
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcetylation of BLM protein regulates its function in response to DNA damage
Yankun Wang and
Jianyuan Luo *
*
Department of Medical Genetics, Peking University Health Science Center, 38 Xueyuan Road, Beijing, China 100191. E-mail: wangyankun@bjmu.edu.cn; luojianyuan@bjmu.edu.cn
First published on 5th December 2017
Abstract
Bloom syndrome is an autosomal recessive disease with phenotypes of cancer predisposition and premature aging caused by mutations of the blm gene. BLM belongs to the RecQ DNA helicase family and functions in maintaining genomic stability. In this study, we found that several lysine residues of BLM were acetylated in cells. The dynamic acetylation levels of BLM were regulated by CBP/p300 and SIRT1. We further identified that five lysines, K476, K863, K1010, K1329, and K1411, are the major acetylation sites. Treating cells with different DNA damage agents found that acetylation of BLM was different in response to etoposide and hydroxyurea, suggesting that BLM acetylation may have multiple functions in DNA repair.
Introduction
Bloom syndrome protein (BLM), coded by the blm gene, is a 1417 amino acid protein. Mutations or deletions of the blm gene lead to Bloom Syndrome (BS).1 It is an inherited autosomal-recessive disease characterized by the increase of sister chromatid exchange, predisposition to cancers, and chromosomal aberrations.2,3 BLM belongs to the RecQ family of DNA helicases which contain a core helicase domain.4,5 Mutations in WRN and RecQL4, the other members of the RecQ helicase family, also lead to two other premature aging and cancer-prone syndromes, Werner and Rothmund–Thomson syndromes, respectively.6,7The functional role of BLM is to maintain genomic stability and it has been shown to be involved in a number of DNA metabolism processes.8 BLM is one of the most essential regulator proteins in the repair of DNA double strand breaks (DSBs).9 DSBs usually cause serious damage to cells due to severe loss of genetic materials. In human cells, there are two pathways to repair DSBs: homologous recombination repair (HR) and non-homologous end joining repair (NHEJ).10,11 BLM participates in both pathways. In the pathway of HR, BLM protects DNA polymerases and the HR precursor structure reaching the stalled DNA forks and provides a platform for exonucleases and other DNA repair proteins. Further in the repair process, BLM produces longer stretches of ssDNA with the cooperation of EXO1 and DNA2 and helps recruiting RAD51 to the damage site and consequently exchanges with the RPA.12,13 Furthermore, when HR repair is finished, BLM dissolutes the D-loop sequence generated by the homologous recombination and makes DNA back to integrated condition.14 On the other hand, BLM interacts with 53BP1 and completes the repair in the NHEJ pathway.11 It has been found that BLM is sensitive to multiple stress factors, including hydroxyurea (HU), etoposide and ionizing radiation (IR) which all lead to DSBs.10,15 Although the survival rate is significantly decreased in BLM deficient cells, they showed different states after dealing with these stress factors.16,17 The complex regulatory mechanism for BLM protein that functions in DNA double-strand break repairs still remains elusive.
Integrity of genome maintenance is primarily reflected in DNA damage and repair.18 There are amounts of harmful factors that lead to defense responses stimulating DNA damage in vivo. DNA damage response (DDR) depends on a complicated signaling pathway regulated by DNA repair proteins, and coordinates with specific factors.19 Post-translational modifications play crucial roles in regulating this machinery. For example, ubiquitination of H2A/H2A.X is a precondition for 53BP1 and BRCA1 recruited to DNA strands.20,21 Post-translational modifications were also a pivotal approach for BLM functioning when chromosomes were aberrant.22 Phosphorylation, ubiquitination, and SUMOylation of BLM have been shown to regulate the special signaling pathways of DDR and interactions between its partners. BLM restarts an origin of replication after HU or IR treatment via phosphorylation by ATM/ATR at Thr99 and Thr122.23,24 Ubiquitination of BLM at Lys105, 225, and 259 mediated by RNF8/RNF168 leads to timely degradation upon failing to repair the damage.25 SUMOylation of BLM is necessary for the interaction between BLM and RAD51 and promotes HR repair.26
Acetylation is one of the popular post-translational modifications controlling diverse cellular processes.27 Recent studies indicated that acetylation on nonhistone proteins takes critical effects on DNA repair regulation. It has been reported that acetylation modulates the functions of proteins involved in the repair of DNA damage.28 When cells overexpressed enzymatic inactive acetyltransferase Tip60, reparation of double strand breaks becomes defective.29 Tip60 acetylates ATM leads to ATM Ser1981 autophosphorylation and activation in response to DNA damage.30 NBS1 acetylation inhibits its phosphorylation and sensitizes cells to ionizing irradiation,31 and SIRT1 mediated deacetylation of NBS1 is important for the function of NBS1 in DSBs repair. WRN acetylation helps in the translocation from nucleoli to DNA damage foci in early stage after DNA damage while in late stage, SIRT1 mediated deacetylation increases its enzymatic activities and facilitates DNA repair.32 Deacetylation of XPA also plays a positive role in the repair of UV-induced DNA damage.33 RPA1 acetylation enhances its interaction with XPA and promotes nucleotide excision repair (NER),34,35 and SIRT1 mediated deacetylation also enhances the interaction between TopBP1 and Rad9 and activates the ATR-Chk1 pathway.36
BLM is an indispensable protein in DNA metabolism. It is necessary to understand the function of its post-translational modifications in DNA repair. It has been found that acetylation on lysine residues take great effects on WRN and RECQL4, members of the same family as BLM, in genomic stability maintenance.32,37 It suggests that acetylation modification may be an important way to regulate RecQ family proteins. To date, acetylation of BLM has not been reported. In this study, we provided evidences that BLM can be acetylated and the main enzymes for acetylation regulation are CBP/p300 and SIRT1. We also identify five lysine residues as the major acetylation sites of BLM. Treating cells with different DNA damage agents revealed that acetylation of BLM was different in response to etoposide and hydroxyurea, suggesting BLM acetylation may play multiple functions in DNA repair.
