses (TGA) were finished on a Delta Series TA-SDT Q600 (USA) in a N2 atmosphere in the temperature range between room temperature and 800 °C at a heating rate of 10 °C min−1 using Al2O3 crucible. 1H NMR and 13C NMR spectra were recorded on a Bruker NMR AVANCE III 400 or 500 spectrometers. The powder X-ray diffraction was collected on a Rigaku D/MaX 2550 PC with Cu-Kα radiation. The reaction products were quantified (GC data) by a Fuli Gas Chromatography equipped with a HP-5 capillary column and a flame ionization detector, and identified by QP2010 GC-MS.Synthesis
Ti(Phen)(OC2H5)2Cl2:titanium(IV) chloride (110 μL, 1 mmol) was slowly added to a previously prepared ethanol solution (10 mL) containing 1,10-phenanthroline (0.198 g, 1 mmol). Then the resulting solution was allowed to slowly evaporate at room temperature and about two days later colourless block-shaped single crystals were obtained. Yield: 57.0%. Elem. anal. calcd for the synthesized compound 1: C, 42.17%; H, 4.6%; N, 7.20%. Found: C, 42.55%; H, 4.5%; N, 6.98%. UV-Vis [λmax; nm (ε; L cm−1 mol−1) in methanol; Fig. S5, ESI†]: 230 (57420), 263 (35100). IR (KBr pellet, cm−1; Fig. S6, ESI†): 3336(s), 2928(m), 1654(w), 1626(w), 1582(w), 1519(m), 1424(s), 1222(w), 1136(w), 1118(w), 1104(s), 1065(s), 921(m), 870(m), 845(s), 735(m), 724(s), 646(m), 602(m), 429(m). 1H NMR [DMSO-d6, 25 °C, 400 MHz; δ (ppm)] (Fig. S7, ESI†): 9.25 (d, J = 6.3 Hz, 2H), 8.89 (d, J = 8.2 Hz, 2H), 8.25 (s, 2H), 8.08 (dd, J = 8.2, 4.7 Hz, 2H), 3.44 (q, J = 7.0 Hz, 4H), 1.05 (t, J = 7.0 Hz, 6H). 13C NMR [DMSO-d6, 25 °C, 126 MHz, δ (ppm)] (Fig. S8, ESI†): 148.25, 140.54, 139.45, 129.32, 127.34, 125.13, 55.96, 18.44.
Thianthrene 5-oxide: a soln of MCPBA (878 mg, 5.08 mmol) in CH2Cl2 (15 mL) was added to a stirred solution of thianthrene (1.0 g, 4.62 mmol) in CH2Cl2 (15 mL) cooled in an ice bath under N2. After 1 h, the mixture was extracted with sat. aq. NaHCO3 (3 × 15 mL). The organic layer was washed with H2O (2 × 25 mL), then dried (MgSO4) and concentrated under vacuum. The sample was then chromatographed on silica gel utilizing petroleum ether/ethyl acetate (6:1) as eluent to give thianthrene oxide in a good yield. 1H NMR [CDCl3-d6, 25 °C, 500 MHz; δ (ppm)]: 7.93 (d, J = 7.8 Hz, 2H), 7.63 (d, J = 7.7 Hz, 2H), 7.56 (t, J = 7.6 Hz, 2H), 7.43 (t, J = 7.5 Hz, 2H).
