
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular fractionation of a soil fulvic acid (FA) and competitive sorption of trace metals (Cu, Zn, Cd, Pb) in hematite–solution systems: effect of the FA-to-mineral ratio†
Guillaume Fleury *,
Mirella Del Nero and
Remi Barillon
*,
Mirella Del Nero and
Remi Barillon
Institut Pluridisciplinaire Hubert Curien, UMR7178 Université de Strasbourg, CNRS, 23 rue du Loess, 67037 Strasbourg, France. E-mail: guillaume.fleury@iphc.cnrs.fr; Tel: +33 3 88 10 61 68
First published on 5th September 2017
Abstract
Understanding of the interactions occurring between fulvic acids (FAs) and trace metals in mineral–solution systems is a major issue for cycles of organic matter and micro-pollutants in surface media. Batch experiments and molecular-scale investigations were conducted to address the mechanisms regulating the molecular fractionation of a terrestrial FA and the competition between Cu, Zn, Cd and Pb during sorption onto hematite, a mineral ubiquitous in soils, with a focus on effects of FA-to-mineral ratio (r). A main result is that r controlled both the identity of the FA molecules preferentially sorbed on hematite and the sequence of the FA-promoted sorption of metals, at acidic pH. Data at moderate r evidenced that the sorption degree of a FA molecule increased with molecular acidity, supporting surface complex formation at hematite sites involving preferentially the most acidic and oxygenated FA molecules. FA-promoted sorption of strong Lewis acids such as Pb and Cu was favored (relative to Cd or Zn) by sorbed FA bearing multiple oxygenated functionalities. In contrast, preferential uptake of condensed aromatics and low oxygenated aliphatics/not-condensed aromatics prevailed at high r. A reduced FA-promoted sorption of Cu, which contrasted with an increased FA-promoted sorption of Cd, was observed too. Complex interactions must be invoked (competitive effects, hydrophobic forces, hydrogen bonding) to explain the striking results obtained in highly-competitive FA-concentrated systems. Our data highlight that (coupled) retention/mobilization of reactive organic molecules and of toxic metals like Cd and Pb is a function of the FA-to-metal oxide ratio of soils.
1. Introduction
Humic substances (HSs) like humic acids and fulvic acids (FAs) are ubiquitous in all natural surface systems as mixtures of many organic compounds with various compositions and origins. HSs have been widely reported to have the ability to bind metals and hydrophobic compounds1–3 and to sorb on the surfaces of (nano)minerals.4,5 The latter process is expected to play an important role in the surface properties and sorption capacities of relevant minerals of soils such as Fe-/Al-oxyhydroxides or clays, as well as on the concentration and chemical reactivity of dissolved organic molecules of surface waters. HS–mineral surface interactions are therefore of interest for several environmental issues, e.g., the geo-cycle of organic carbon, dissemination of natural or engineered nanoparticles in aquatic systems, and mobility and bioavailability of trace metal elements (TMEs) that have natural or anthropogenic origins and are potentially toxic at excess concentrations (e.g., Cu and Zn) or at trace levels (e.g., Cd or Pb). In particular, many field studies6–10 have highlighted that HSs exert major control on the migration/retention behavior of TMEs in natural surface systems.Considerable experimental work has been done over past decades to identify parameters and processes regulating the distribution of a TME between mineral surface and solution in ternary metal–HS–mineral systems, with the final aim of better understanding the impact of HS on the fate of trace metals in soils. It has been widely reported that the sorption of TMEs (Cu, Pb, Co, lanthanides…) on surfaces of metal (hydr)oxides and clays is reduced by the presence of HS at high pH values due to complexation of the metal by dissolved HS11 and is promoted by the sorption of HS at low pH values due to specific interactions between organic matter and trace metal cations at mineral surfaces.11,12 Experimental and spectroscopic evidences were given for complexation of metals with the functional groups of sorbed organic matter12–14 and/or formation of ternary surface–metal–HS complexes.15,16 A few experimental studies have been devoted to the competitive sorption of TMEs in ternary metal–HS–mineral systems, too.12,13,15,17 Elliott et al.17 showed that the presence of HS is likely to change the affinity order of divalent metals for soil surfaces. The authors found that the affinity order of Cu, Pb, Cd and Zn for acidic soils corresponds to the order of pKs for the first metal hydrolysis products (i.e. Pb > Cu > Zn > Cd) in the absence of HS, whereas the retention of Cd is favored over that of Zn in the presence of HS. This was in agreement with experimental studies showing that the stability constants of dissolved metal–HS complexes increase in the order Pb and Cu > Cd > Zn1–3 (this order being explained by differences in the sizes and electronic structures of the metal cations), supporting existence of specific interactions between metals and HS sorbed at surfaces of soil minerals. pH is not the only parameter reported to govern effect of HS on metal sorption, which has been shown to depend on solution characteristics such as ionic strength and concentration of metal13,18,19 or on surface properties of mineral,20 too. In contrast, the effects of the characteristics of HS on the sorption of TMEs in ternary metal–HS–mineral systems have seldom been investigated. An extensive batch study by Zuyi et al.19 supported that the nature of the HS, its fractionation by sorption, and its coverage on the surface of mineral are key parameters of sorption of an inorganic cation. Further work providing in-depth insights into the effects of these parameters on HS sorption is needed for exploring main mechanisms controlling the sorption of a HS and for better understanding of their impacts on the retention behavior of TMEs. Such an approach conducted in well-constrained mineral–solution systems taken as models of soil sub-systems is of particular interest for advancing towards a better knowledge of (coupled) cycles of TMEs and organics in more-or-less humic geochemical systems.