Materials and method
Cells and reagents
HeLa, HEK293T cells were obtained from the American Type Culture Collection and cultured at 37 °C and 5% CO2 in Dubelcco's modified Eagle's medium, supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin.The immunoblots were processed according to standard procedures using primary antibodies directed to BLM (Bethyl, A300-110A), β-actin (Santa Cruz, sc-47728), HA (CST, 3724), Flag (Sigma, F3165), SIRT1 (Santa Cruz, sc-74504), Myc (MILLIPORE, 05-724) and ac-K (CST, 9441L).
Plasmid construction
FLAG-tagged-BLM plasmids were a gift from Prof. Jiadong Wang (Peking University Health Center). SIRT1, CBP, p300, pCAF, TIP60, and hMOF were described previously.32 Other genes were PCR from cDNAs, and subcloned into the pcDNA3.1 expression vector as entry clones. The FLAG or HA tags were constructed at the N-terminal. All deletion mutants were generated using the PCR mutagenesis method and verified by sequencing.Cell transfection
Cell transfection was performed using PEI transfection reagent. Cells were 40–60% confluent before transfection. Dilute 1–5 μg DNA in proper volume of opti MEM (Invitrogen), combine PEI transfection reagent and mix well. The mass ratio of DNA and PEI was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4. Incubate mixture for 20 minutes. Add the complexes to the medium and change fresh medium within 6 hours after transfection.
:4. Incubate mixture for 20 minutes. Add the complexes to the medium and change fresh medium within 6 hours after transfection.
Immunoprecipitation and western blotting
Indicated plasmids encoding FLAG-tagged, HA-tagged, and Myc-tagged are transiently co-transfected into HEK293T cells and harvested cells were lysed by using FLAG lysis buffer (50 Mm Tris–HCl [pH 8.0], 137 Mm NaCl, Triton X-100 1%), sarkosyl 0.2%, glycerol 1%, 1 mM Na3VO4, containing 1 mM DTT, 1 mM PMSF and 1% protease inhibitor(sigma) or BC200 buffer (50 mM Tris–HCl pH 7.3, 200 mM NaCl, 10% glycerol, 0.2% TritonX-100). For acetylation assay, the cells were lysed in FLAG lysis buffer supplemented with 10 μM TSA and 5 mM nicotinamide. The whole proteins were collected and incubated with anti-FLAG M2 beads (Sigma, M8823) overnight at 4 °C, and eluted with either glycine, or flag peptide or SDS-loading buffer accordingly. Samples were boiled in 4× SDS loading buffer and resolved on SDS-PAGE. Membranes were blocked in 5% milk in TBST buffer (TBS with Tween 20) and then probed with antibodies as indicated.RNAi for SIRT1
The oligonucleotide sequences targeting SIRT1.GST pulldown assay
GST fusion proteins were expressed in Rossata cells. After immobilized on Glutathione Sepharose 4B beads, GST fusion proteins were incubated with purified SIRT1 protein for 2 hours. The samples were subjected to SDS-PAGE and analyzed by western blotting and GST fusion proteins were visualized by Coomassie blue staining.Coomassie blue staining
Rinse the gel for 30 minutes with deionized water. Stain the gel with enough Gel code Blue Stain Reagent (Invitrogen) to cover the gel, when bromophenol blue were thoroughly disappeared. Wash the gel with deionized water to remove the background.Co-immunoprecipitation assay
