
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic catalytic effects of oxygen and nitrogen functional groups on active carbon electrodes for all-vanadium redox flow batteries†
Min Eui Leea,
Hyoung-Joon Jin *a and
Young Soo Yun*b
*a and
Young Soo Yun*b
aWCSL (World Class Smart Lab) of Green Battery Lab., Department of Polymer Science and Engineering, Inha University, Incheon 22212, South Korea. E-mail: hjjin@inha.ac.kr
bDepartment of Chemical Engineering, Kangwon National University, Samcheok 25913, South Korea. E-mail: ysyun@kangwon.ac.kr
First published on 6th September 2017
Abstract
All-vanadium redox flow batteries (VRFBs) have attracted a great deal of attention as large-scale energy storage devices. However, VRFBs suffer from unfavorable energy efficiency, originating from large polarization effects that influence the redox reaction of the vanadium ions. Although the catalytic effects of the oxygen and nitrogen functional groups in carbon-based electrodes on the redox reactions of the vanadium ion have been documented in several reports, the specific catalytic effects are not understood due to the lack of systematic studies. In this study, pyroproteins having dual functionality (i.e., nitrogen and oxygen functional groups) were coated onto the surface of conventional carbon felts (the composite is hereafter termed P-CFs) and their catalytic effects on the V2+/V3+ redox reaction were characterized and compared with those of carbon felts possessing a coating layer with only oxygen or nitrogen functional groups. The synergistic catalytic effects of nitrogen and oxygen in the pyridonic structure of the P-CFs led to the lowest anodic and cathodic peak potential separation of ∼0.17 V among all the evaluated samples, having various chemical configurations with oxygen or nitrogen only. This study provides insight for the design of carbon-based active electrode materials in VRFBs and their feasibility was demonstrated in VRFB full-cells.
Introduction
Owing to the depletion of fossil fuels and environmental issues, renewable energy technologies such as solar, wind, biomass, and tidal power have attracted much attention globally.1,2 The clean energy from these renewable resources can be naturally replenished on the human timescale. However, power generation by such systems is uncontrolled and may be operative at a time when no additional power is required. To achieve a stable, on-demand energy supply, large-scale electrical energy storage (EES) systems are indispensable.3 The design of redox flow batteries (RFBs) can be flexibly tailored for target applications, and RFBs offer long-term electrochemical stability given that the active redox-couples with no host structure are dissolved in two separate electrolytes that are individually stored in containers of the desired size, making RFBs promising candidates as large-scale EESs.4–6 The operating voltage of RFBs is tunable depending on the redox couples used, some of which are operational within the water splitting voltage window (∼1.23 V) owing to their large overpotential.7,8 However, when different types of redox couples are utilized as the two separated electrolytes, self-discharge originating from ion-crossover leads to decay of the energy efficiency with successive cycles.9,10 Therefore, all-vanadium redox flow batteries (VRFBs) that employ vanadium ions in both the anolyte and catholyte were proposed by Skyllas-Kazacos and coworkers, who achieved considerably reduced ion-crossover.11,12 In addition, the redox reactions between V2+/V3+ (−0.26 V vs. SHE) and VO2+/VO2+ (1.0 V vs. SHE) have a high redox potential (∼1.26 V), leading to high energy density.13,14 Despite the merits of VRFBs, the decline of the cell efficiency due to intrinsic polarization is regarded as a major limitation of VRFBs, hindering their extensive application.15,16 The cell polarization of VRFBs mainly originates from the charge-transfer resistance between the vanadium ions and the electrode, wherein the V(II)/V(III) and V(IV)/V(V) redox reactions are highly affected by the surface properties of the electrodes.17–21 Carbon felt (CF) has typically been used as an electrode for VRFBs because of its three-dimensional macroporous structure that facilitates mass transport, high electrical conductivity, and chemical stability in strongly acidic electrolytes; CF is also amenable to mass production.21–24 However, owing to the poor electrochemical activity and kinetic reversibility, several studies have focused on alleviating activation polarization of CF by surface modification with species such as redox-active heteroatoms and/or