X-ray crystallographic determination
Diffraction data of the titanium complex were collected at 293 K for compound 1 on an Oxford Diffraction Xcalibur CCD diffractometer using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). Absorption correction was applied using multiscan technique. The structure was solved by direct method and refined by full-matrix least-squares method using the program SHELXL-97 within WINGX.19 All non-hydrogen atoms were easily found from the Fourier difference maps and refined with anisotropic temperature parameters. The graphics was drawn by the ORTEP. The detailed crystallographic data and structure refinement parameters for the compound were recorded in Table S2.† Selected bond distances and angles for them are listed in Table S3 (see the ESI†).Catalytic oxidation of methyl phenyl sulfide
In a typical experiment, sulfides (1.00 mmol) and catalyst (2.5 × 10−3 mmol) in methanol (5 mL) was stirred at 298 K temperature for 10 min and then 30% hydrogen peroxide (1.00 mmol) was introduced as the oxidant. At the end of specified time, aliquots of 100 μL reaction mixture were taken at regular intervals, treated with in 300 μL of methanol and then analyzed by GC/MS and GC.General procedure for oxidation of p-ClC6H4SCH3 and C6H5SOCH3
Catalyst (2.5 × 10−3 mmol), p-ClC6H4SCH3 (1 mmol), C6H5SOCH3 (1 mmol), H2O2 (1 mmol) and methanol (5 mL) were added to a glass tube; then the catalytic reaction was performed at 25 °C for 15 min. After the reaction was completed, the resulting mixture was analyzed by GC-MS and GC.General procedure for the oxidation of thianthrene 5-oxide
Catalyst (2.5 × 10−3 mmol), thianthrene 5-oxide (1 mmol), H2O2 (1 mmol) and methanol (5 mL) were added to a glass tube; then the catalytic reaction was performed at 25 °C for 15 min. After the reaction was completed, the resulting mixture was analyzed by HPLC-MS and HPLC.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Zhejiang Provincial Natural Science Foundation of China (LQ16B030004) and the Natural Science Foundation (Y201224604) for the Education Ministry of Zhejiang Province.Notes and references
- (a) J. Li, L. Wang, Y. Q. Ye, X. Fu, Q. Ren, H. Zhang and Z. Deng, Eur. J. Pharm. Sci., 2016, 85, 47 CrossRef CAS PubMed; (b) G. Sipos, E. E. Drinkel and R. Dorta, Chem. Soc. Rev., 2015, 44, 3834 RSC; (c) X.-T. Zhou and H.-B. Ji, Catal. Commun., 2014, 53, 29 CrossRef CAS; (d) R. Afrasiabi, F. Jalilian, B. Yadollahi and M. R. Farsani, Inorg. Chem. Commun., 2014, 50, 113 CrossRef CAS; (e) R. Fernandez de la Pradilla, C. Simal, R. H. Bates, A. Viso and L. Infantes, Org. Lett., 2013, 15, 4936 CrossRef CAS PubMed; (f) A. Viso, R. Fernandez de la Pradilla, M. Urena, R. H. Bates, M. A. del Aguila and I. Colomer, J. Org. Chem., 2012, 77, 525 CrossRef CAS PubMed; (g) E. Wojaczynska and J. Wojaczynski, Chem. Rev., 2010, 110, 4303 CrossRef CAS PubMed; (h) W. Al-Maksoud, S. Daniele and A. B. Sorokin, Green Chem., 2008, 10, 447 RSC; (i) J. M. Shin, Y. M. Cho and G. Sachs, J. Am. Chem. Soc., 2004, 126, 7800 CrossRef CAS PubMed; (j) B. M. Choudary, C. R. V. Reddy, B. V. Prakash, M. L. Kantam and B. Sreedhar, Chem. Commun., 2003, 754 RSC.
- (a) S. Xun, W. Zhu, Y. Chang, H. Li, M. Zhang, W. Jiang, D. Zheng, Y. Qin and H. Li, Chem. Eng. J., 2016, 288, 608 CrossRef CAS; (b) S. O. Ribeiro, D. Julião, L. Cunha-Silva, V. F. Domingues, R. Valença, J. C. Ribeiro, B. de Castro and S. S. Balula, Fuel, 2016, 166, 268 CrossRef CAS; (c) C. M. Granadeiro, L. S. Nogueira, D. Julião, F. Mirante, D. Ananias, S. S. Balula and L. Cunha-Silva, Catal. Sci. Technol., 2016, 6, 1515 RSC; (d) J. P. Ferreira, R. Viveiros, A. Loureno, M. S. da Silva, A. Rosatella, T. Casimiro and C. A. M. Afonso, RSC Adv., 2014, 4, 54948 RSC; (e) W. Zhu, P. Wu, Y. Chao, H. Li, F. Zou, S. Xun, F. Zhu and Z. Zhao, Ind. Eng. Chem. Res., 2013, 52, 17399 CrossRef CAS; (f) K. Bahrami, M. M. Khodaei, V. Shakibaian, D. Khaledian and B. H. Yousefi, J. Sulfur Chem., 2012, 33, 155 CrossRef CAS; (g) Y. Hu, Q. He, Z. Zhang, N. Ding and B. Hu, Chem. Commun., 2011, 47, 12194 RSC.