In this study, we addressed the effects of a key parameter, the fulvic acid-to-mineral ratio on the mechanisms involved in the sorption of a terrestrial fulvic acid onto hematite taken as a model of iron oxihydroxides existing in soils, and on the competitive sorption of Cu, Zn, Cd and Pb at trace levels. It has long been known that the sorption of a HS on the surface of a metal oxihydroxide results in a chemical fractionation of HS between mineral surface and solution21–23 – as HS are heterogeneous mixtures of thousands of organic compounds of various compositions, structures and reactivity of functional groups – that is to say that some compounds of HS are preferentially sorbed on the surface. Recently, descriptions of the mechanisms and molecular parameters governing the sorptive fractionation of an aquatic FA, an HA and two terrestrial FAs were provided by using ultra-high resolution mass spectrometry combined with electrospray ionization (ESI-FTMS) in studies of the identity of the HS compounds which are preferentially sorbed (and not sorbed) on an aluminum oxide surface.24–26 Galindo and Del Nero24 and Fleury et al.26 have provided molecular scale evidence that, at acidic pH, the sorption degree of a FA molecule in a CO2 series is positively correlated with its molecular acidity, for the highly reactive, oxygen-functionalized compounds sorbed via surface ligand exchange such as aliphatics, not-condensed aromatics (NCAs), and polycyclic aromatic compounds (PACs) of high atomic O/C ratio. Fleury et al.26 have also revealed in their investigations using FAs rich in condensed aromatics, that both molecular acidity and hydrophobic character are involved for PACs and low O/C NCAs, and that sorption is the result of surface ligand exchange reactions or hydrophobic interactions according to the atomic O/C ratio of the molecules. Their results compared well with those reported for the sorption of the soluble fraction of an humic acid on alumina,25 where there was observed a heightened contribution of the hydrophobic interactions due to the highest content in highly condensed aromatic compounds of this terrestrial humic substance. All these findings imply that sorptive fractionation mechanisms taking place on an alumina surface are similar for HA and FA, with extent of the various mechanisms being largely determined by the amount, nature and reactive ability of the molecules present in HS. However, no study has been focusing so far on the effect of FA-to-iron oxihydroxide ratio on the mechanisms involved in the sorptive fractionation of FA, nor on the FA-driven competitive sorption behavior of TMEs. It has long been known that mobility of TMEs in aquatic systems, sediments and soils is largely dependent on their interactions with mineral surfaces such as iron (hydr)oxides and clays.27 Iron oxihydroxides are widespread in soils, sediments and aquatic systems,28 where they are typically nanosized29 and occur in many forms ranging from amorphous ferrihydrite to crystalline goethite and hematite.30 They are widely reported to be excellent adsorbents for various anions, for organic matter31,32 and for heavy metals33,34 including cadmium, copper, lead and zinc.35–37 They are therefore expected to influence largely the mobility of these species in various ecosystem compartments,38 owing to their high specific surface area and high surface reactivity. Hematite has moreover been used in many studies of NOM/Fe-oxihydroxides/metal model systems.39
The approach used here for investigations of PPH–hematite–metal systems combined macroscopic studies of the (co)sorption of a terrestrial FA (noted PPH) and divalent metals with descriptions at the molecular scale of the FA sorption. Sorption isotherms of Cu, Zn, Cd and Pb onto hematite were derived from batch experiments of competitive metal sorption as a function of pH and at different PPH-to-mineral ratios. On-line electrophoretic mobility (EM) measurements of mineral suspensions were also performed for gaining insights into the surface charges of the particles. The use of a LTQ Orbitrap XL hybrid mass spectrometer with high resolution and high mass accuracy allowed detecting thousands of dissolved compounds constitutive of PPH, deriving the elemental composition of most of them, and identifying sorbing/not sorbing molecules by comparing ESI(−) mass spectra recorded before and after contact with the mineral surface of PPH solutions at acidic pH (∼4). Macroscopic and molecular-scale data were combined to elucidate the mechanisms and the chemical identity of the organic molecules involved in FA sorption and gain insights into the competitive sorption of TMEs in systems at various FA surface coverage of an iron oxide.
2. Materials and methods
2.1 Materials
All the solutions were prepared using reagent grade chemicals and ultra-pure water (purity >18 MΩ cm). The fulvic acid noted “PPH” was extracted from a sample of soil developed over sandstone under a beech forest and collected near La Petite-Pierre (Vosges Mountains, France) following the International Humic Substances Society (IHSS) standard method for isolation of soil fulvic acids. This method excerpted from Swift40 uses XAD-8 resin adsorption to isolate fulvic acids. Stock solutions of PPH were prepared and stored frozen in the dark until used. Hematite nanoparticles were provided by US Research Nanomaterials, with a chemical purity >99.5%, a 30 nm particle size and a surface area of 40 ± 20 m2 g−1.2.2 FA sorptive fractionation in binary FA–hematite systems
Batch experiments of PPH sorption onto hematite were carried out following the procedure described in a previous publication26 at pH 3.9 ± 0.2 and at different FA-to-mineral ratios, r, leading to low or high FA surface coverage, s (r = 0.1, 0.6, or 1.4 mg C per m2 leading to s = 0.1, 0.3 and 0.7 mg C per m2, respectively). No attempt was made to fix the ionic strength of the initial experimental solutions, as background salts were previously observed to alter considerably the ESI-MS response. After shaking of the 10 mL samples for 24 hours at 298 K, the final pH of the suspensions were measured. The suspensions were then centrifuged at 8000 rpm during 2.5 hours for solution–colloid separation and the supernatants were collected. Aliquots were taken for measurements of dissolved organic carbon (DOC) using a Shimadzu TOC-VCPH analyzer, and all the remaining supernatant solutions were adjusted (using a 0.01 M HCl solution) to a pH value of 3.5 prior to ESI(−)-MS analysis. On-line measurements of the electrophoretic mobility (EM) of the particles in suspensions were also performed on aliquots of samples collected before centrifugation using dynamic light scattering (Malvern Zetasizer Nano ZS instrument). Values reported for EM correspond to mean values calculated from three measurement replicates.An additional series of PPH batch sorption experiments was carried at different pH values (3 < pH < 6.5) and at r = 1.4, in order to determine the amount of PPH sorbed at the surface of hematite as a function of pH. Same procedure as that described above was used, and all remaining supernatant solutions were used for measurements of dissolved organic carbon (DOC) using a Shimadzu TOC-VCPH analyzer.
A Thermo Scientific LTQ Orbitrap hybrid mass spectrometer was used for the analysis of FA solutions in the ESI negative ionization mode. Analytical parameters are described in detail in a previous publication.26 The samples were infused directly into the ESI source with a spray voltage of 3.5 kV at a flow rate of 10 μL min−1. The source capillary was held at −50 V and 275 °C, and a voltage of −240 V was applied to the tube lens. For each acquisition, 100 scans (2 s per scan) were co-added using Xcalibur software. The Orbitrap MS was externally calibrated in negative ion mode on the 50–2000 m/z range using a LTQ/FT-Hybrid ESI Calmix solution, for a mass accuracy better than 3 ppm. All MS spectra were acquired on two separate ranges, 120–400 m/z and 400–798 m/z, to improve mass accuracy in the low mass region and signal-to-noise (S/N) ratio for high m/z ions. Chemical formulae were assigned to m/z values of peaks detected with S/N >4 using the Xcalibur software. All possible formulae attributable to a given odd m/z value were calculated by considering 12C, 1H and 16O atoms (with upper limits for the number of atoms equal to 200, 600 and 50, respectively) and by rejecting all formulae having theoretical masses differing by more than 3 ppm from the measured mass. For even m/z values (odd masses), peaks corresponding to species including one 13C isotope were rejected. Because of the low nitrogen content of HS, the remaining species were considered to contain at most one 14N atom, in addition to 12C, 1H and 16O atoms.