HEK293T cells were lysed in BC100 buffer (50 mM Tris–HCl pH 7.3, 100 mM NaCl, 10% glycerol, 0.2% TritonX-100) for 1 h. The whole cell lysates were collected and incubated with protein A/G agarose beads together with anti-BLM beads 4 °C overnight, then eluted with SDS loading buffer. The samples were subjected to SDS-PAGE and analyzed by western blotting.In vitro acetylation and deacetylation assay
In order to purify the acetylated BLM protein, FLAG-BLM was transfected into HEK293T cells with CBP. SIRT1 and CBP proteins were also purified from HEK293T cells. Indicated purified proteins and reagents were added in the reaction and the mixtures were incubated at 37 °C for 1 h. The acetylation incubation buffer contained 30 mM HEPES, pH 8.0, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride and 17 nM Ac-CoA. The deacetylation incubation buffer contained the 50 μM NAD+ co-factor (Sigma) (when indicated), 10% glycerol, 4 mM MgCl2, 100 mM NaCl. Reaction mixtures were subjected to SDS-PAGE and analyzed by western blotting.BLM acetylation and deacetylation assay in cells
FLAG-BLM was transfected with indicated plasmids for 36 h before harvest and then lysed in FLAG lysis buffer. When indicated, 10 μM TSA, and 5 mM nicotinamide were used in lysis buffer to prevent proteins from deacetylation. After immunoprecipitated indicated proteins, samples were subjected to SDS-PAGE and analyzed by western blotting with anti-FLAG antibodies or anti-acetylated lysine.Results
BLM is acetylated by CBP in cells
To investigate whether BLM could be acetylated in cells, FLAG-BLM and different acetyltransferases plasmids were transfected into HEK293T. BLM acetylation levels were examined by western blot with anti-acetylated lysine antibody. As shown in Fig. 1A, the BLM acetylation was observed in cells that were co-transfected with CBP, p300 and pCAF (Fig. 1A, lanes 2, 3 and 4, upper panel), but not in cells co-transfected with Tip60 or hMOF (Fig. 1A, lanes 5 and 6, upper panel).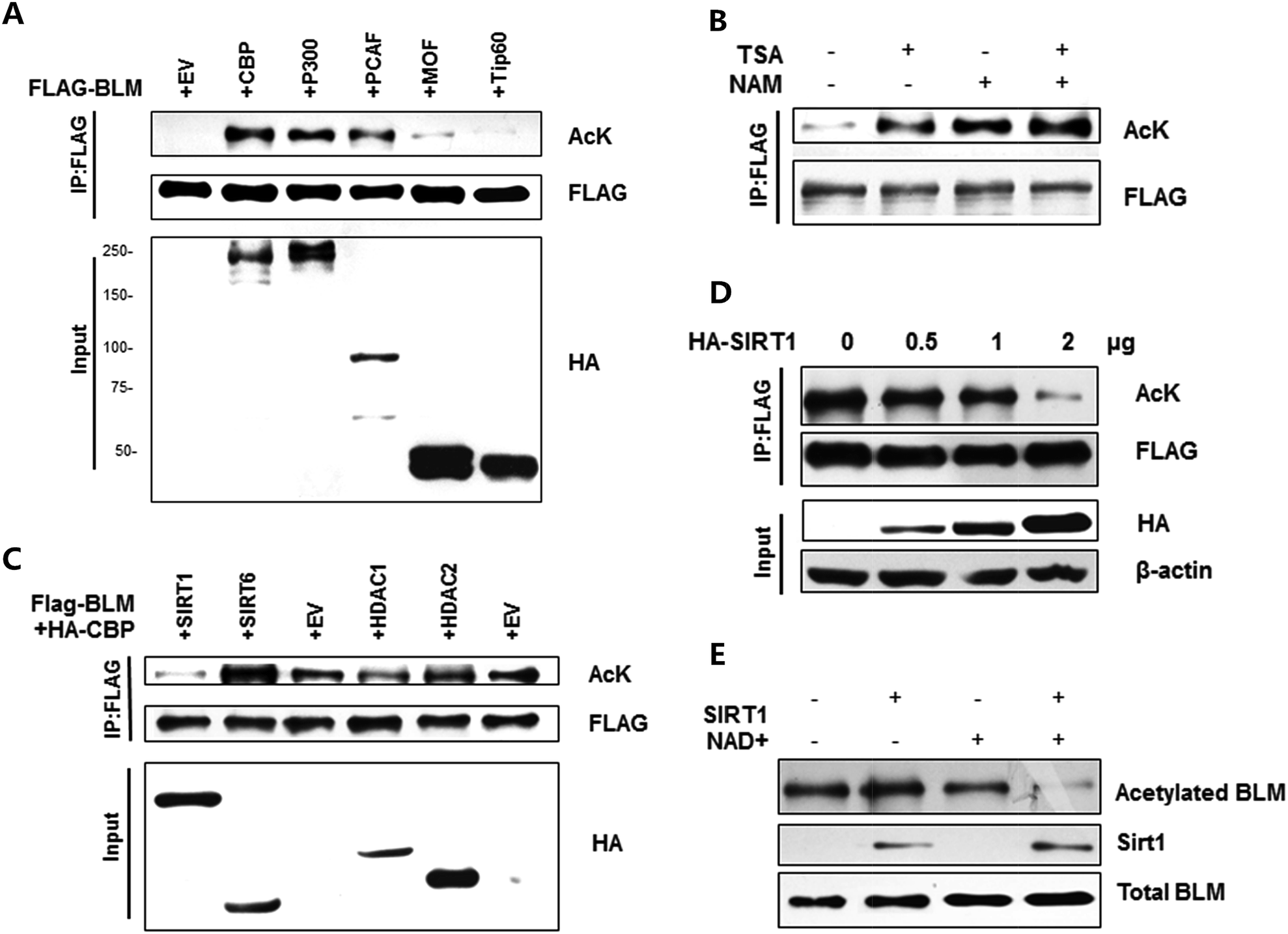 | ||
| Fig. 1 Acetylation of BLM is regulated by CBP and SIRT1. (A) BLM can be acetylated by CBP and p300. HEK293T cells were transfected with indicated acetyltransferases. BLM acetylation levels were examined. (B) BLM acetylation levels increased with TSA and nicotinamide treatment. HEK293T cells transfected with FLAG-BLM and HA-CBP were either treated with vehicle or nicotinamide or TSA or both of them for 6 h. BLM acetylation levels were examined. (C) BLM can be deacetylated by SIRT1 and HDAC1. HEK293T cells were transfected with indicated HDACs and CBP. BLM acetylation levels were then examined. (D) BLM deacetylation assay in the cell. HEK293T cells were co-transfected with FLAG-BLM and CBP with different concentrations of SIRT1 or controls as indicated. BLM acetylation levels were then examined. (E) In vitro deacetylation assay for BLM. SIRT1 and acetylated BLM protein were purified in vivo. The indicated reaction mixtures were incubated at 37 °C for 1 h and then BLM acetylation levels were then examined. | ||
Acetylation of BLM was also detected when cells were treated with HDAC inhibitors nicotinamide and TSA. When cells were treated with TSA alone, we detected increased BLM acetylation (Fig. 1B, lane 2). When cells were treated with nicotinamide alone, we detected stronger BLM acetylation (Fig. 1B, lane 3). When cells were treated with both TSA and nicotinamide, we detected the strongest BLM acetylation (Fig. 1B, lane 4). These results suggested that the major deacetylase for BLM could be class III deacetylase.
