
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comments on “Experimental and theoretical studies of the nanostructured {Fe3O4@SiO2@(CH2)3Im}C(CN)3 catalyst for 2-amino-3-cyanopyridine preparation via an anomeric based oxidation”, RSC Adv., 2016, 6, 50100–50111, and “The first computational study for the oxidative aromatization of pyrazolines and 1,4-dihydropyridines using 1,2,4-triazolinediones: an anomeric-based oxidation”, RSC Adv., 2016, 6, 102280–102291
Sadegh
Salehzadeh

Faculty of Chemistry, Bu-Ali Sina University, Hamedan, Iran. E-mail: saleh@basu.ac.ir
First published on 18th August 2017
Abstract
The papers mentioned in the title of the present comment article contain some calculations/results that are in disagreement with some basic concepts of chemistry. The calculations include (i) the calculation of an energy difference, more than 1000 J mol−1, between one pair of enantiomers, (ii) the calculation of an energy difference between the Lewis structures (resonance forms), and (iii) the calculation of bond orders in different pathways of resonance process.
Introduction
Herein, I want to discuss some results reported in two recently published papers by Zolfigol et al.,1,2 in order to clarify for readers which calculations contain inaccuracies that are in disagreement with basic concepts. The mentioned authors have previously reported3 a short correction on ref. 2, which is not enough.(1) Calculating an energy difference between one pair of enantiomers.
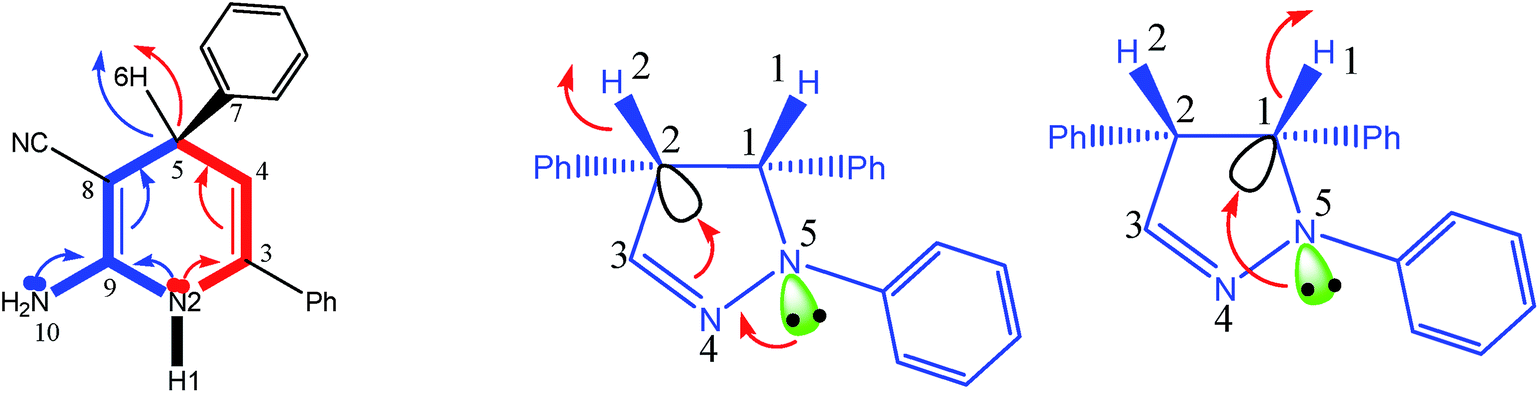
We know that each chiral center in a molecule is assigned a prefix (R or S), according to whether its configuration is right- or left-handed. The R and S stereoisomers are non-superimposable mirror images. Thus the R and S isomers have identical chemical and physical properties in an achiral environment, as they are mirror images of each other. They have the same stability, both kinetically and thermodynamically, and the number and the type of atoms, the type and the strength of chemical bonds, the type and the strength of intermolecular atom/orbital interactions and the geometry and the symmetry of these molecules all are the same. Indeed, the energies of enantiomers can be different, in the femtojoule to picojoule per mole range, only because of a parity violation.4 However, recently Zolfigol et al.1 during the study on the intermediate molecule shown in Fig. 1 have reported that the S isomer is, about 0.29 kcal