For identified compounds, calculation of the aromaticity index (A.I.) as defined by Koch and Dittmar41 allowed to distinguish between three categories of compounds: PACs (0.67 < A.I.), NCAs (0.5 < A.I. < 0.67), and compounds with a pronounced aliphatic character (A.I. < 0.5). The chemical formulae were represented in Van Krevelen (VK) diagrams which plot the hydrogen/carbon (H/C) ratio versus the oxygen/carbon (O/C) ratio of the compounds. To derive the order of relative affinities of PPH compounds for the mineral surface, we used the parameter I defined by Galindo and Del Nero24 and Fleury et al.26 which is equal to the ratio of the normalized peak intensity of an ion in supernatant (after sorption) on the normalized peak intensity of the ion in initial solution (before sorption), with the normalization being made to the total ion current (TIC) of the considered solution. The lower the value of I of an ion, the higher its relative affinity for the mineral surface: ions with I = 0 are totally sorbed on the mineral surface, while ions with I ≠ 0 are distributed between surface and solution.
It is well-known that the intensity of an ion on a mass spectrum not only depends on its concentration but also on its ionization efficiency and on the matrix.26 The effect on the relative peak intensities of the ions (and thus on their values of I) induced only by the decrease in solution concentration upon PPH sorption at the surface of hematite was studied in a previous publication,26 in order to differentiate it from the effect on I of the sorptive fractionation process itself. The results of this study showed that a value of I lower than 0.7 (or 0.9 for the range 400–798 m/z range at pH 3.8) indicates a rather good sorption of an ion on the mineral surface, while ions showing values comprised between 0.7 and 1 (or 0.9 and 1.2 for the range 400–798 m/z range at pH 3.8) are poorly sorbed or not sorbed at all. Ions showing values higher than 1 (or 1.2) are not sorbed at all during the experiment.
2.3 Metal sorption experiments in ternary metal–FA–hematite systems
Sorption isotherms of Cu, Zn, Cd and Pb onto hematite were obtained from series of multi-elemental batch experiments carried out as a function of pH (3 < pH < 7), with and without fulvic acids. Hematite suspensions were prepared in individual PEHD tubes with a mineral-to-solution ratio of 0.375 g L−1. For experiments performed in the presence of fulvic acid, PPH was then added to obtain FA-to-mineral ratios of 0.10 ± 0.04 mg C per m2 and 1.4 ± 0.1 mg C per m2. Aliquots of a multi-elemental stock solution prepared from nitrate standards were introduced simultaneously in the tubes, to obtain initial concentrations of 0.8 ± 0.2 μM for each of Cu, Zn, Cd and Pb. The pH values of the solutions were adjusted using 0.1 M HNO3 and NaOH solutions. After shaking of the 10 mL samples for 24 h at 298 K, final pH values were measured. The suspensions were centrifuged at 8000 rpm during 2.5 hours for solution–colloid separation and the supernatants were collected. Aliquots were taken for DOC measurements and remaining supernatant solutions were analyzed by Inductively Coupled Plasma Mass Spectrometry (ICP-MS). No experimental replicates were made. Error bars shown on graphics correspond to analytical standard deviations calculated on the mean analytical values.On-line measurements of the electrophoretic mobility (EM) of the particles in suspensions were performed on aliquots of samples collected before centrifugation using dynamic light scattering (Malvern Zetasizer Nano ZS instrument), following the same procedure as previously (cf. Section 2.2). Values reported for EM correspond to mean values calculated from three measurement replicates.
3. Results
3.1 Sorption behavior of FA in hematite–solution systems
 | ||
| Fig. 1 Amount of PPH sorbed at pH 3.9 on the surface of hematite as a function of FA-to-mineral ratio of experiment, along with previous data acquired by our team26 for other systems of interest (cf. text). | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K) of carboxyl groups (COOH) and phenolic groups (Ph) of PPH calculated using the procedure of Ritchie et Perdue43 and concentrations of hematite surface groups ([S]surf, cf. text)
K) of carboxyl groups (COOH) and phenolic groups (Ph) of PPH calculated using the procedure of Ritchie et Perdue43 and concentrations of hematite surface groups ([S]surf, cf. text)
| Mineral | r ratio (mg C per m2) | FA sorption (%) | [COOH]FA (μeq L−1) | logKCOOH |
[Ph]FA (μeq L−1) | logKPh |
[S]surf (μmolsites L−1) |
|---|---|---|---|---|---|---|---|
| Hematite | 0.1 | 100 | 33.6 | 1.1 | 12.1 | 9.27 | 190 |
| 0.6 | 54 | 144 | 51.8 | ||||
| 1.4 | 52 | 336 | 121 |
 | ||
| Fig. 2 Effects of sorption of PPH and/or of competitive sorption of trace level concentrations of Pb, Cu, Zn and Cd (at 0.8 ± 0.2 μM for each metal) on the surface charge of hematite as revealed by EM data recorded in suspensions of 0.375 g L−1 of hematite at different pHs. | ||