nanocomposites with metal-based electrochemical catalysts.15,16,20,21,23–26 Although metal-based catalysts such as Pt,27 Mn3O4,28 WO3,29 CeO2,30 and ZrO2 (ref. 31) exhibit high redox-activities with vanadium redox couples, these species exhibit poor stability under acidic conditions and/or require a relatively complex process with expensive precursors, which is undesirable for large-scale EESs. On the other hand, the introduction of redox-sensitive functional groups onto the surface of CF through facile surface modification using well-developed chemistry leads to significant mitigation of the overpotential originating from the major activation polarization process.20,21,23,25,32–36 Kim et al. reported that CF with oxygen functional groups (e.g., hydroxyl or carboxylic acid groups) introduced onto the surface via combined corona discharge and hydrogen peroxide was suitable as a redox host for the VO2+/VO2+ couples.35 It was also reported that the redox activity could be improved by co-doping nitrogen atoms and oxygen groups onto the surface of CF.19,36 Despite these results and though the effects of nitrogen, oxygen, and their hybrid groups on carbon-based electrodes have been demonstrated in several reports, systematic studies of their catalytic effects remain insufficient.17,19,20,23,26,32,33,36–38 Therefore, it is difficult to understand which types of heteroatoms or chemical configurations are most effective, what is the relationship between the heteroatom content and the catalytic effect, and whether there is a synergistic effect between the oxygen and nitrogen functional groups. Furthermore, it is noteworthy that the catalytic reactions of the redox-active heteroatoms with V2+/V3+ and VO2+/VO2+ progress independently, leading to different catalytic effects on both sides.23 In other words, separate surface modification is needed for the anode and the cathode in order to enhance the catalytic effects. Nevertheless, most of the literature has focused on development of the cathode for the VO2+/VO2+ redox couples, whereas there are few reports concerning the anode.15,39,40 Considering that the theoretical redox voltage of the V2+/V3+ couples is 0.26 V lower than that of the hydrogen evolution reaction (0 V vs. SHE) in acidic aqueous electrolyte, competition between the desired reaction and water splitting is inevitable; this may have a negative impact on the reaction kinetics and efficiency.39In this study, thin pyroprotein coating layers containing numerous oxygen and nitrogen heteroatoms are introduced onto the surface of CF (nitrogen and oxygen co-doped sample; P-CFs), and their catalytic effects on the redox reaction of V2+/V3+ couples for VRFBs are investigated. Further, to confirm the inter-relation between the catalytic effects of the surface species, oxygen or nitrogen-doped carbonaceous coating layers are prepared on the surface of CF via glucose (oxygen-doped sample; O-CFs) or melamine (nitrogen-doped sample, N-CFs) adsorption with subsequent thermal treatment. It is demonstrated that the pyroprotein coated CFs exhibits a significantly lower anodic and cathodic peak potential separation (ΔEp) of ∼0.17 V, compared with those of O-CFs and N-CFs (∼0.32 and ∼0.24 V, respectively); even with the P-CF sample having a lower heteroatom content than those of the O-CFs and N-CFs, a superior ΔEp value is achieved. These results clearly demonstrate the synergistic catalytic effects of the oxygen and nitrogen heteroatoms on the V2+/V3+ redox reactions. Furthermore, full-cell VRFBs based on P-CFs exhibit enhanced energy efficiency, rate capability, and stable cycling behavior compared with those based on CFs.
Experimental section
Preparation of regenerated silk fibroin solution
Cocoons from B. mori silkworms were obtained from the Uljin Farm, South Korea. To extract the glue-like sericin protein and impurities, the silkworm cocoons were boiled for 25 min in an aqueous solution of 0.02 M Na2CO3 (i.e., degumming process). After washing with deionized (DI) water several times, the resulting silk fibroin was dried at room temperature for 3 days. The regenerated silk protein solutions were prepared as described elsewhere.41 Briefly, the degummed silk fibroin was dissolved in an aqueous solution of 9.3 M LiBr (99%; Sigma-Aldrich, St. Louis, MO, USA) at 60 °C for 6 h. The silk protein solution was dialyzed in water using a Slide-a-Lyzer dialysis cassette (MWCO 3500; Pierce) for 2 days. The final concentration of the regenerated silk fibroin (RSF) in the aqueous solution was approximately 8 wt%.Preparation of P-CFs, O-CFs, and N-CFs