mol−1 (≈1200 J mol−1), more stable than the R one. Also they have claimed that while the S isomer is more stable than R one, the R isomer due to the lower bond order of its C–H bond (0.91 against 0.93) has a higher rate of oxidation reaction. Furthermore, the authors have proved that the transition state (TS) for R isomer is, about 4.5 kcal mol−1 (≈19000 J mol−1), more stable than S one while we remember that these chiral molecules have reacted with an achiral ion.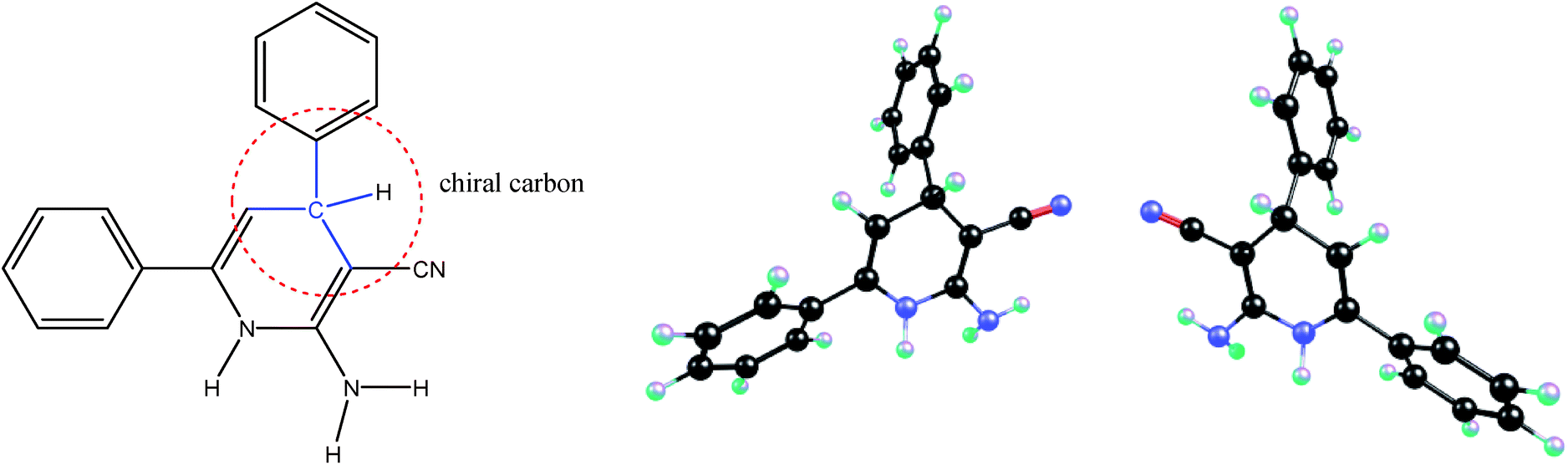 | ||
| Fig. 1 The schematic representation of chiral carbon in the compound studied by Zolfigol et al. (left), and molecular structures of its R and S stereoisomers shown here (right). | ||
Apart from this fact that it is unlikely to think a 0.02 difference in the value of bond orders can be a good reason for higher reactivity of one compound over other one, the question is what is the origin of above differences in values of bond orders, degree of stability of above R and S isomers and corresponding TSs reported by Zolfigol et al.?
Indeed, in each of above R and S molecules with respect to NH, NH2 and phenyl groups we can consider different conformers or geometrical isomers (authors have mentioned that the R and S isomers have the cis and trans isomers without giving/comparing their relative energies).1 If the authors did not compare the same conformers/isomers of R and S stereoisomers, then we can easily find why their S isomer is more stable than R one. In the right side of Fig. 1, I have compared the same conformers of R and S isomers. If we compare the same conformers of R and S stereoisomers, that are really the mirror images of each other, then we can prove that they have the same energy/stability. Thus, the observation of an energy difference between the R and S isomers shows that we have not compared the same conformers (i.e. not two mirror images) or we have not been able to successfully optimize one of them. Authors, instead of comparing the stability of R and S (which surely is the same), should try to find the best conformer with a minimized energy for one of them (either R or S). Furthermore, it has not been necessary to check the transition state (TS) of reaction for both R and S isomers, while the interacted molecule with these chiral molecules had been an achiral compound and both TSs should have the same energy.
(2) Calculating an energy difference between the Lewis structures (resonance forms).