 | ||
| Fig. 3 VK diagrams for PPH compounds sorted as a function of their relative affinities at pH 3.9 for the surface of hematite at r = 0.1 mg C per m2 (a, b), r = 0.6 mg C per m2 (c, d), and r = 1.4 mg C per m2 (e, f) and identified in the mass ranges 120–400 m/z and 400–798 m/z, respectively. | ||
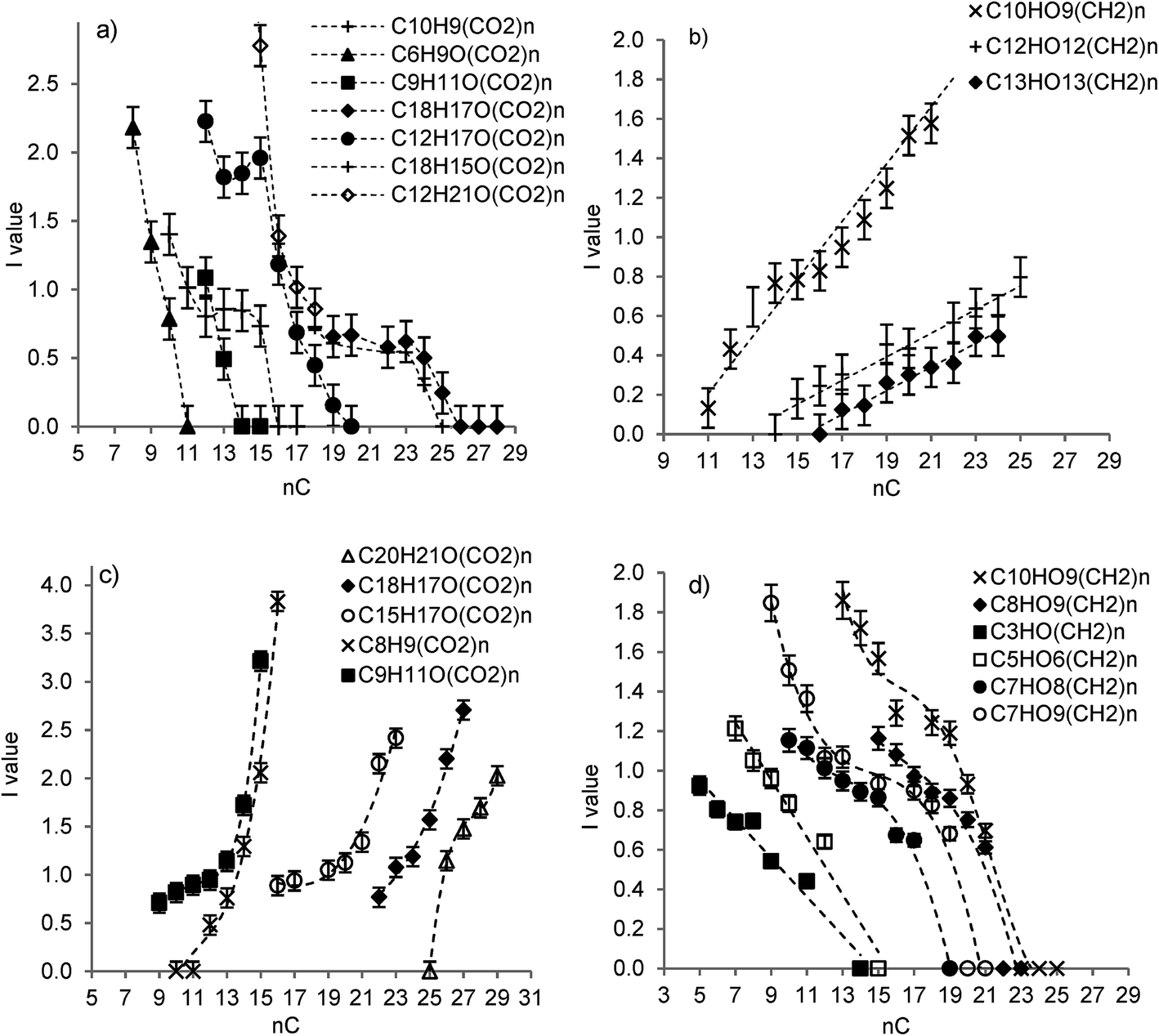 | ||
| Fig. 4 Evolution of the value of I as a function of number of carbon atoms (nC) of aliphatic and NCA compounds (with O/C >0.2) in –CO2 and –CH2 series of PPH, after sorption of the FA in hematite–solution systems at pH 3.9 and at r = 0.6 mg C per m2 (a and b, respectively) or r = 1.4 mg C per m2 (c and d, respectively). | ||
3.2 Sorption isotherms of metals in FA–hematite–solution systems
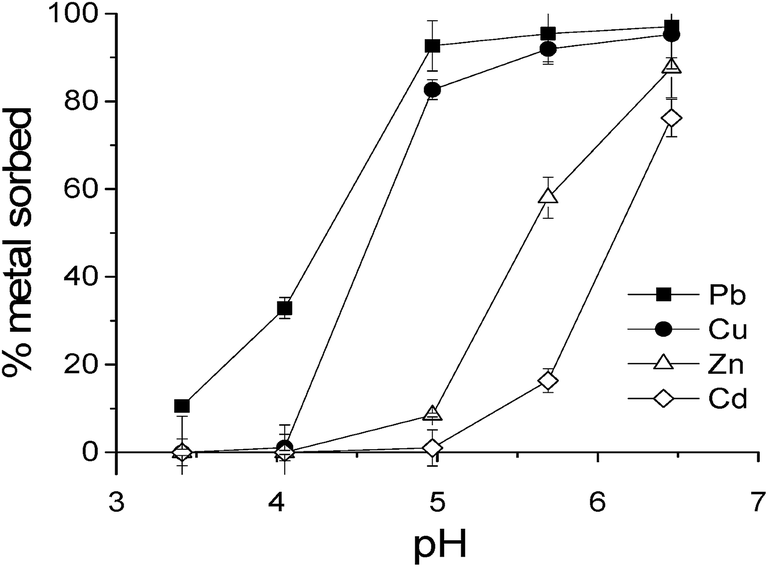 | ||
| Fig. 5 Results of experiments of the competitive sorption of Pb, Cu, Zn and Cd at trace level concentrations (at 0.8 ± 0.2 μM for each metal) in suspensions of 0.375 g L−1 hematite at different pHs. | ||
 | (1) |
 | ||
| Fig. 6 Results of experiments of the competitive sorption of Pb, Cu, Zn and Cd at trace level concentrations (at 0.8 ± 0.2 μM for each metal) in suspensions of 0.375 g L−1 hematite at different pHs, in the absence of PPH or for initial PPH-to-hematite ratios of 0.1 mg C per m2 or 1.4 mg C per m2. | ||
| r ratio (mg C per m2) | KCu (L mol−1) | KZn (L mol−1) | KCd (L mol−1) |
|---|---|---|---|
| 0.1 | 1.25 × 105 | 1.65 × 103 | 1.85 × 103 |
| 1.4 | 5.08 × 103 | 2.45 × 103 | 5.72 × 103 |
4. Discussion
A main finding of this study is that the FA-to-hematite ratio is a key parameter governing trends in sorption on hematite observed at acidic pH both of the molecules constitutive of a terrestrial fulvic acid and of divalent metals at trace level concentration. In particular, its effect on the sorptive fractionation of a fulvic acid had never been explored before. The ESI-MS data presented here provide new knowledge at molecular level that advances the in-depth description of the interactions occurring in complex FA–metal oxide–TME–solution systems.4.1 FA fractionation behavior
Significant differences were found between the fractionation patterns obtained for the sorption of PPH at a high value and at a low value of the FA-to-hematite ratio, respectively, which addresses the underlying mechanisms. All the results obtained at the molecular scale for the hematite–solution system at moderate value of r and at moderate FA surface coverage indicate that the sorption behavior of PPH at pH 4 is mostly the result of a highly selective mechanism, which is a surface ligand exchange involving the high affinity ferrinol sites at the mineral surface and carboxyl groups of PPH molecules. The main molecular parameter governing selectivity is molecule acidity, as shown by the nC–I relations existing within CO2 and CH2 homologous series whatever the class of compounds to which a series belongs. Significance of the existence of such relationships has been widely discussed in our previously published studies.25,26,63 The numerous homologous series accounting for the large number of different basic structures in PPH, and the wide range of I values observed, complied well with the concept that a FA is made up by small subunits held together by weak forces. It evidenced moreover that surface ligand exchange competed efficiently the cohesion forces of the supramolecular assembly to induce a disruption of the FA structure during the process of sorption. Uptake of the least oxygenated PACs and NCAs is probably driven by hydrophobic interactions between their hydrophobic moieties and those of the strongly sorbed PACs.26,63Molecular acidity is not the only parameter to be taken into account to explain the PPH sorption behavior at high FA-to-hematite ratio. The dramatic FA fractionation schemes observed during FA sorption compare well with published studies reporting that hydrophobic fractions of FAs and/or the organic molecules with high contents in aromatic moieties activated by oxygenated functionalities are preferentially sorbed on the surfaces of Fe-oxide and Al-oxide minerals as compared to aliphatic fractions.21,23,64,65 An important observation from our ESI-MS data is the much lower number of totally sorbed PPH molecules observable in the experiment at high value of r than in the experiment at moderate value of r, despite a higher FA coverage. These results attest for a strong competition of the PPH molecules for the sorption on the hematite surface at high FA-to-mineral ratio. We propose that two parameters beside molecular acidity contribute to the fractionation pattern in such a highly competitive and FA-concentrated system, namely, the amounts of ferrinol surface sites available for coordination of FA carboxyl groups and the rate of hematite dissolution.