- (a) B. M. Trost and M. Rao, Angew. Chem., Int. Ed., 2015, 54, 5026 CrossRef CAS PubMed; (b) F. Colobert, V. Valdivia, S. Choppin, F. R. Leroux, I. Fernandez, E. Alvarez and N. Khiar, Org. Lett., 2009, 11, 5130 CrossRef CAS PubMed; (c) J. Legros, J. R. Dehli and C. Bolm, Adv. Synth. Catal., 2005, 347, 19 CrossRef CAS; (d) S. Kobayashi, M. Sugiura and C. Ogawa, Adv. Synth. Catal., 2004, 346, 1023 CrossRef CAS; (e) S. Kobayashi, C. Ogawa, H. Konishi and M. Sugiura, J. Am. Chem. Soc., 2003, 125, 6610 CrossRef CAS PubMed; (f) I. Fernandez and N. Khiar, Chem. Rev., 2003, 103, 3651 CrossRef CAS PubMed; (g) M. C. Carreno, Chem. Rev., 1995, 95, 1717 CrossRef CAS.
- (a) H.-B. Ji, T. T. Wang, L.-F. Wang and Y.-X. Fang, React. Kinet. Catal. Lett., 2007, 90, 259 CrossRef CAS; (b) M. M. Lakouraj, M. Tajbakhsh, F. Shirini and M. V. A. Tamami, Synth. Commun., 2005, 35, 775 CrossRef CAS; (c) Y. J. Chen and J. Y. Shen, Tetrahedron Lett., 2005, 46, 4205 CrossRef CAS; (d) L. Xu, J. Cheng and M. L. Trudell, J. Org. Chem., 2003, 68, 5388 CrossRef CAS PubMed.
- (a) K. Kaczorowska, Z. Kolarska, K. Mitka and P. Kowalski, Tetrahedron, 2005, 61, 8315 CrossRef CAS; (b) B. Karimi, M. Ghoreishi-Nezhad and J. H. Clark, Org. Lett., 2005, 7, 625 CrossRef CAS PubMed; (c) R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977 RSC; (d) K. Sato, M. Hyodo, M. Aoki, X. Q. Zheng and R. Noyori, Tetrahedron, 2001, 57, 2469 CrossRef CAS.
- (a) S. Hussain, D. Talukdar, S. K. Bharadwaj and M. K. Chaudhuri, Tetrahedron Lett., 2012, 53, 6512 CrossRef CAS; (b) N. Mizuno and K. Kamata, Coord. Chem. Rev., 2011, 255, 2358 CrossRef CAS; (c) Y. Wu, J. Liu, X. Li and A. S. C. Chan, Eur. J. Org. Chem., 2009, 2607 CrossRef CAS.
- (a) P. Le Maux and G. Simonneaux, Chem. Commun., 2011, 47, 6957 RSC; (b) H. Egami and T. Katsuki, Synlett, 2008, 1543 CAS; (c) H. Egami and T. Katsuki, J. Am. Chem. Soc., 2007, 129, 8940 Search PubMed.
- (a) M. M. Najafpour, M. Amini, M. Holynska, M. Zare and E. Amini, New J. Chem., 2014, 38, 5069 RSC; (b) H. Srour, J. Jalkh, P. Le Maux, S. Chevance, M. Kobeissi and G. Simonneaux, J. Mol. Catal. A: Chem., 2013, 370, 75 CrossRef CAS; (c) A. Pordea, D. Mathis and T. R. Ward, J. Organomet. Chem., 2009, 694, 930 CrossRef CAS.