P-CFs, O-CFs, and N-CFs were prepared by simple solution-coating of pristine CFs (XF-30A, thickness: 4.3 mm, Toyobo, Japan), followed by thermal treatment at the desired temperature. The CF was dip-coated in an aqueous 0.5 wt% solution of RSF, glucose (99.5%, Sigma-Aldrich), or melamine (99%, Sigma-Aldrich) and dried in a vacuum oven at 30 °C for 3 h. The RSF, glucose, or melamine-coated CFs were heated at different temperatures (400, 600, 800, 1000, and 1200 °C) in a tube furnace. A heating rate of 5 °C min−1 and an Ar flow rate of 200 mL min−1 were applied; the samples were held at the final temperature for 2 h.Characterization
The morphologies of the samples were observed using field-emission scanning electron microscopy (FE-SEM, S-4300SE, Hitachi, Japan). X-ray diffraction (XRD, Rigaku DMAX-2500) analysis of the samples was performed using Cu-Kα radiation (wavelength λ = 0.154 nm) at 40 kV and 100 mA. Raman spectra were acquired using a continuous-wave, linearly polarized laser (514.4 nm wavelength, 2.41 eV, 16 mW power). The laser beam was focused by a 100× objective lens, resulting in a spot with a diameter of ∼1 μm. The acquisition time and number of circulations used to collect each spectrum were 10 s and 5, respectively. The chemical compositions of the samples were examined by XPS (AXIS-HIS, Kratos Analytical, Japan) using monochromated Mg-Kα radiation (hν = 1486.6 eV).Electrochemical characterization
The electrochemical performance of the samples and their flow-cell devices were characterized with the aid of a potentiostat (PGSTAT302N, Autolab), Wonatech automatic battery cycler, and single-cell test equipment (Standard Cell – Blue, Standard Energy Co., Ltd., Republic of Korea). For the half-cell experiments, a hand-made, three-electrode configuration system was used,42 in which P-CFs, O-CFs, or N-CFs with a diameter of 5 mm (area: 0.196 cm2) excess CF, and Ag/AgCl were used as the working, counter, and reference electrodes, respectively. VOSO4 (0.1 M, 99.5%, Sigma-Aldrich) was dissolved in 2 M H2SO4 (96.5%, Sigma-Aldrich) and used as the electrolyte for the half-cell tests. For the full-cell experiments, a single cell was assembled with copper current collectors, bipolar plates, and P-CF//CF or CF//CF electrode pair, and cation exchange membranes (Nafion 115, DuPont Co., Ltd., USA). As a pre-treatment, the membrane was immersed in DI water for 24 h. The working electrodes with an area of 9 cm2 were used as both the cathode and anode. The electrolyte was prepared by dissolving 1.6 M VOSO4 in 4 M H2SO4 solution and the electrolyte volume in each of the catholyte and anolyte tanks was 60 mL.Results and discussion
The morphologies of the CFs and P-CFs-400 samples (where the heat-treatment temperature is indicated at the end of the nomenclature, e.g. −400, −600, etc.) are shown in Fig. 1(a) and (b). Both samples have a similar macroporous structure and smooth topologies, suggesting homogeneous coating of the pyroprotein layer on the surface of the CFs. It was also confirmed that N-CFs-400 and O-CFs-400 exhibited no morphological differences compared with CFs and P-CFs (Fig. S1†). The surface carbon microstructure of the samples was characterized by acquiring the Raman spectra, which showed characteristic D and G bands of hexagonal carbon structure centered at ∼1350 and ∼1580 cm−1, respectively. The D and G bands originate from the intrinsic phonon mode of the infinite aromatic ring with A1g symmetry that is activated by the structural disorder and the hexagonal carbon structure related to the E2g vibration mode of the sp2-hybridized C atoms, respectively. Hence, the D to G intensity ratio (ID/IG) indicates the approximate size of the poly-hexagonal carbon structures. The ID/IG ratios of P-CFs-400, N-CFs-400, and O-CFs-400 were ∼1.02, ∼1.00, and ∼1.01, which are higher than that (0.86) of CFs, indicating that the surface carbon structures of the P-CFs-400, N-CFs-400, and O-CFs-400 samples have more defects. In addition, the G bands of CFs were distinctively shifted to higher frequencies (blue-shift) in the case of P-CFs-400, N-CFs-400, and O-CFs-400. The blue-shift of the G band suggests that the Raman-inactive non-zero phonon density of states above the G band become active owing to phonon confinement caused by the created defects.43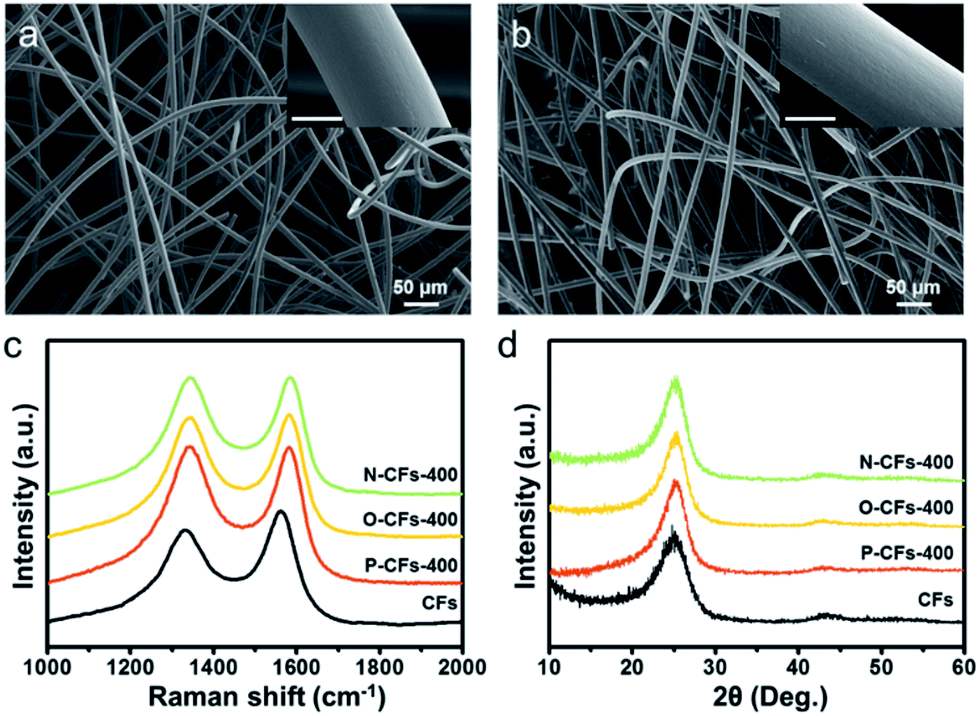 | ||
| Fig. 1 FE-SEM images of (a) CFs and (b) P-CFs-400 (inset: FE-SEM images, scale bar: 5 μm). (c) Raman spectra and (d) XRD patterns of CFs, P-CFs-400, O-CFs-400, and N-CFs-400. | ||