A first hypothesis is that the strong competition for coordination at the ferrinol surface sites is in favor of the condensed aromatic structures carrying oxygenated functionalities (cf. values of I in Fig. 3e and f) because the latter may represent the most acidic of all organic components of terrestrial FA. Synergistic effects are expected to be induced by a high occupancy of the ferrinol sites by the most acidic PACs bearing multiple oxygenated functionalities. Their presence in the first sorption layer formed on the surface of hematite likely provides significant amounts of hydrophobic domains available for further interactions with the hydrophobic moieties of PACs of low O/C present in the solution, as well as with NCAs and aliphatics of intermediate and low O/C. Moreover, the large extent of deprotonation of the carboxyl groups (i.e., of those not engaged directly in surface bonding) of the oxygenated PACs in the first layer of sorbed FA results in high negative charges at hematite–solution interface (cf. Fig. 2). As a consequence, the dielectric constant of water at the surface of hematite may be apparently lower than that of free water (because electric fields induced by surface charge influence its value as shown by Booth66), which favors the adsorption of hydrophobic molecules. Moreover, the negative surface charges may prevent the approach in the close proximity of hematite surface of dissolved molecules which are highly acidic (deprotonated) such as NCAs and aliphatics of high O/C ratio, due to electrostatic repulsion. All these effects may represent one of the causes for the increase of sorption degree with increase of number of CH2 of a molecule in a –CH2 series (i.e., an increase of its aliphatic character and a decrease of its acidity) that was observed for NCAs and aliphatics in experiment at high r (Fig. 4d). Consistently, they may account for the observation that the degree of sorption of a PPH molecule in a CO2-series is inversely correlated to its number of carboxyl groups, with such a trend being explained by a decrease in molecule acidity with nCO2.67 In summary, all the effects described above might have led to higher degrees of sorption (likely via H-bonding or hydrophobic effects) for the least acidic and least oxygenated molecules in these classes of compounds than for the most acidic ones.
A second hypothesis is that the highly oxygenated NCAs and aliphatics of PPH are preferentially kept in solution because they tend to participate in aqueous complex formation with Fe3+ released by dissolution of hematite. The inverse and linear correlation observed between degree of sorption of a PPH molecule (NACs and aliphatics) and nCO2 at high r (Fig. 4c) may thus be interpreted by a partial control of PPH fractionation due to the increasing participation of a PPH molecule to chemical binding in solution with an increase of its acidity, through reactions of aqueous complex formation involving its oxygenated (carboxyl) groups and aqueous Fe. Previous studies have indeed shown that Fe ions participate to formation of strong metallo-organic complexes with HAs, either in solution or at mineral surfaces.68,69 Moreover, it has long been known that presence of organic matter increases solubility and rates of dissolution of Fe-oxides.70 Differences in sorptive fractionation trends of PPH observed with increasing FA-to-hematite ratio may thus be well accounted for by competitive effects between Fe3+ metallic centers at hematite surface and dissolved Fe3+ ions for coordination of the highly oxygenated NCAs and aliphatics bearing multiple functionalities. Actually, both characteristics of high FA-to-hematite ratio systems, i.e., high dissolution rates of hematite and limited amounts of ferrinol surface sites available for ligand exchange with the high amounts of oxygenated functional groups of FA, may concur to the strong fractionation observed between the highly-sorbed oxygenated PACs and the poorly-sorbed oxygenated NCAs and aliphatics, owing to small differences in acidity and/or relative affinities for surface and aqueous Fe of these molecules which all have potentially strong metal binding properties.
4.2 Competitive sorption behavior of the metals
Few studies have reported so far experimental results on the competitive sorption of TMEs in ternary metal–HS–mineral systems. We provide evidence that the presence of a terrestrial FA not only changes the affinity order of the divalent metals for hematite surface at acidic pH but also that the affinity order is highly dependent on FA-to-hematite ratio and FA surface coverage. Processes leading to the change reported here in the order of metal sorption at high FA-to-mineral ratio are of primary interest in the mobility/retention behavior of metals in soils rich in humic substances.The promotion sequence by PPH of metal sorption onto hematite (Pb > Cu > Cd > Zn) reported at low r (where PPH is totally sorbed) and at low pH (where no metal sorption occurs in the absence of PPH) was in good agreement with the results of Elliott et al.17 In such conditions, it clearly reflects the overall order in metal–PPH–surface binding and it is similar to the order in stability constants reported for metal–humic substance complexes in several studies.2,58 Some discrepancies however exist regarding relative stability of metal–fulvate complex between Pb and Cu. Unlike in the aforementioned studies, several authors have found a higher value of stability constant for Cu than for Pb.1,71 It must be stressed that the reported values for metal–humic substances complexation are dependent on many experimental parameters, e.g., pH, ionic strength, composition of humic/fulvic acid and metal loading, which often makes them not being directly comparable. For example, Town and Filella62 made a collection of published lead-humic substances equilibrium quotients and they highlighted by taking into account metal ion loading that a linear relationship exists between metal complexation capacity of organic matter and logK value. This relationship evidenced that apparent stronger binding sites of humic substances are utilized at lower metal ion loadings and that progressively weaker sites contribute to metal complexation at higher loadings.62 Nonetheless, there is a general agreement that metal–humic substance constants are higher for Cu and Pb than for Cd, then Zn, e.g., Pb- and Cu-humate/fulvate complexes are much more stable than Cd–Zn-humate/fulvate complexes. On the basis of the literature, we can hypothesize similar general trends in the order of co-adsorption of the metals with PPH on the surface of the hematite (observed at low r) and the order of binding of the metals to dissolved PPH, which is: Pb and Cu > Cd > Zn. A change in the sequence of promotion of the adsorption of metals by FA can therefore not be explained only by a simple scheme of competition between the aqueous complexation and the surface complexation of the metals with bulk FA. Explaining the change in the promotion sequence with r observed in this study at pH 4–5, in particular the promoted sorption of Cd and Zn over that of Cu, requires considering the intervention of processes cooperating or competing with sorption (depending on the metal under consideration) which are closely linked to characteristics of the high FA-on-hematite ratio system, e.g., solution containing dissolved FA and Fe3+ ions (high hematite dissolution rate), amounts of ferrinol sites available for coordination of carboxyl groups of FA and subsequent fractionation behavior of PPH, specific properties of the heavily FA-loaded surface.