Identify major deacetylases for BLM
Next, we investigated which deacetylases are responsible for BLM deacetylation. According to the results shown in Fig. 1B, we chose several deacetylases, which are mainly responsible for nuclear protein deacetylation for the test. As shown in Fig. 1C, the acetylation levels of BLM protein decreased strongly after expression with SIRT1 (lane 1). In addition, HDAC1 could also acetylate BLM, but is not as strong as SIRT1 (lane 4), suggesting that SIRT1 is the major deacetylase for BLM.We proceeded to investigate the regulation of BLM deacetylation by SIRT1. As shown in Fig. 1D, BLM acetylation levels were high when cells were co-transfected with BLM and CBP (Fig. 1D, lane 1); however, the levels of BLM acetylation progressively decreased after increased expression of SIRT1 (Fig. 1D, lanes 2–4). These results demonstrate that BLM is a particular substrate for SIRT1 deacetylase activity. We further verified BLM deacetylation by SIRT1 through the in vitro deacetylation assay. As shown in Fig. 1E, the acetylation levels sharply decreased after adding the NAD+ and SIRT1, indicating that BLM can be deacetylated by SIRT1 (lane 4) and is NAD+ dependent (lane 4 vs. lane 2). These findings show that SIRT1 is a key mediator in the regulation of BLM acetylation.
BLM interacts with SIRT1 both in vitro and in vivo
We further investigated the interaction between SIRT1 and BLM. First, a transient transfection assay was performed. Both or either of the differently tagged BLM and SIRT1 were transfected in HEK293T cells. As shown in Fig. 2A and B, immunoprecipitation with the FLAG(BLM) antibody co-immunoprecipitated HA-SIRT1 (Fig. 2A, lane 3, upper panel) and the FLAG(SIRT1) antibody co-immunoprecipitated Myc-BLM as well (Fig. 2B, lane 3, upper panel). Endogenous BLM and SIRT1 interaction was also examined. As shown in Fig. 2C, endogenous BLM clearly co-immunoprecipitated with endogenous SIRT1 protein (lane 3).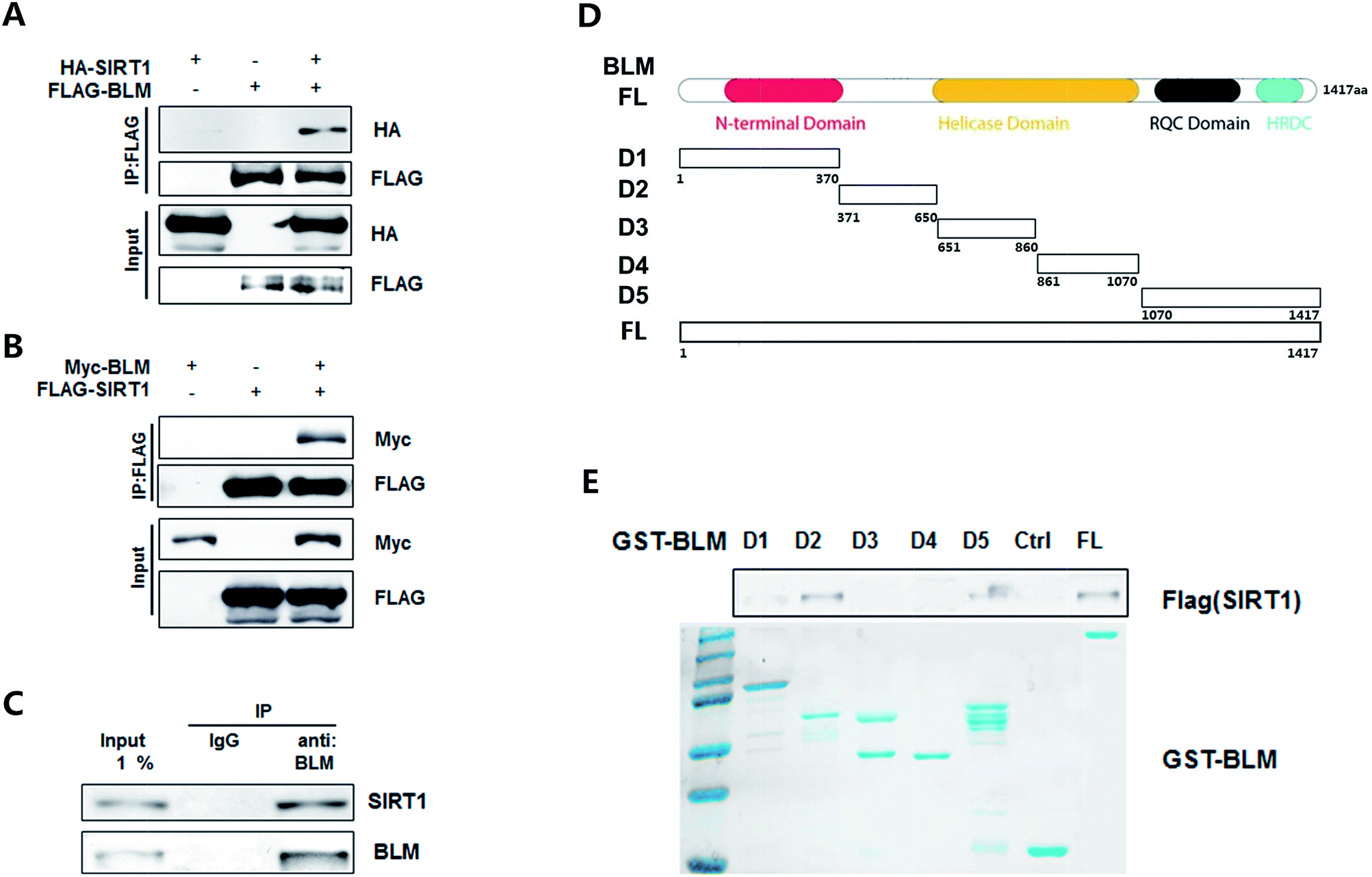 | ||
| Fig. 2 BLM interacts with SIRT1 both in vivo and in vitro (A) BLM interacts with SIRT1. HEK293T cell lysates expressed with FLAG-BLM and HA-SIRT1 and subjected to western blotting with anti-HA and FLAG antibodies. (B) SIRT1 interacts with BLM. HEK293T cell lysates expressed with FLAG-SIRT1 and Myc-BLM and subjected to western blotting with anti-Myc and FLAG antibodies. (C) Endogenous co-IP assay for BLM and SIRT1. HEK293T cell lysates were subjected to immunoprecipitation with control IgG and anti-BLM antibodies. The immunoprecipitates were then blotted with the indicated antibodies. (D) Schematic diagram of full length and parts of regions of BLM used in this study. (E) GST pulldown assay for different regions or full length of BLM with SIRT1. GST-BLM proteins were purified from bacteria. SIRT1 protein was purified from HEK293T cells and they were incubated with GST beads and the elutes were subjected to western blotting with SIRT1. | ||