These results also confirm that the properties of the surface carbon in the samples were altered by introducing the coating layers onto the surface of the CFs. In contrast, the X-ray diffraction (XRD) patterns of the samples showed similar broad peaks at 25.3°, corresponding to the graphitic (002) structure, suggesting that the bulk carbon structures remained unchanged. The modified surface properties of P-CFs-400, N-CFs-400, and O-CFs-400 were characterized using X-ray photoelectron spectroscopy (XPS), as shown in Fig. 2. The O 1s spectra of P-CFs-400 and O-CFs-400 show similar peaks corresponding to chemical structures with C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds (Fig. 2(a)), although the quantity of oxygen and the bonding ratios were different for the samples (Fig. 2(b) and (c)). The C/O ratio of O-CFs-400 was 4.5, which is much smaller than those of CF (15.8) and P-CF (8.6). The oxygen content of O-CFs and P-CFs gradually decreased with an increase in the heat-treatment temperature (HTT) from 400 to 1200 °C (see Fig. S2 and S3†). The N 1s spectra of P-CFs-400 and N-CFs-400 show similar peaks of pyridinic-N and N-oxide structures at ∼398.3 and ∼403.2 eV, respectively (Fig. 2(d)). Further, the major chemical bonds gave rise to peaks at ∼400.6 and ∼400.1 eV for P-CFs-400 and N-CFs-400, respectively. It is noteworthy that although the main peaks of P-CFs-400 and N-CFs-400 were located in similar positions, the peaks indicate different chemical structures. Due to the presence of edge sites of poly-hexagonal carbon structures with numerous oxygen and nitrogen, the main peak of P-CFs-400 could originate from the pyridonic-N structure, whereas for N-CFs-400 containing numerous nitrogen species and negligible oxygen, the major peak corresponds to the pyrrolic-N structure. Except the chemical configuration of the major peak, there was no distinct difference in the nitrogen bonding of P-CFs-400 and N-CFs-400 (Fig. 2(e)). As shown in Fig. 2(f), the CFs sample had a negligible nitrogen content corresponding to a C/N ratio of 84.5, whereas the P-CFs-400 sample had a considerably high nitrogen content (C/N ratio of 12.1), which is about 7 times higher than that of CFs (Fig. 2(f)). Moreover, the nitrogen content of N-CFs-400 is around 1.5 times higher than that of P-CFs-400, corresponding to a C/N ratio of 7.5. The nitrogen content of P-CFs and N-CFs also decreased with a gradual increase in the HTT from 400 to 1200 °C (see Fig. S3†). The specific C/O and C/N ratios of the samples are summarized in Tables S1–S3.† Additionally, the modified surfaces of P-CFs-400, N-CFs-400, and O-CFs-400 exhibited good wettability, which is in clear contrast with that of CFs (Fig. S4†).
O bonds (Fig. 2(a)), although the quantity of oxygen and the bonding ratios were different for the samples (Fig. 2(b) and (c)). The C/O ratio of O-CFs-400 was 4.5, which is much smaller than those of CF (15.8) and P-CF (8.6). The oxygen content of O-CFs and P-CFs gradually decreased with an increase in the heat-treatment temperature (HTT) from 400 to 1200 °C (see Fig. S2 and S3†). The N 1s spectra of P-CFs-400 and N-CFs-400 show similar peaks of pyridinic-N and N-oxide structures at ∼398.3 and ∼403.2 eV, respectively (Fig. 2(d)). Further, the major chemical bonds gave rise to peaks at ∼400.6 and ∼400.1 eV for P-CFs-400 and N-CFs-400, respectively. It is noteworthy that although the main peaks of P-CFs-400 and N-CFs-400 were located in similar positions, the peaks indicate different chemical structures. Due to the presence of edge sites of poly-hexagonal carbon structures with numerous oxygen and nitrogen, the main peak of P-CFs-400 could originate from the pyridonic-N structure, whereas for N-CFs-400 containing numerous nitrogen species and negligible oxygen, the major peak corresponds to the pyrrolic-N structure. Except the chemical configuration of the major peak, there was no distinct difference in the nitrogen bonding of P-CFs-400 and N-CFs-400 (Fig. 2(e)). As shown in Fig. 2(f), the CFs sample had a negligible nitrogen content corresponding to a C/N ratio of 84.5, whereas the P-CFs-400 sample had a considerably high nitrogen content (C/N ratio of 12.1), which is about 7 times higher than that of CFs (Fig. 2(f)). Moreover, the nitrogen content of N-CFs-400 is around 1.5 times higher than that of P-CFs-400, corresponding to a C/N ratio of 7.5. The nitrogen content of P-CFs and N-CFs also decreased with a gradual increase in the HTT from 400 to 1200 °C (see Fig. S3†). The specific C/O and C/N ratios of the samples are summarized in Tables S1–S3.† Additionally, the modified surfaces of P-CFs-400, N-CFs-400, and O-CFs-400 exhibited good wettability, which is in clear contrast with that of CFs (Fig. S4†).