A metal ion is expected to participate in formation of two main types of ternary inner-sphere metallo-organic sorption complexes on the surface of a mineral, namely, the metal tends to bind to functional groups of sorbed organic molecules to form surface–FA–metal complexes, and/or it makes a bridge between the surface and the sorbed organic molecules by binding directly to oxygen atoms of the hydroxyls groups present on hematite to form stable ternary surface–metal–FA complexes. Sorbed PPH molecules may occupy sorption sites of Cu under conditions of high r where the surface coverage of hematite by PPH is high (s = 0.7 mg C per m2), preventing thereby the formation of surface–Cu–FA complexes. Although possible, such a process cannot account alone for the decrease in Cu sorption with r. Another important process to take into account is that Pb and Cu compete for the same sorption sites. This is revealed by our data of the single (Fig. S4†) or of the competitive sorption (Fig. 6) of Cu on hematite showing that the presence of Pb decreases the FA-promoted sorption of Cu at low FA-to-mineral ratio. In the multi-metal experiments at low r, a competition between Pb and Cu and PPH molecules for ferrinol sites is to be ruled out because ferrinols are available in large quantities with respect to the concentration of metals or functional groups of FA (cf. Table 1). Hence, it may be inferred that Cu participates in the absence of Pb to the formation of ternary surface–organic ligand–Cu complexes implying sorption sites present in very small quantities at hematite surface, probably multidentate sites. In multi-metal systems, Pb is likely to compete successfully for binding at such sites, owing to its marked affinity reported for the stronger binding sites of humic substances.62 This is consistent with data on metal-humate, metal salicylate and metal benzoate of Kostic et al.2 showing that there is a strong competition between Pb(II) ions and Cu(II) ions in the binding for carboxyl and phenolic binding sites of humate macromolecules. In the experiment at high r, both Pb and the Fe3+ ions released at acidic pH by the promoted dissolution of hematite are likely to compete against binding of Cu at the multidentate sites of the sorbed PPH fraction. It has been suggested in a published study of the effects of Fe competition on binding of rare earth elements to humic acid68 that Fe3+ has a marked affinity for the few strong HA multidentate sites, as Fe was shown to be a strong competitor for heavy rare earth elements in low pH conditions at which Fe3+ binds to HA. Iron implication provides a valid explanation for the decrease in the FA-promoted Cu sorption on hematite observed here at acid pH (4–5) when the value of r is changed from low to high in the experiment, despite the number of functional groups of PPH sorbed is potentially increased concomitantly. Competitive phenomena between Pb, Fe and Cu for the small quantities of the multidentate sites of the sorbed organic matter would thus explain that the FA-promoted Cu sorption decreases with the presence of Pb or with a high FA-on-hematite ratio, favoring certainly the complexation of Cu with dissolved FA molecules at low pH. Moreover, the dissolved molecules in the studied system at high r have potentially strong binding properties for metals (cf. paragraph 4.1). Consistently, the sorption of Cu onto hematite observed at near-neutral pH value in the absence of FA is significantly reduced in the presence of dissolved PPH. The extent to which the sorptive fractionation of FA regulates the distribution of FA multidentate sites between the hematite surface and the solution is an important topic for further research to elucidating mineral–metal–humic interactions in soils rich in humic substances and the detailed effect of a difference in composition/reactivity between adsorbed and dissolved fractions of FA on the reduction of the FA-promoted Cu sorption at acidic pH.
The higher affinity of Cd and Zn for hematite observed at high r than at low r (at acidic pH 4–5) would necessarily imply differences in the reactivity and characteristics existing between a poorly PPH-loaded and heavily PPH-loaded surface of the hematite. First, as underlined before, the large extent of deprotonation of the carboxyl groups of the oxygenated PACs sorbed in a first sorption layer results in a heavily negatively charged surface of hematite, for a high value of r (cf. Fig. 2). This may favor the further adsorption of Cd2+ and Zn2+ as outer-sphere metal–fulvate surface complexes and/or the electrostatic attraction exerted on the metal ions by the strongly charged surface by FA may promote their placement at a certain distance of the solution–hematite interface in a diffuse layer of counter-ions compensating the surface charges. Second, the sorption of Zn and Cd is possibly favored at high r due to the higher degree of sorption of PPH molecules of intermediate O/C to which these metals tend to associate preferentially, owing to their low Lewis acidity. Further studies are needed to evaluate the exact role of preferential adsorption onto Fe oxides of NACs or aliphatic compounds of FA which display intermediate O/C ratios on the inversion of the order of the promotion of sorption of Cu and Cd (Zn) in conditions of high FA-to-mineral ratio.
5. Conclusions
The results presented here highlight that the fulvic acid-to-mineral ratio is a key parameter controlling the competitive sorption of both the organic molecules constitutive of a terrestrial fulvic acid and of divalent trace metals on the surface of hematite.In particular, our ESI-MS data provide new knowledge at molecular level of the sorptive fractionation of a FA that improves greatly our understanding of the mechanisms of the FA–Fe oxide–solution interactions. Whereas molecular acidity is the main parameter driving the sorption of a FA molecule in weakly FA-concentrated hematite–solution systems, this molecular parameter has the reverse effect of promoting the mobility of aliphatics and not-condensed aromatics in the highly competitive FA-concentrated systems. In the latter conditions, a series of complex interactions are to be considered, – e.g., strong competitive effects between most acidic molecules, hydrophobic effects, hydrogen bonding – to explain the preferential sorption of all PACs and of the low oxygenated FA molecules. These results emphasize that a thorough study of the chemical characteristics of individual HS components, such as acidity, hydrophobicity, and functional groups, and of relationships between molecular characteristics and sorptive fractionation of HS, is powerful for acquiring detailed knowledge of HS sorption mechanisms; knowledge on which our comprehensive understanding of the behavior of complex mixtures such as HS in geochemical systems can be built.