After the careful study of recent published paper of Zolfigol research group2 I found that they have reported an energy difference between the resonance forms of an ion (compound a-1H in their paper).We know that the structures shown in Fig. 2 are the different Lewis structures, or in other words two possible resonance forms, for “one ion” and not two different ions/isomers. However, in Fig. 2 and 3 of ref. 2 cited here, two different optimized structures are reported for above resonance forms. Interestingly, in contrast to a real enolic form, herein a proton is not bonded to oxygen atom and only with their imagination an electron/charge is completely moved from nitrogen atom to oxygen atom, so they have named it in Fig. 3 of ref. 2 as enolate form. We know that there are some basic principles in the resonance theory. First of all, resonance structures are not real, they just show possible Lewis structures for a compound. Resonance structures are not in equilibrium with each other, so we use the resonance sign (↔) between them. Resonance structures are not isomers. Isomers have different arrangement of both atoms and electrons. Resonance forms differ only in arrangement of electrons, where atoms are in the same positions. In ref. 2, I believe the authors have misrepresented the principles of resonance theory.
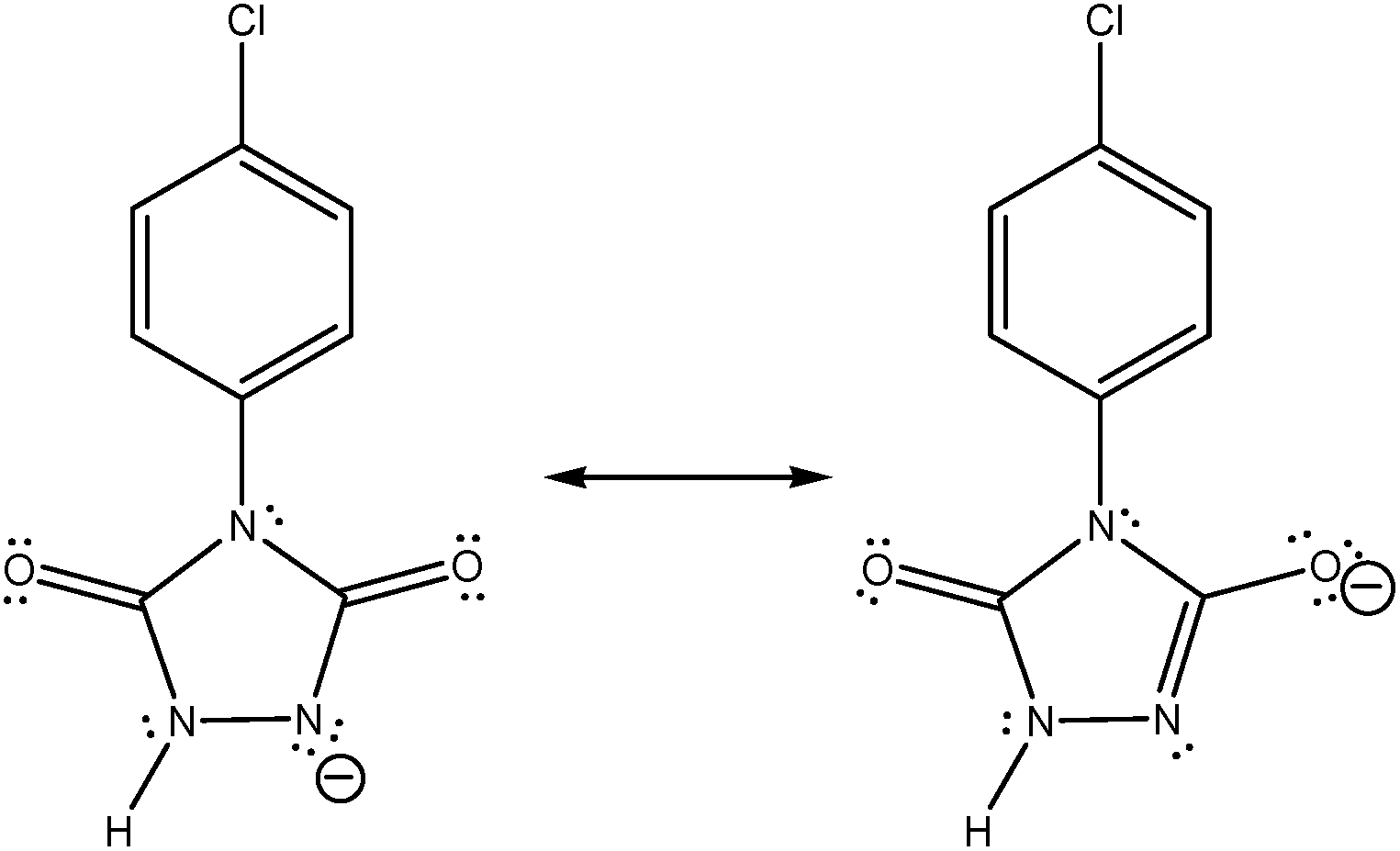 | ||
| Fig. 2 The resonance forms (and not the real structures) of compound a-1H which are introduced in ref. 2 as keto (left) and enolic (right) forms that are in equilibrium. | ||
In Fig. 2 and 3 of their paper,2 the authors have put an equilibrium sign (⇌) between the resonance forms. Secondly, I note that the authors have reported (both in the text and in Fig. 3) an energy difference of about 0.1 kcal mol−1 between the above unreal structures. Thirdly, in the page 102![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 287, they have mentioned that they have reported the bond lengths of enolic form, which is not really an enolic form and is a resonance form for ketonic form (see the left side of Fig. 2 presented here). Interestingly, for another investigated oxidant (b), that has an alkyl group instead of p-chlorophenyl moiety of the oxidant (a), they have found that keto–enol forms have the same energy (see Fig. 5 and also right column in the page 102287 of ref. 2). In Fig. 3, I have shown two possible resonance forms for both the ketonic and enolic tautomers of compounds a-1H or b-1H in Zolfigol's proposed mechanism. The