Furthermore, we tested the direct interaction between these two proteins and the specific region of BLM mediating this interaction. The BLM protein was divided into five pieces (Fig. 2D) and the GST fusion proteins were generated and immobilized on GST-agarose. As shown in Fig. 2E, SIRT1 bound to immobilized GST-BLM FL but not to immobilized GST alone. Moreover, SIRT1 also bound to the D2 and D5 regions, and no binding to other regions were detected. Thus, these findings indicated that SIRT1 interacts with BLM both in vitro and in vivo and binds to the BDHCT and C-terminal region of BLM.
Identification of BLM acetylation sites
In order to identify the acetylation sites of BLM, we purified acetylated BLM protein after transfection of FLAG-tagged BLM with CBP in HEK293T cells. After immunoprecipitation with M2 beads, the IP products were subjected to SDS-PAGE and Coomassie blue staining. The BLM bind was cut out for mass-spectrometry analysis. Acetylation signals were detected in 50 lysines of BLM and five of them have high scores >10, (Fig. 3A). They are K105, K129, K476, K1010, and K1329. | ||
| Fig. 3 Mapping the BLM acetylation sites (A) lists of BLM potential acetylated site identified by mass spectrometry analysis. Results show scores greater than 10. (B) In vitro acetylation assay for different regions or full length of BLM with CBP. GST-BLM proteins was purified from bacteria, CBP protein was purified from HEK293T cells, components were incubated at 37 °C for 1 h and then the BLM acetylation levels were examined. (C) FLAG-BLM-Single mutant or WT was transfected with CBP into HEK293 cells. BLM acetylation levels were then examined. (D) BLM-WT or double mutant or triple mutant or quadruple mutant or 5KR mutant was transfected with CBP into cells. BLM acetylation levels were then examined. | ||
To map out the major acetylation sites of BLM, we performed the in vitro acetylation assay with different region of BLM. The same set of GST-BLM proteins described in Fig. 2 was used. As shown in Fig. 3B. Acetylation signals were detected at GST-D2, GST-D4, and GST-D5, suggesting that the major acetylation sites of BLM are located at these three regions.
According to these two assays above in conjunction with the data from the database,38 we chose five lysine residues as the candidate sites. To confirm BLM acetylation sites in cells, we mutated these putatively acetylated lysines to arginines and tested acetylation levels of BLM. Firstly, single mutations were made on all five lysines. As shown in Fig. 3C, each of the single mutations showed reductions in the overall level of BLM acetylation to varying degrees. Next, we examined double mutation (K1010/K1411), triple mutant (K476/K863/K1411 and K1010/K1329/K1411), quadruple mutant (K476/K863/K1329/K1411) and 5 KR mutants in cells (Fig. 3D). It showed further decreased BLM acetylation levels in these mutants, suggesting that acetylation of BLM mainly induced at these five lysines.