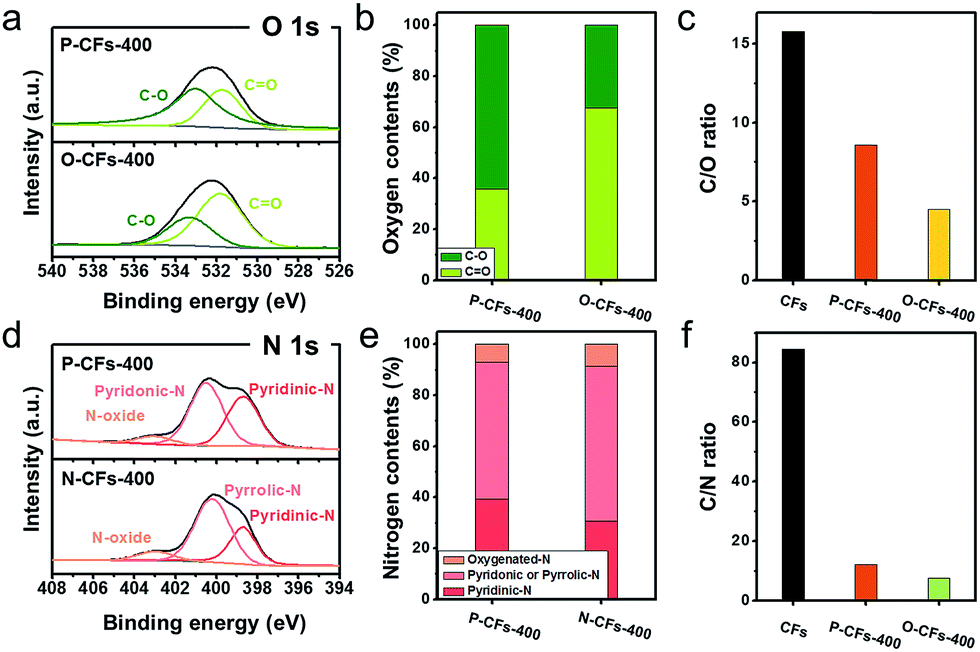 | ||
| Fig. 2 XPS analysis for P-CFs-400, O-CFs-400, and N-CFs-400. (a) High-resolution O 1s spectra, (b) oxygen chemical composition and (c) content of oxygen functional groups for P-CFs-400 and O-CFs-400. (d) High-resolution N 1s spectra, (e) nitrogen chemical composition and (f) content of nitrogen functional groups for P-CFs-400 and N-CFs-400. | ||
The electrochemical catalytic activity of CFs, P-CFs-400, O-CFs-400, and N-CFs-400 was evaluated vs. Ag/AgCl in the voltage window of 0 to −1.0 V by using a hand-made three-electrode configuration system with 0.1 M VOSO4 dissolved in 2 M H2SO4 solution as the electrolyte (Fig. S5†). The cyclic voltammograms (CVs) of the samples characterized at 5 mV s−1 present distinct redox peaks with different ΔEp values and peak current densities (Fig. 3(a)). P-CFs-400 gave rise to the lowest ΔEp value (∼170.9 mV) of all the characterized samples, including N-CFs-400 and O-CFs-400 with ΔEp values of 238.0 and 352.4 mV, respectively. These results reveal that the catalytic effects of active carbon materials on the redox reaction of V2+/V3+ couples are not just dependent on the quantitative values of the active chemical species because O-CFs-400, having the highest heteroatom content, gave rise to a relatively poor ΔEp value. In the specific case of O-CFs-400, although the oxygen content declined significantly with increasing HTT, the ΔEp values obtained at the same sweep rate of 5 mV s−1 showed a moderate increase from 323.4 to 359.1 mV, corresponding to 11% (Table S4†). The overall ΔEp values of O-CFs were similar to that (363.0 mV) of CFs (Table S4†). In contrast, the ΔEp values of N-CF declined continuously from 238.0 to 352.4 mV as the C/N ratio increased, corresponding to an ∼82% decrease (Table S5†). Therefore, from a qualitative point of view, the superiority of nitrogen groups compared with oxygen groups can be verified from these results. In addition, the ΔEp value of P-CFs is much smaller than them of previously reported results (Table S7†). However, in order to account for the highest performance of P-CFs-400 with a relatively low nitrogen content compared with N-CFs, a new interpretation of the catalytic effect on the V2+/V3+ redox reaction is required. Considering the chemical structures of N-CFs and P-CFs, the better electrochemical performance of P-CFs may be induced by the pyridonic structure of P-CFs-400. In particular, a clear difference was observed in the ex situ O 1s spectra of P-CFs-400 versus O-CFs-400 during the redox reaction, as shown in Fig. S6.† In the XPS O 1s spectra of P-CFs-400 acquired after the reduction reaction at −0.8 V, a strong shift (of about 1.1 eV) of the O 1s peak toward lower binding energy was observed, whereas the XPS O 1s spectra of O-CFs acquired under the same conditions showed a weak shift (about 0.3 eV) (Fig. S7†). This result suggests a strong increase in the electron density of the oxygen groups of P-CFs-400, which could be accompanied by the redox reaction of the V2+/V3+ couple. Although the oxygen chemical configurations of P-CFs-400 and O-CFs-400 are similar, the peaks of the oxygen groups of P-CFs-400 were more strongly shifted, which is not explained by a redox reaction involving just oxygen groups. Although it remains inconclusive, as shown in the schematic illustration of Fig. 4, the nitrogen beside the oxygen on the edge site of the poly-hexagonal carbon structures possibly can work in tandem with the oxygen in the redox reaction of V2+/V3+ couples. The synergistic catalytic effects of nitrogen and oxygen have been observed in alkali cation redox reactions.44,45