optimized structure of each isomer is not the same with one of the two illustrated resonance forms for it, but is somewhat similar to both of them. Thus into the optimized structure of these isomers none of the atoms have the charge of −1 and none of the bonds has bond order of 2. Thus, the resonance forms in the Zolfigol's paper2 that authors have named them as keto and enolic (enolate in Fig. 3 of ref. 2) forms, only exist on the paper and after optimization of the structure of compounds a-1H and b-1H both of them, with respect to the oxygen atom in the left side of these compounds, are ketonic (Fig. 3, X1 and X2). Thus only if through a tautomerization process the proton of NH group in these compounds transferred and attached to the oxygen atom in the left side of molecule, then we will have an enolic tautomer (Fig. 3, Y1 and Y2). Obviously, the stabilities of above structural isomers shown in Fig. 3 cannot be the same or very similar.
287, they have mentioned that they have reported the bond lengths of enolic form, which is not really an enolic form and is a resonance form for ketonic form (see the left side of Fig. 2 presented here). Interestingly, for another investigated oxidant (b), that has an alkyl group instead of p-chlorophenyl moiety of the oxidant (a), they have found that keto–enol forms have the same energy (see Fig. 5 and also right column in the page 102287 of ref. 2). In Fig. 3, I have shown two possible resonance forms for both the ketonic and enolic tautomers of compounds a-1H or b-1H in Zolfigol's proposed mechanism. The optimized structure of each isomer is not the same with one of the two illustrated resonance forms for it, but is somewhat similar to both of them. Thus into the optimized structure of these isomers none of the atoms have the charge of −1 and none of the bonds has bond order of 2. Thus, the resonance forms in the Zolfigol's paper2 that authors have named them as keto and enolic (enolate in Fig. 3 of ref. 2) forms, only exist on the paper and after optimization of the structure of compounds a-1H and b-1H both of them, with respect to the oxygen atom in the left side of these compounds, are ketonic (Fig. 3, X1 and X2). Thus only if through a tautomerization process the proton of NH group in these compounds transferred and attached to the oxygen atom in the left side of molecule, then we will have an enolic tautomer (Fig. 3, Y1 and Y2). Obviously, the stabilities of above structural isomers shown in Fig. 3 cannot be the same or very similar.