The data on the competitive sorption of divalent metals in hematite–FA solution systems support that a potentially highly toxic metal as Pb is strongly retained on Fe-oxides owing to its peculiar affinity for a few multidendate sorption sites of sorbed FA molecules bearing multiple oxygenated functionalities. In contrast, it is suggested that sorption of Cu on such sites is competed successfully at acidic pH by the presence and the preferential sorption of Pb, and by that of Fe3+ ions in conditions of high FA-to-mineral ratio favoring the enhanced dissolution of iron oxides and the subsequent release of Fe in solution. Competitive effects between surface complexation of Cu and aqueous complexation of Cu involving the highly acidic NACs and aliphatics left in solution in FA-concentrated systems may also concur to a decrease in FA-promoted retention of Cu on the metal oxide. Unlike for Cu, the highly FA-rich systems display enhanced ability for retardation of Cd, another metal highly toxic, owing to the characteristics of the heavily FA-loaded mineral surfaces, including a possible role of the specific retention of the FA compounds (aliphatics and NACs) of intermediate O/C ratios in these conditions. Further studies combining macroscopic and molecular-scale information are needed to precise the exact role of a possible difference in metal affinity order between the FA molecules preferentially sorbed (PACs and less oxygenated FA molecules) and those preferentially left in solution (acidic and oxygenated NACs and aliphatics) on the competitive behavior of divalent metals in high FA-to-mineral ratio conditions. Valuable insights into the role of sorptive fractionation pattern of HS on the competitive sorption of TEMs on iron oxides may also be provided by combined macroscopic and molecular investigations using supramolecular assemblies such as humic acids, which are known to display at low HS-to-metal oxide ratio a heightened contribution of hydrophobic interactions as compared to FA, due to their highest content in PACs.25 More information on relationships between HS molecular characteristics, HS fractionation and competitive metal sorption on iron oxides is of interest to better comprehend the coupled cycles of metals and organic matter in geochemical surface systems rich in Fe-oxyhydroxides, owing to their high specific surface area and high surface reactivity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank IPHC and the Alsace Region for their financial support. We also gratefully acknowledge the reviewers for their helpful comments and for improvement of the manuscript.References
- A. K. Pandey, S. D. Pandey and V. Misra, Ecotoxicol. Environ. Saf., 2000, 47, 195–200 CrossRef CAS PubMed.
- I. Kostic, T. Andjelkovic, R. Nikolic, A. Bojic, M. Purenovic, S. Blagojevic and D. Andjelkovic, J. Serb. Chem. Soc., 2011, 76, 1325–1336 CrossRef CAS.
- R. Yang and C. M. G. van den Berg, Environ. Sci. Technol., 2009, 43, 7192–7197 CrossRef CAS PubMed.
- M. Ochs, B. Cosovic and W. Stumm, Geochim. Cosmochim. Acta, 1994, 58, 639–650 CrossRef CAS.
- M. A. Schlautman and J. J. Morgan, Geochim. Cosmochim. Acta, 1994, 58, 4293–4303 CrossRef CAS.
- S.-Z. Lee, L. Chang, H.-H. Yang, C.-M. Chen and M.-C. Liu, J. Hazard. Mater., 1998, 63, 37–49 CrossRef CAS.
- D. C. Adriano, Trace Elements in the Terrestrial Environment, Springer-Verlag GmbH, Berlin, Heidelberg, 2001 Search PubMed.
- E. F. Covelo, M. L. Andrade and F. A. Vega, J. Food, Agric. Environ., 2004, 2, 244–250 CAS.
- E. Tipping, A. J. Lawlor, S. Lofts and L. Shotbolt, Environ. Pollut., 2006, 141, 139–150 CrossRef CAS PubMed.
- F. A. Vega, E. F. Covelo and M. L. Andrade, J. Colloid Interface Sci., 2006, 298, 582–592 CrossRef CAS PubMed.
- I. Christl and R. Kretzschmar, Geochim. Cosmochim. Acta, 2001, 65, 3435–3442 CrossRef CAS.
- J. A. Davis, Geochim. Cosmochim. Acta, 1984, 48, 679–691 CrossRef CAS.
- M. Bäckström, M. Dario, S. Karlsson and B. Allard, Sci. Total Environ., 2003, 304, 257–268 CrossRef.
- X. L. Tan, X. K. Wang, H. Geckeis and T. Rabung, Environ. Sci. Technol., 2008, 42, 6532–6537 CrossRef CAS PubMed.
- A. Violante, M. Ricciardella and M. Pigna, Water, Air, Soil Pollut., 2003, 145, 289–306 CrossRef CAS.
- Q. Fan, D. Shao, Y. Lu, W. Wu and X. Wang, Chem. Eng. J., 2009, 150, 188–195 CrossRef CAS.
- H. A. Elliott, M. R. Liberati and C. P. Huang, J. Environ. Qual., 1986, 15, 214–219 CrossRef CAS.
- I. Heidmann, I. Christl and R. Kretzschmar, Geochim. Cosmochim. Acta, 2005, 69, 1675–1686 CrossRef CAS.
- T. Zuyi, C. Taiwei, D. Jinzhou, D. XiongXin and G. Yingjie, Appl. Geochem., 2000, 15, 133–139 CrossRef CAS.
- J. M. Zachara, C. T. Resch and S. C. Smith, Geochim. Cosmochim. Acta, 1994, 58, 553–566 CrossRef CAS.
- M. Meier, K. Namjesnik-Dejanovic, P. A. Maurice, Y.-P. Chin and G. R. Aiken, Chem. Geol., 1999, 157, 275–284 CrossRef CAS.
- P. Reiller, B. Amekraz and C. Moulin, Environ. Sci. Technol., 2006, 40, 2235–2241 CrossRef CAS PubMed.
- Q. Zhou, P. A. Maurice and S. E. Cabaniss, Geochim. Cosmochim. Acta, 2001, 65, 803–812 CrossRef CAS.
- C. Galindo and M. Del Nero, Environ. Sci. Technol., 2014, 48, 7401–7408 CrossRef CAS PubMed.
- C. Galindo and M. Del Nero, RSC Adv., 2015, 5, 73058–73067 RSC.
- G. Fleury, M. Del Nero and R. Barillon, Geochim. Cosmochim. Acta, 2017, 196, 1–17 CrossRef CAS.
- W. Stumm and J. J. Morgan, Aquatic chemistry: an introduction emphasizing chemical equilibria in natural waters, Wiley, 1981 Search PubMed.
- J. Buffle, R. R. De Vitre, D. Perret and G. G. Leppard, Geochim. Cosmochim. Acta, 1989, 53, 399–408 CrossRef CAS.
- U. Schwertmann and R. Taylor, in Minerals in Soils Environments, ed. J. B. Dixon and S. R. Weed, 2nd edn, 1989 Search PubMed.
- V. Barrón and J. Torrent, in Minerals at the Nanoscale, ed. F. Nieto, K. J. T. Livi and R. Oberti, Mineralogical Society of Great Britain and Ireland, London, 2013, pp. 297–336 Search PubMed.
- J. A. Davis, Geochim. Cosmochim. Acta, 1982, 46, 2381–2393 CrossRef CAS.