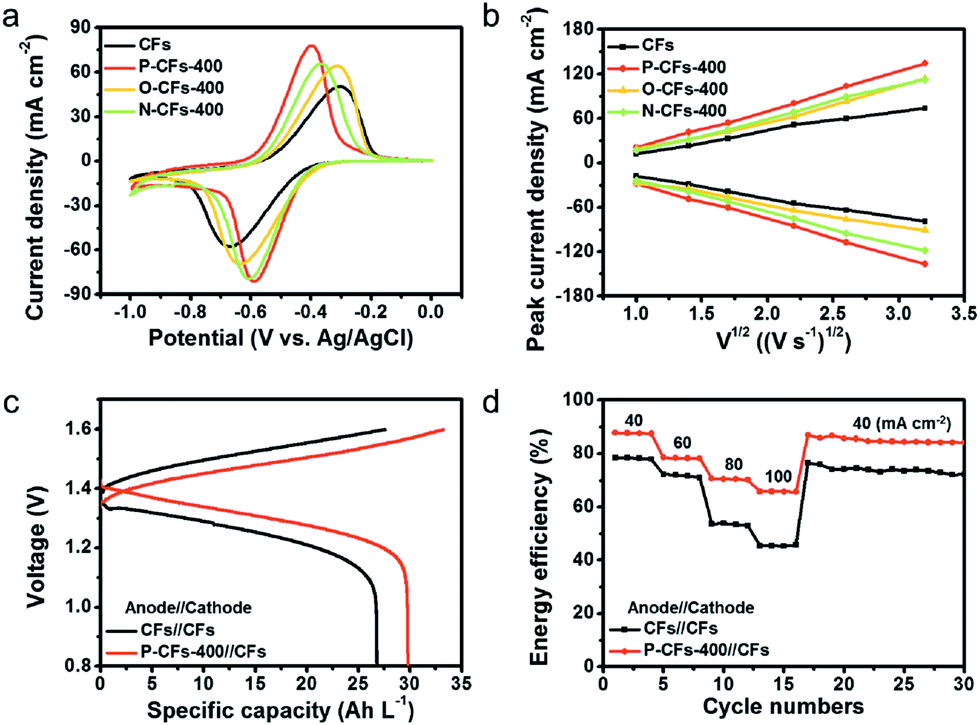 | ||
| Fig. 3 Electrochemical performance characterized using half-cell and full-cell configurations. (a) CV curves at a sweep rate of 5 mV s−1 and (b) peak current densities at different sweep rates from 1 to 10 mV s−1 for CFs, P-CFs-400, O-CFs-400, and N-CFs-400 over a voltage window of −1.0 to 0 V vs. Ag/AgCl in an electrolyte of 0.1 M VOSO4 dissolved in 2 M H2SO4; half-cell was used. VRFB single-cell operation based on CFs//CFs and P-CFs-400//CFs: (c) galvanostatic charge/discharge profiles at a current density of 60 mA cm−2 and (d) energy efficiency for continuous charging–discharging cycles at different current densities over a voltage window of 0.8–1.6 V in an electrolyte of 1.6 M VOSO4 dissolved in 4 M H2SO4. | ||
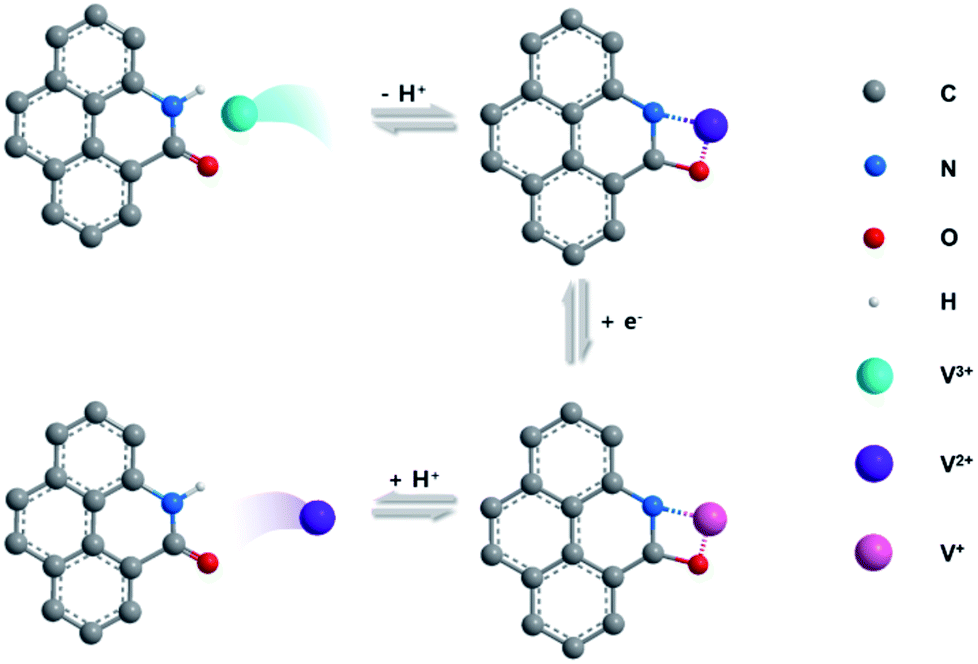 | ||
| Fig. 4 Schematic image exhibiting possible mechanism of redox reaction of V2+/V3+ redox couple on pyridonic structure of P-CFs. Oxygen and nitrogen functional groups can provide a catalytic effect for the redox reaction by their synergistic behavior. | ||
Furthermore, the slopes of the square root of the sweep rate versus oxidation and reduction peak current density plots have larger value for P-CFs-400 than for O-CFs-400, N-CFs-400, and CFs, indicating better mass transfer in P-CFs-400 (Fig. 3(b)). Moreover, the catalytic effects of the samples on the VO2+/VO2+ couple are completely different from their catalytic effects on the redox reaction of the V2+/V3+ couple (Tables S4–S6†). These results also suggest that the catalytic effects of the surface functional groups on the anolyte and catholyte are different. Electrochemical impedance spectroscopy (EIS) data support our claims, as shown in Fig. S7.† For the redox reaction of V2+/V3+ couple, P-CFs-400 exhibited the lowest charge transfer resistance in all the tested samples including CFs, O-CFs-400 and N-CFs-400. In contrast, P-CFs-400, O-CFs-400 and N-CFs-400 showed the similar charge transfer resistance on the redox reaction of the VO2+/VO2+ couple. Additionally, the cycling stability of P-CFs-400 was tested by repetitive CV tests at a sweep rate of 10 mV s−1 (Fig. S8†). During 200 cycles, the CV curves of P-CFs-400 were slightly changed for the redox reactions of both V2+/V3+ and VO2+/VO2+ couples, indicating good cycling stability of P-CFs-400. The feasibility of using P-CFs-400 as the anolyte was further tested in full-cell VRFB systems (Fig. S8†), which were assembled with CFs as the counter electrode for the catholyte, and the electrochemical performance was compared with that of all the CFs systems. The galvanostatic charge–discharge profiles of P-CFs-400//CFs characterized in the operating voltage window of 0.8–1.6 V at a current density of 60 mA cm−2 showed lower overpotentials of ∼50 and ∼60 mV in the charge and discharge profiles, respectively, compared with those of CFs//CFs (Fig. 3(c)). Therefore, under the same given conditions, P-CFs-400//CFs showed an increased capacity of ∼29.8 A h L−1 compared with that (∼26.8 A h L−1) of CFs//CFs, and the gap was more pronounced with an increase in the current density, as shown in Fig. S8(a).† In addition, the voltage efficiency of P-CFs-400//CFs was significantly higher than that of CFs//CFs (Fig. S8(b)†). As a result, the catalytic effects of the pyridonic-N groups led to improved energy efficiency of P-CFs-400//CFs by reducing the polarization for the redox reaction of the V2+/V3+ couple (Fig. 3(d)). Although the energy efficiency of P-CFs-400//CFs is slightly higher than them of the previously reported values, the results shows the catalytic effects of P-CFs-400 in the anodic voltage region (Table S8†).46,47