The BLM acetylation is increased in response to DNA damage
Next, we investigated whether acetylation of BLM affects the function of BLM in the DNA damage response. The acetylation of BLM was detected after treating the cells with etoposide (Fig. 4A) or HU (Fig. 4B). The acetylation of BLM significantly increased in response to DNA damage induced by etoposide, but decreased by HU in a dose dependent manner (Fig. 4A and B). These results suggest that different types/causes of DNA damage may trigger different BLM responses. We further treated cells with HU at differing times. As shown in Fig. 4C, the acetylation levels were decreased when induced by HU in a time dependent manner. Moreover, the decline of BLM acetylation by DNA damage was abolished after SIRT1 knockdown (Fig. 4D, lane 5 vs. lane 4). In addition, the acetylation of BLM increased by knocking down the SIRT1 (Fig. 4D, lane 4 vs. lane 2). The findings on acetylation of BLM were different in response to etoposide and hydroxyurea, suggesting BLM acetylation may play multiple functions in DNA repair.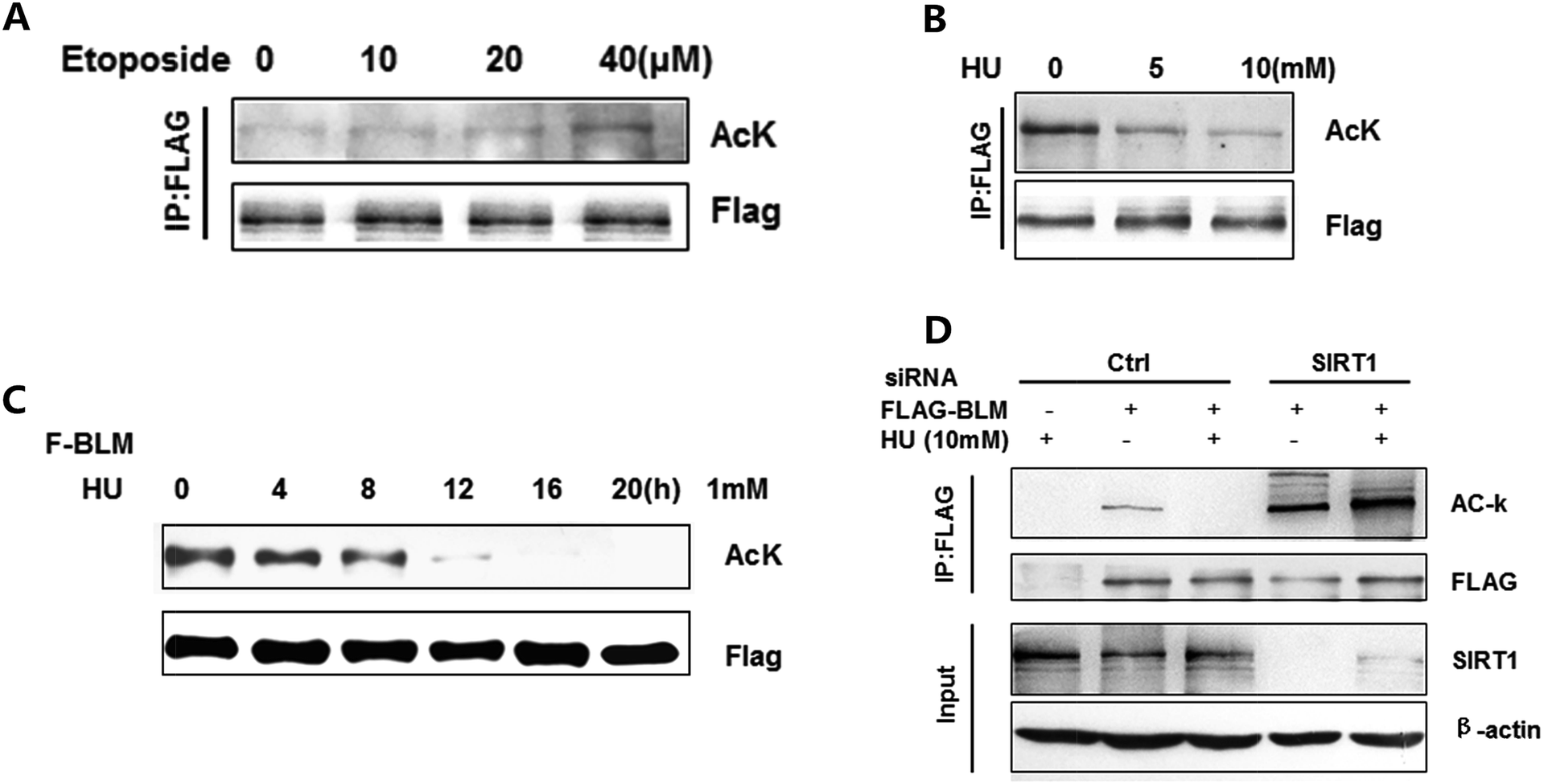 | ||
| Fig. 4 BLM acetylation levels changed in response to DNA damage (A) acetylated BLM was detected after treating cells with different concentrations of etoposide. FLAG-BLM transfected HEK293T cells were treated with etoposide for the indicated concentrations and subjected to western blotting with anti-acetylated lysine and FLAG antibodies. (B) Acetylated BLM was detected after treating cells with different concentrations of HU. FLAG-BLM transfected HEK293T cells were treated with HU for the indicated concentrations and BLM acetylation levels were then examined. (C) Acetylated BLM was detected after treating cells with HU for different times. Cells were treated with HU for the indicated periods and BLM acetylation levels were then examined. (D) Acetylated BLM was detected after knocking down SIRT1 protein and treating cells with HU. FLAG-BLM were transfected with control or SIRT1 siRNA in HEK293T cells and subjected to western blotting with indicated antibodies. | ||
Discussion
Acetylation modification plays a key role in the regulation of DNA repair and acts on many DNA repair related proteins.39–41 SIRT1 is identified as one of the major deacetylases to catalyze this process. For example, SIRT1 deacetylated WRN at 6 lysine residues, promoting its translocation from nucleoli to the damage foci when DNA was impaired by cytotoxic substances, thus inhibiting its degradation.37 The stability, activity, and location of p53 and Ku70 were also mediated by SIRT1.42,43 Our results show that it holds a close relationship with BLM as well. SIRT1 can deacetylate and interact with BLM in vivo and in vitro under both normal conditions and in response to DNA damage (Fig. 2 and 4). Therefore, the functional regulation of BLM by SIRT1 is worth further exploration and investigation.In our study, we identified five lysines as the major acetylation sites of BLM. These five lysines were located at the BDHCT domain, helicase domain, RQC domain, and HRDC domain respectively, indicating that acetylation of BLM plays an important role in the spread spectrum of its function in genomic stability maintenance. For instance, RQC and HRDC domains are highly conserved in the RecQ family. Duplex DNA strands bind to both of these domains, and are unwounded by the helicase activities of BLM. In addition, these two domains can cooperate with each other when the DNA are damaged and form the Holliday forked structure completing the migration.44 By contrast, the BDHCT-box associated domain is a unique domain for the BLM protein and the functions of this region are not clear. It was speculated that this region may involve in DNA replication and repair in an Mg+ and ATP dependent manners. Interestingly, mutations in HRDC domain were seen as one of the major reasons leading to the Bloom syndrome.45 This information implied that the acetylation of BLM may play crucial roles in DNA replication, and DNA damage response and repair.