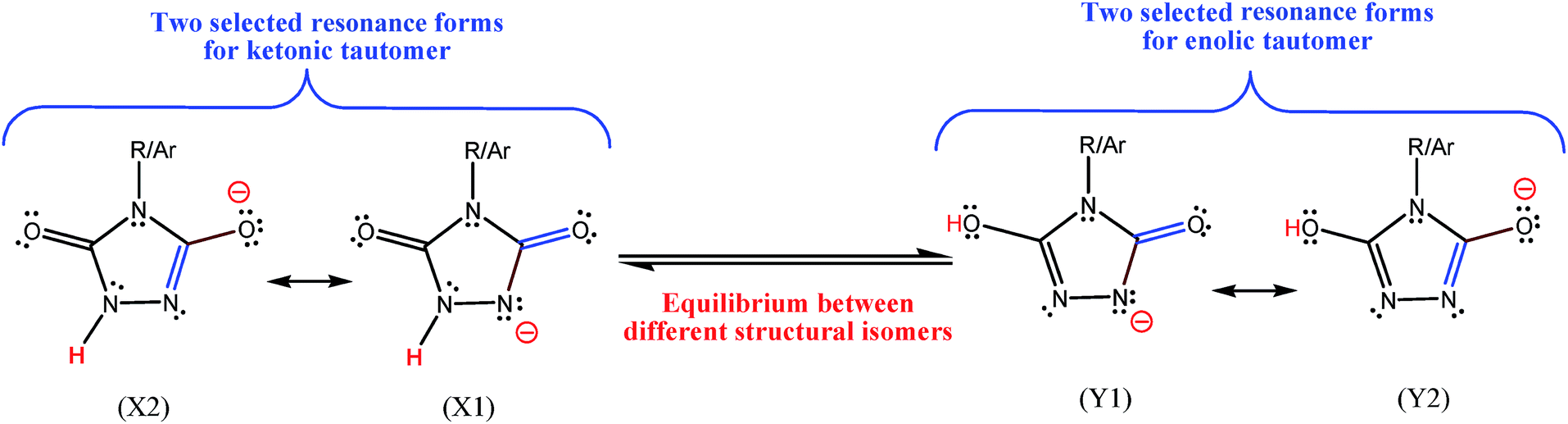 | ||
| Fig. 3 Two selected resonance forms for each of two structural isomers correctly proposed here for compounds a-1H or b-1H which are incorrectly reported in ref. 2. Ar = p-chlorophenyl, compound a-1H, and R = n-propyl, compound b-1H in ref. 2. The formal charge of all atoms, except the indicated atom, in the resonance forms is zero. | ||
(3) Calculating the bond orders in different pathways of resonance process.
As can be seen in Table 5 of ref. 1 and Table 1 of ref. 2, authors have calculated the bond orders for different electron delocalization pathways in both R and S stereoisomers. The pathways shown in Fig. 4 of present paper are exactly those that have been studied in above cited papers. Authors have assumed that before removal of any atom from the studied molecule (for example a hydride ion from a C–H group in their studied compounds) and only with drawing the pathway of resonance process on the paper they can easily calculate the changes in bond orders into the chemical structure of molecule.1 Then with comparison the value of calculated bond orders (Mulliken bond orders are abbreviated as MBO pathways) they can find which pathway of electron delocalization is suitable and thus which atom will be removed from the molecule.2 As can be seen in Table 5 of ref. 1, they have proposed that, where all atoms are fixed, the movement of electrons (or may be the anomeric effect), beginning from NH versus NH2 group has different effect on bond orders of both R and S molecules. For each bond in both above cases (pathways), most of bond orders have the very similar or even the same values, indicating that all data are related to a single structure or to two very similar structures that are not optimized correctly. The data in Table 5 of ref. 1, show that usually the difference between the bond orders is 0.01 or 0.02. Also, as expected, the value of bond orders for N2–C9 and C4–C5 (see the left side of present Fig. 4) for both the R and S molecules and for both pathways are 0.99 and 0.97, respectively. Also the calculated bond order of C5–C8 for both pathways is the same and for R and S isomer is 0.97 and 0.98, respectively. Furthermore, the values of bond orders for single and double bonds shown in Fig. 4 clearly show that the π electrons do not move through the indicated pathways. Thus all above data clearly indicate that their efforts in studying different pathways of electron delocalization and also Table 5 of ref. 1 (similarly Table 1 of ref. 2) are misleading. Indeed, in the optimized structure of one molecule all orbital interactions and different ways of electron delocalization exist at the same time. For example, both the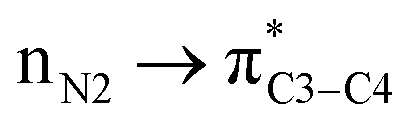 and
and 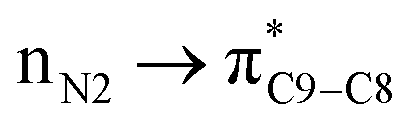 donor–acceptor interactions discussed in ref. 1 exist at same time and not in two separate and detectable/traceable pathways. Thus we cannot calculate different bond lengths and/or different bond orders for one bond in one molecule while we consider different pathways for delocalization of electrons and/or for anomeric effect in our mind.