- D. J. Ramon and M. Yus, Chem. Rev., 2006, 106, 2126 CrossRef CAS PubMed.
- P. Pitchen, E. Dunach, M. N. Deshmukh and H. B. Kagan, J. Am. Chem. Soc., 1984, 106, 8188 CrossRef CAS.
- F. Difuria, G. Modena and R. Seraglia, Synthesis, 1984, 325 CrossRef CAS.
- (a) S. Superchi, P. Scafato, L. Restaino and C. Rosini, Chirality, 2008, 20, 592 CrossRef CAS PubMed; (b) B. Jiang, X.-L. Zhao, J.-J. Dong and W.-J. Wang, Eur. J. Org. Chem., 2009, 987 CrossRef CAS; (c) P. Gogoi, T. Kotipalli, K. Indukuri, S. Bondalapati, P. Saha and A. K. Saikia, Tetrahedron Lett., 2012, 53, 2726 CrossRef CAS.
- (a) G. Pescitelli, L. Di Bari and P. Salvadori, J. Organomet. Chem., 2006, 691, 2311 CrossRef CAS; (b) J. M. Brunel, Chem. Rev., 2005, 105, 857 CrossRef CAS PubMed; (c) Y. Chen, S. Yekta and A. K. Yudin, Chem. Rev., 2003, 103, 3155 CrossRef CAS PubMed.
- (a) K. P. Bryliakov and E. P. Talsi, Eur. J. Org. Chem., 2008, 3369 CrossRef CAS; (b) K. P. Bryliakov and E. P. Talsi, Eur. J. Org. Chem., 2011, 4693 Search PubMed; (c) E. P. Talsi and K. P. Bryliakov, Appl. Organomet. Chem., 2013, 27, 239 CrossRef CAS.
- (a) M. Mba, L. J. Prins, C. Zonta, M. Cametti, A. Valkonen, K. Rissanen and G. Licini, Dalton Trans., 2010, 39, 7384 RSC; (b) M. K. Panda, M. M. Shaikh and P. Ghosh, Dalton Trans., 2010, 39, 2428 RSC; (c) E. P. Talsi, T. V. Rybalova and K. P. Bryliakov, ACS Catal., 2015, 5, 4673 CrossRef CAS; (d) G. Licini, M. Mba and C. Zonta, Dalton Trans., 2009, 5265 RSC.
- (a) E. Takahashi, K. Kamata, Y. Kikukawa, S. Sato, K. Suzuki, K. Yamaguchi and N. Mizuno, Catal. Sci. Technol., 2015, 5, 4778 RSC; (b) L. Wang, G. Xiong, J. Su, P. Li and H. Guo, J. Phys. Chem. C, 2012, 116, 9122 CrossRef CAS; (c) F. Bonino, A. Damin, G. Ricchiardi, M. Ricci, G. Spano, R. D'Aloisio, A. Zecchina, C. Lamberti, C. Prestipino and S. Bordiga, J. Phys. Chem. B, 2004, 108, 3573 CrossRef CAS.
- K. P. Bryliakov, Mini-Rev. Org. Chem., 2014, 11, 87 CrossRef CAS.
- F. P. Ballistrer, G. A. Tomaselli, R. M. Toscano, V. Conte and F. D. Furid, J. Am. Chem. Soc., 1991, 113, 6209 CrossRef.
- (a) G. M. Sheldrick and T. R. Schneider, in Macromolecular Crystallography, Pt B, ed. C. W. Carter and R. M. Sweet, 1997, vol. 277, pp. 319–343 Search PubMed; (b) G. M. Sheldrick, Macromolecular Crystallography, Pt B, 1997, vol. 45, pp. 849–854 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 1554126. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra06883b |
| This journal is © The Royal Society of Chemistry 2017 |