Our findings on HU and etoposide bringing two opposite changes on BLM acetylation (Fig. 4) raised great interests in understanding the behavior. HU and etoposide are common agents for DNA damage. Both of them could lead to DSBs,14,16 but the mechanisms were completely different. HU is an inhibitor for ribonucleotide diphosphate reductase and leads to DNA synthesis inhibition. The etoposide is an inhibitor for topoisomerase II, which leads to transmit duplex breaks in DNA. Therefore, the explanation of this phenomenon will be associated with the mechanism of action. First, two agents may trigger different DNA damage responses. Second, BLM plays many different roles in DNA repair and it may take different effects after stimulated by two agents. Moreover, the condition of cells also changed after treating with two agents for different concentrations or times. Due to these uncertain conditions, functions of BLM could develop in different directions. In any case, these results indicate that the acetylation of BLM influences the response for DNA damage.
Conclusions
In this study, we have provided the first evidence that BLM could be acetylated in cells and is mainly mediated by CBP/p300 and SIRT1 at five lysine residues (Fig. 1 and 2). Acetylation of BLM was different in response to etoposide and hydroxyurea, suggesting BLM acetylation may play multiple functions in DNA repair (Fig. 4).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Dr Jiadong Wang from Peking University for providing BLM plasmid. This study was funded by the National Natural Science Foundation of China, No. 81471405, No. 81270427, No. 81671389, and the 973 Program No. 2013CB530801.Notes and references
- J. German, Medicine, 1993, 72, 393–406 CrossRef CAS PubMed
.
- R. K. Chakraverty and I. D. Hickson, BioEssays, 1999, 21, 286–294 CrossRef CAS PubMed
- N. A. Ellis, J. Groden, T. Z. Ye, J. Straughen, D. J. Lennon, S. Ciocci, M. Proytcheva and J. German, Cell, 1995, 83, 655–666 CrossRef CAS PubMed
- K. L. Puranam and P. J. Blackshear, J. Biol. Chem., 1994, 269, 29838–29845 CAS
- M. Seki, H. Miyazawa, S. Tada, J. Yanagisawa, T. Yamaoka, S. Hoshino, K. Ozawa, T. Eki, M. Nogami and K. Okumura, et al., Nucleic Acids Res., 1994, 22, 4566–4573 CrossRef CAS PubMed
- C. E. Yu, J. Oshima, Y. H. Fu, E. M. Wijsman, F. Hisama, R. Alisch, S. Matthews, J. Nakura, T. Miki, S. Ouais, G. M. Martin, J. Mulligan and G. D. Schellenberg, Science, 1996, 272, 258–262 CAS
- S. Kitao, A. Shimamoto, M. Goto, R. W. Miller, W. A. Smithson, N. M. Lindor and Y. Furuichi, Nat. Genet., 1999, 22, 82–84 CrossRef CAS PubMed
- A. J. van Brabant, R. Stan and N. A. Ellis, Annu. Rev. Genomics Hum. Genet., 2000, 1, 409–459 CrossRef CAS PubMed
- D. L. Croteau, V. Popuri, P. L. Opresko and V. A. Bohr, Annu. Rev. Biochem., 2014, 83, 519–552 CrossRef CAS PubMed
- S. Sengupta, S. P. Linke, R. Pedeux, Q. Yang, J. Farnsworth, S. H. Garfield, K. Valerie, J. W. Shay, N. A. Ellis, B. Wasylyk and C. C. Harris, EMBO J., 2003, 22, 1210–1222 CrossRef CAS PubMed
- A. Grabarz, J. Guirouilh-Barbat, A. Barascu, G. Pennarun, D. Genet, E. Rass, S. M. Germann, P. Bertrand, I. D. Hickson and B. S. Lopez, Cell Rep., 2013, 5, 21–28 CrossRef CAS PubMed
- P. Schwertman, S. Bekker-Jensen and N. Mailand, Nat. Rev. Mol. Cell Biol., 2016, 17, 379–394 CrossRef CAS PubMed
- D. V. Bugreev, X. Yu, E. H. Egelman and A. V. Mazin, Genes Dev., 2007, 21, 3085–3094 CrossRef CAS PubMed
- J. Renkawitz, C. A. Lademann and S. Jentsch, Nat. Rev. Mol. Cell Biol., 2014, 15, 369–383 CrossRef CAS PubMed
- J. M. Sidorova, K. Kehrli, F. Mao and R. Monnat Jr, DNA Repair, 2013, 12, 128–139 CrossRef CAS PubMed
- S. Sengupta, S. P. Linke, R. Pedeux, Q. Yang, J. Farnsworth, S. H. Garfield, K. Valerie, J. W. Shay, N. A. Ellis, B. Wasylyk and C. C. Harris, EMBO J., 2003, 22, 1210–1222 CrossRef CAS PubMed