donor–acceptor interactions discussed in ref. 1 exist at same time and not in two separate and detectable/traceable pathways. Thus we cannot calculate different bond lengths and/or different bond orders for one bond in one molecule while we consider different pathways for delocalization of electrons and/or for anomeric effect in our mind.
(4) Discussion regarding electron transfer.
There is also another inaccuracy which is also related to our knowledge of basic concepts of chemistry. We know that the concept of “electron transfer” is absolutely different with that of “donor–acceptor interactions inside the molecules”. We use the term “electron transfer” where electron(s) move from one atom/molecule/ion to another one, in a redox reaction. However, the authors of above papers have named the donor–acceptor interactions such as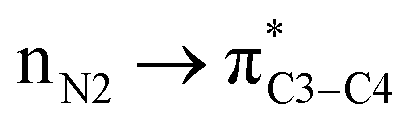 and/or the delocalization of electron(s) inside the molecule, as an electron transfer (specially see the ref. 1). We note that both the above orbitals (nN2 and
and/or the delocalization of electron(s) inside the molecule, as an electron transfer (specially see the ref. 1). We note that both the above orbitals (nN2 and 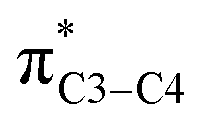 ) belong to one molecule and therefore electron has not transferred from one atom/ion/molecule to another one. Furthermore, writing the
) belong to one molecule and therefore electron has not transferred from one atom/ion/molecule to another one. Furthermore, writing the 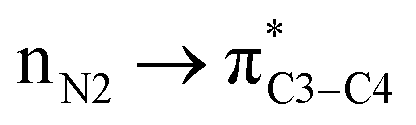 (nN2 to
(nN2 to 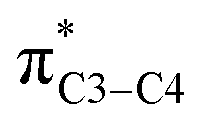 ) does not mean that the complete transfer of electron(s) occurs from one orbital to another one. Thus it is misleading to use the term “electron transfer” for description of above type of orbital interactions inside the molecules.
) does not mean that the complete transfer of electron(s) occurs from one orbital to another one. Thus it is misleading to use the term “electron transfer” for description of above type of orbital interactions inside the molecules.
Conclusions
Three issues with two recently published papers were reviewed with respect to basic concepts of chemistry. Firstly, it was discussed that R and S stereoisomers in an achiral environment, have the same energy (without considering the possible negligible effect of parity violation effect), bond orders, reactivity, etc. secondly, it was explained that resonance structures are not real, are not isomers and are only possible Lewis structures. On the other hand, keto–enol forms both are real, are not different resonance forms of one compound and are two structural isomers with different stabilities. Thus when we want to compare the relative stabilities of keto–enol forms, we have to check whether two unreal resonance forms are not considered instead of two real isomers. Thirdly, it was discussed that because the resonance structures are not real, we cannot calculate/report different bond orders for different resonance structures, as we only have one real structure. Furthermore, it was explained that the orbital interactions and/or electron delocalisation inside the molecule are absolutely different with the electron transfers between the atoms/ions/molecules.Conflicts of interest
There are no conflicts to declare.Acknowledgements
I am grateful to the editor-in-chief and editorial board members of RSC advances, as well as the people responsible in the Royal Society of Chemistry for their patience and the time they spent on responding to my letters.Notes and references
- M. A. Zolfigol, M. Kiafar, M. Yarie, A. Taherpour and M. Saeidi-Rad, RSC Adv., 2016, 6, 50100–50111 RSC
.
- M. Kiafar, M. A. Zolfigol, M. Yarie and A. Taherpour, RSC Adv., 2016, 6, 102280–102291 RSC
- M. Kiafar, M. A. Zolfigol, M. Yarie and A. Taherpour, RSC Adv., 2016, 6, 107295 RSC
- M. Quack, Angew. Chem., Int. Ed., 2002, 41, 4618–4630 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2017 |