- R. Wagai and L. M. Mayer, Geochim. Cosmochim. Acta, 2007, 71, 25–35 CrossRef CAS.
- M. Del Nero, S. Salah, T. Miura, A. Clément and F. Gauthier-Lafaye, Radiochim. Acta, 1999, 87, 135–150 CrossRef CAS.
- J. L. Jerden and A. K. Sinha, J. Geochem. Explor., 2006, 91, 56–70 CrossRef CAS.
- S. K. Singh, V. Subramanian and R. J. Gibbs, Crit. Rev. Environ. Control, 1984, 14, 33–90 CrossRef.
- A. Tessier, F. Rapin and R. Carignan, Geochim. Cosmochim. Acta, 1985, 49, 183–194 CrossRef CAS.
- C. A. Johnson, Geochim. Cosmochim. Acta, 1986, 50, 2433–2438 CrossRef CAS.
- R. M. Cornell and U. Schwertmann, in The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses, Wiley-VCH Verlag GmbH & Co., 2nd edn, 2003 Search PubMed.
- R. J. Murphy, J. J. Lenhart and B. D. Honeyman, Colloids Surf., A, 1999, 157, 47–62 CrossRef CAS.
- R. S. Swift, in Methods of soil analysis. Part 3. Chemical methods, Soil Science Society of America, Madison, WI, 1996, pp. 1018–1020 Search PubMed.
- B. P. Koch and T. Dittmar, Rapid Commun. Mass Spectrom., 2006, 20, 926–932 CrossRef CAS.
- C. L. Peacock and D. M. Sherman, Geochim. Cosmochim. Acta, 2004, 68, 2623–2637 CrossRef CAS.
- J. D. Ritchie and E. M. Perdue, Geochim. Cosmochim. Acta, 2003, 67, 85–96 CrossRef CAS.
- H. Fu, X. Quan, S. Chen, H. Zhao and Y. Zhao, J. Environ. Sci., 2005, 17, 43–47 CAS.
- S. Ghosh, Z.-Y. Wang, S. Kang, P. C. Bhowmik and B. S. Xing, Pedosphere, 2009, 19, 21–30 CrossRef CAS.
- J. D. Filius, J. C. Meeussen, D. G. Lumsdon, T. Hiemstra and W. H. van Riemsdijk, Geochim. Cosmochim. Acta, 2003, 67, 1463–1474 CrossRef CAS.
- R. Wershaw, Environ. Sci. Technol., 1993, 27, 814–816 CrossRef.
- R. R. Engebretson and R. von Wandruszka, Environ. Sci. Technol., 1994, 28, 1934–1941 CrossRef CAS PubMed.
- A. Piccolo, Soil Sci., 2001, 166, 810–832 CrossRef CAS.
- G. Plancque, B. Amekraz, V. Moulin, P. Toulhoat and C. Moulin, Rapid Commun. Mass Spectrom., 2001, 15, 827–835 CrossRef CAS PubMed.
- R. D. Harter, Soil Sci. Soc. Am. J., 1983, 47, 47–51 CrossRef CAS.
- M. M. Benjamin and J. O. Leckie, J. Colloid Interface Sci., 1981, 79, 209–221 CrossRef CAS.
- L. Bochatay, P. Persson, L. Lövgren and G. E. Brown Jr, J. Phys. IV, 1997, 7, C2–C819 CrossRef.
- D. Rodda, J. Wells and B. Johnson, J. Colloid Interface Sci., 1996, 184, 564–569 CrossRef CAS PubMed.
- A. P. Robertson and J. O. Leckie, Environ. Sci. Technol., 1998, 32, 2519–2530 CrossRef CAS.
- I. Christl and R. Kretzschmar, Geochim. Cosmochim. Acta, 1999, 63, 2929–2938 CrossRef CAS.
- D. A. Dzombak and F. M. Morel, J. Colloid Interface Sci., 1986, 112, 588–598 CrossRef CAS.
- E. M. Logan, I. D. Pulford, G. T. Cook and A. B. MacKenzie, Eur. J. Soil Sci., 1997, 48, 685–696 CrossRef CAS.
- P. Zhou, H. Yan and B. Gu, Chemosphere, 2005, 58, 1327–1337 CrossRef CAS PubMed.
- A. Manceau, M.-C. Boisset, G. Sarret, J.-L. Hazemann, M. Mench, P. Cambier and R. Prost, Environ. Sci. Technol., 1996, 30, 1540–1552 CrossRef CAS.
- S. Pompe, K. Schmeide, M. Bubner, G. Geipel, K. H. Heise, G. Bernhard and H. Nitsche, Radiochim. Acta, 2000, 88 DOI:10.1524/ract.2000.88.9-11.553.
- R. M. Town and M. Filella, Sci. Total Environ., 2002, 300, 143–154 CrossRef CAS PubMed.
- C. Galindo and M. Del Nero, Environ. Sci. Technol., 2014, 48, 7401–7408 CrossRef CAS PubMed.
- F. Claret, T. Schäfer, J. Brevet and P. E. Reiller, Environ. Sci. Technol., 2008, 42, 8809–8815 CrossRef CAS PubMed.
- K. Kaiser, Org. Geochem., 2003, 34, 1569–1579 CrossRef CAS.
- F. Booth, J. Chem. Phys., 1951, 19, 391–394 CrossRef CAS.
- C. R. Evanko and D. A. Dzombak, Environ. Sci. Technol., 1998, 32, 2846–2855 CrossRef CAS.
- R. Marsac, M. Davranche, G. Gruau, A. Dia, M. Pédrot, M. Le Coz-Bouhnik and N. Briant, Chem. Geol., 2013, 342, 119–127 CrossRef CAS.
- C. Catrouillet, M. Davranche, A. Dia, M. Bouhnik-Le Coz, R. Marsac, O. Pourret and G. Gruau, Chem. Geol., 2014, 372, 109–118 CrossRef CAS.
- B. Zinder, G. Furrer and W. Stumm, Geochim. Cosmochim. Acta, 1986, 50, 1861–1869 CrossRef CAS.
- D. S. Gamble, M. Schnitzer and I. Hoffman, Can. J. Chem., 1970, 48, 3197 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Complementary macroscopic data of pH-dependency of PPH sorbed onto hematite at r = 1.4 mg C per m2 (Fig. S1), complementary molecular-scale data: ESI(−)-MS spectra of PPH native solution and of supernatants of PPH sorption experiments onto hematite at pH 3.9 (Fig. S2) and VK diagrams for PPH native solution (Fig. S3), and comparative results of experiments of single and competitive Cu sorption onto 0.375 g L−1 hematite for an initial PPH-to-mineral ratio of 0.1 mg C per m2 (Fig. S4). See DOI: 10.1039/c7ra06838g |
| This journal is © The Royal Society of Chemistry 2017 |