- S. So, N. Adachi and H. Koyama, DNA Cell Biol., 2007, 26, 517–525 CrossRef CAS PubMed
- J. Bartek and J. Lukas, Curr. Opin. Cell Biol., 2007, 19, 238–245 CrossRef CAS PubMed
- M. Goldstein and M. B. Kastan, Annu. Rev. Med., 2015, 66, 129–143 CrossRef CAS PubMed
- A. Fradet-Turcotte, M. D. Canny, C. Escribano-Diaz, A. Orthwein, C. C. Leung, H. Huang, M. C. Landry, J. Kitevski-LeBlanc, S. M. Noordermeer, F. Sicheri and D. Durocher, Nature, 2013, 499, 50–54 CrossRef CAS PubMed
- M. Stucki, J. A. Clapperton, D. Mohammad, M. B. Yaffe, S. J. Smerdon and S. P. Jackson, Cell, 2005, 123, 1213–1226 CrossRef CAS PubMed
- S. Bohm and K. A. Bernstein, DNA Repair, 2014, 22, 123–132 CrossRef CAS PubMed
- Y. Zhao, J. R. Brickner, M. C. Majid and N. Mosammaparast, Trends Cell Biol., 2014, 24, 426–434 CrossRef CAS PubMed
- S. L. Davies, P. S. North, A. Dart, N. D. Lakin and I. D. Hickson, Mol. Cell. Biol., 2004, 24, 1279–1291 CrossRef CAS PubMed
- S. Tikoo, V. Madhavan, M. Hussain, E. S. Miller, P. Arora, A. Zlatanou, P. Modi, K. Townsend, G. S. Stewart and S. Sengupta, EMBO J., 2013, 32, 1778–1792 CrossRef CAS PubMed
- K. J. Ouyang, M. K. Yagle, M. J. Matunis and N. A. Ellis, Front. Genet., 2013, 4, 167 Search PubMed
- E. Verdin and M. Ott, Nat. Rev. Mol. Cell Biol., 2015, 16, 258–264 CrossRef CAS PubMed
- M. Kruszewski and I. Szumiel, DNA Repair, 2005, 4, 1306–1313 CrossRef CAS PubMed
- T. Ikura, V. V. Ogryzko, M. Grigoriev, R. Groisman, J. Wang, M. Horikoshi, R. Scully, J. Qin and Y. Nakatani, Cell, 2000, 102, 463–473 CrossRef CAS PubMed
- Y. Sun, X. Jiang, S. Chen, N. Fernandes and B. D. Price, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13182–13187 CrossRef CAS PubMed
- Z. Yuan and E. Seto, Cell Cycle, 2007, 6, 2869–2871 CrossRef CAS PubMed
- K. Li, A. Casta, R. Wang, E. Lozada, W. Fan, S. Kane, Q. Ge, W. Gu, D. Orren and J. Luo, J. Biol. Chem., 2008, 283, 7590–7598 CrossRef CAS PubMed
- W. Fan and J. Luo, Mol. Cell., 2010, 39, 247–258 CrossRef CAS PubMed
- M. Zhao, R. Geng, X. Guo, R. Yuan, X. Zhou, Y. Zhong, Y. Huo, M. Zhou, Q. Shen, Y. Li, W. Zhu and J. Wang, Cell Rep., 2017, 20, 1997–2009 CrossRef CAS PubMed
- H. He, J. Wang and T. Liu, Cell Rep., 2017, 20, 2010–2025 CrossRef CAS PubMed
- T. Liu, Y. H. Lin, W. Leng, S. Y. Jung, H. Zhang, M. Deng, D. Evans, Y. Li, K. Luo, B. Qin, J. Qin, J. Yuan and Z. Lou, Mol. Cell, 2014, 56, 681–695 CrossRef CAS PubMed
- T. Dietschy, I. Shevelev, J. Pena-Diaz, D. Hühn, S. Kuenzle, R. Mak, M. F. Miah, D. Hess, M. Fey, M. O. Hottiger, P. Janscak and I. Stagljar, J. Cell Sci., 2009, 122, 1258–1267 CrossRef CAS PubMed
- S. Zhao, W. Xu, W. Jiang, W. Yu, Y. Lin, T. Zhang, J. Yao, L. Zhou, Y. Zeng, H. Li, Y. Li, J. Shi, W. An, S. M. Hancock, F. He, L. Qin, J. Chin, P. Yang, X. Chen, Q. Lei, Y. Xiong and K. L. Guan, Science, 2010, 327, 1000–1004 CrossRef CAS PubMed
- M. Shogren-Knaak, H. Ishii, J. M. Sun, M. J. Pazin, J. R. Davie and C. L. Peterson, Science, 2006, 311, 844–847 CrossRef CAS PubMed
- Y. Zhao, J. R. Brickner, M. C. Majid and N. Mosammaparast, Trends Cell Biol., 2014, 24, 426–434 CrossRef CAS PubMed
- E. Lozada, J. Yi, J. Luo and D. K. Orren, Biogerontology, 2014, 15, 347–366 CrossRef CAS PubMed
- T. Ghazi, S. Nagiah, C. Tiloke, N. Sheik Abdul and A. A. Chuturgoon, J. Cell. Biochem., 2017, 118, 3866–3874 CrossRef CAS PubMed
- M. Roth, Z. Wang and W. Y. Chen, OncoTargets, 2016, 7, 50195–50214 Search PubMed
- E. Staub, P. Fiziev, A. Rosenthal and B. Hinzmann, BioEssays, 2004, 26, 567–581 CrossRef CAS PubMed
- Z. Liu, M. J. Macias, M. J. Bottomley, G. Stier, J. P. Linge, M. Nilges, P. Bork and M. Sattler, Structure, 1999, 7, 1557–1566 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2017 |