Conclusions
In summary, the surface of CF was dual-coated with oxygen and nitrogen-rich pyroproteins (P-CF) and the catalytic effects of the composites on the redox reaction of the V2+/V3+ couple were compared with those of O-CFs and N-CFs. Whereas the oxygen groups on the surface of CFs have an insignificant catalytic effect on the V2+/V3+ redox reaction, nitrogen groups led to a noticeable decrease of the ΔEp values in a nitrogen-concentration dependent manner, indicating superior catalytic effects of the nitrogen groups compared with oxygen on the V2+/V3+ redox couple. In contrast, P-CFs-400, with a lower nitrogen content than N-CFs-400, had the lowest ΔEp value of ∼170.9 mV among the evaluated samples. This result suggests that the nitrogen and oxygen groups have a synergistic catalytic effect, especially the pyridonic groups, which are the main chemical structures in P-CFs-400. The ex situ XPS O 1s spectra showed a strong peak shift (∼1.1 eV) for P-CFs-400, whereas that of O-CFs-400 exhibited a slight peak shift (∼0.3 eV) despite the similarity of their oxygen configurations. These results support the proposed synergistic catalytic effects of oxygen and nitrogen in the pyridonic-N structure. Furthermore, the feasibility of the P-CF-based electrode was demonstrated in a full-cell VRFB system, where significantly improved energy efficiency was achieved compared with that of all CF-based electrodes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by WCSL (World Class Smart Lab) research grant directed by Inha University.References
- S. Chu and A. Majumar, Nature, 2012, 488, 294 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928 CrossRef CAS PubMed.
- D. Larcher and J.-M. Tarascon, Nat. Chem., 2015, 7, 19 CrossRef CAS PubMed.
- W. Wang, Q. Luo, B. Li, X. Wei, L. Li and Z. Yang, Adv. Funct. Mater., 2013, 23, 970 CrossRef CAS.
- K. Lin, Q. Chen, M. R. Gerhardt, L. Tong, S. B. Kim, L. Eisenach, A. W. Valle, D. Hardee, R. G. Gordon, M. J. Aziz and M. P. Marshak, Science, 2015, 349, 1529 CrossRef CAS PubMed.
- M. Park, J. Ryu, W. Wang and J. Cho, Nat. Rev. Mater., 2016, 2, 16080 CrossRef.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, Bora Raton, FL, USA, 2007 Search PubMed.
- F. F. Abdi, L. Han, A. H. M. Smets, M. Zeman, B. Dam and R. van de Krol, Nat. Commun., 2013, 4, 2195 Search PubMed.
- R. M. Darling, A. Z. Weber, M. C. Tucker and M. L. Perry, J. Electrochem. Soc., 2016, 163, A5014 CrossRef CAS.
- J. Austing, C. N. Kirchner, L. Komsiyska and G. Wittstock, J. Power Sources, 2016, 306, 692 CrossRef.
- M. Skyllas-Kazacos, M. Rychcik, R. G. Robins, A. G. Fane and M. Green, J. Electrochem. Soc., 1986, 133, 1057 CrossRef CAS.
- M. Skyllas-Kazacos and G. Grossmith, J. Electrochem. Soc., 1987, 134, 2950 CrossRef CAS.
- E. Sum and M. Skyllas-Kazacos, J. Power Sources, 1985, 15, 179 CrossRef CAS.
- E. Sum, M. Rychcik and M. Skyllas-Kazacos, J. Power Sources, 1985, 16, 85 CrossRef CAS.
- B. Li, M. Gu, Z. Nie, Y. Shao, Q. Luo, X. Wei, X. Li, J. Xiao, C. Wang, V. Sprenkle and W. Wang, Nano Lett., 2013, 13, 1330 CrossRef CAS PubMed.
- B. Li, M. Gu, Z. Nie, X. Wei, C. Wang, V. Sprenkle and W. Wang, Nano Lett., 2014, 14, 158 CrossRef CAS PubMed.
- M. Park, I.-Y. Jeon, J. Ryu, J.-B. Baek and J. Cho, Adv. Energy Mater., 2015, 5, 1401550 CrossRef.
- Y. Zhou, L. Liu, Y. Shen, L. Wu, L. Yu, F. Liang and J. Xi, Chem. Commun., 2017, 53, 7565 RSC.
- M. Park, J. Ryu, Y. Kim and J. Cho, Energy Environ. Sci., 2014, 7, 3727 CAS.
- J. Kim, H. Lim, J.-Y. Jyoung, E.-S. Lee, J. S. Yi and D. Lee, Carbon, 2017, 111, 592 CrossRef CAS.
- A. M. Pezeshki, J. T. Clement, G. M. Veith, T. A. Zawodzinski and M. M. Mench, J. Power Sources, 2015, 294, 333 CrossRef CAS.
- J. H. Park, J. J. Park, O. O. Park and J. H. Yang, ChemSusChem, 2016, 9, 1 CrossRef.
- K. J. Kim, H. S. Lee, J. Kim, M.-S. Park, J. H. Kim, Y.-J. Kim and M. Skyllas-Kazacos, ChemSusChem, 2016, 9, 1329 CrossRef CAS PubMed.
- T. Liu, X. Li, H. Nie, C. Xu and H. Zhang, J. Power Sources, 2015, 286, 73 CrossRef CAS.
- D. Dixon, D. J. Babu, J. Langner, M. Bruns, L. Pfaffmann, A. Bhaskar, J. J. Schneider, F. Scheiba and H. Ehrenberg, J. Power Sources, 2016, 332, 240 CrossRef CAS.
- J. J. Park, J. H. Park, O. O. Park and J. H. Yang, Carbon, 2016, 110, 17 CrossRef CAS.
- S. Jeong, S. Kim and Y. Kwon, Electrochim. Acta, 2013, 114, 439 CrossRef CAS.
- K. J. Kim, M.-S. Park, J.-H. Kim, U. Hwang, N. J. Lee, G. Jeong and Y.-J. Kim, Chem. Commun., 2012, 48, 5455 RSC.
- D. M. Kabtamu, J.-Y. Chen, Y.-C. Chang and C.-H. Wang, J. Mater. Chem. A, 2016, 4, 11472 CAS.
- M. Jing, X. Zhang, X. Fan, L. Zhao, J. Liu and C. Yan, Electrochim. Acta, 2016, 215, 57 CrossRef CAS.
- H. Zhou, Y. Shen, J. Xi, X. Qiu and L. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 15369 CAS.
- S. Wang, X. Zhao, T. Cochell and A. Manthiram, J. Phys. Chem. Lett., 2012, 3, 2164 CrossRef CAS PubMed.
- S. Park and H. Kim, J. Mater. Chem. A, 2015, 3, 12276 CAS.
- L. Estevez, D. Reed, Z. Nie, A. M. Schwarz, M. I. Nandasiri, J. P. Kizewski, W. Wang, E. Thomsen, J. Liu, J.-G. Zhang, V. Sprenkle and B. Li, ChemSusChem, 2016, 9, 1455 CrossRef CAS PubMed.
- K. J. Kim, S.-W. Lee, T. Yim, J.-G. Kim, J. W. Choi, J. H. Kim, M.-S. Park and Y.-J. Kim, Sci. Rep., 2014, 4, 6906 CrossRef CAS PubMed.
- C. Flox, J. Rubio-García, M. Skoumal, T. Andreu and J. R. Morante, Carbon, 2013, 60, 280 CrossRef CAS.
- L. Wu, Y. Shen, L. Yu, J. Xi and X. Qiu, Nano Energy, 2016, 28, 19 CrossRef CAS.
- M. Park, Y.-J. Jung, J. Kim, H. I. Lee and J. Cho, Nano Lett., 2013, 13, 4833 CrossRef CAS PubMed.
- D. J. Suárez, Z. González, C. Banco, M. Granda, R. Menéndez and R. Santamaría, ChemSusChem, 2014, 7, 914 CrossRef PubMed.
- T.-M. Tseng, R.-H. Huang, C.-Y. Huang, C.-C. Liu, K.-L. Hsueh and F.-S. Shieu, J. Electrochem. Soc., 2014, 161, A1132 CrossRef CAS.
- Y. S. Yun, K.-Y. Park, B. Lee, S. Y. Cho, Y.-U. Park, S. J. Hong, B. H. Kim, H. Gwon, H. Kim, S. Lee, Y. W. Park, H.-J. Jin and K. Kang, Adv. Mater., 2015, 27, 6914 CrossRef CAS PubMed.
- J. H. Park, J. J. Park, O. O. Park, C.-S. Jin and J. H. Yang, J. Power Sources, 2016, 137, 137 CrossRef.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud'homme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36 CrossRef CAS PubMed.
- M. Lee, J. Hong, D.-H. Seo, D. H. Nam, K. T. Nam, K. Kang and C. B. Park, Angew. Chem., Int. Ed., 2013, 52, 8322 CrossRef CAS PubMed.
- Y. S. Yun, D.-H. Kim, S. J. Hong, M. H. Park, Y. W. Park, B. H. Kim, H.-J. Jin and K. Kang, Nanoscale, 2015, 7, 15051 RSC.
- B. Jiang, L. Wu, L. Yu, X. Qiu and J. Xi, J. Membr. Sci., 2016, 510, 18 CrossRef CAS.
- L. Wu, J. Wang, Y. Shen, L. Liu and J. Xi, Phys. Chem. Chem. Phys., 2017, 19, 14708 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1 to S8 and Fig. S1 to S10. See DOI: 10.1039/c7ra08334c |
| This journal is © The Royal Society of Chemistry 2017 |