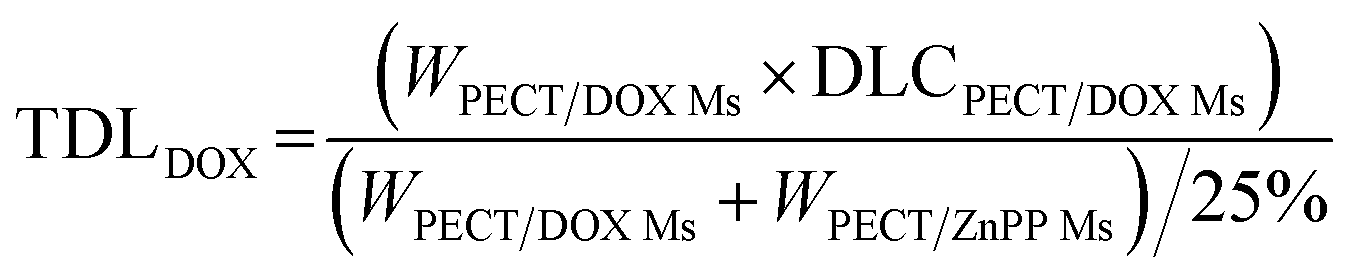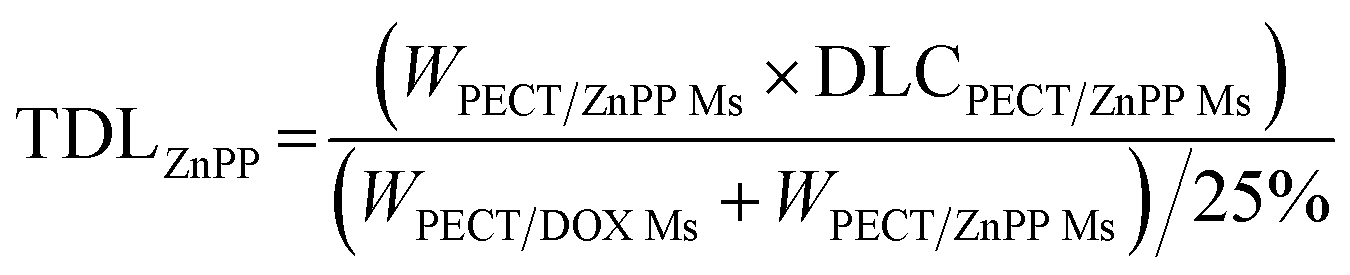DOI:
10.1039/C7RA06357A
(Paper)
RSC Adv., 2017,
7, 34755-34762
Thermosensitive micellar hydrogel for enhanced anticancer therapy through redox modulation mediated combinational effects†
Received
7th June 2017
, Accepted 27th June 2017
First published on 11th July 2017
Abstract
Cancer is increasingly viewed as an eco-system, a community in which tumor cells cooperate with other tumor cells and host cells in their microenvironment. The improved understanding of the intricate relationships in this eco-system has led to revolutionary treatments, which have evolved from relatively nonspecific cytotoxic agents to selective, mechanism-based therapeutics. Herein, from the view of dynamic equilibrium, a synergistic intracellular redox-regulation therapeutic strategy was proposed, in which combinational treatment of chemotherapeutic agents and ROS-elimination inhibitors was expected to effectively kill cancer cells and overcome redox adaptation mechanism associated drug resistance. To this end, a thermosensitive micellar hydrogel was prepared for co-delivery of nanomedicines in situ, which was capable of encapsulating and delivering multiple drugs with diverse therapeutic properties while maintaining the controlled synergistic ratio. Firstly, fluorescence resonance energy transfer (FRET) technology was adopted to track the real-time spatial pattern of drug presentation at a molecular level in this micellar hydrogel. Results suggested that the drug encapsulation in this micellar hydrogel platform proved to be a dynamic equilibrium process, during which free drug movement, drug exchange or penetration between micelles could occur. Furthermore, doxorubicin (DOX) and Zn(II) protoporphyrin IX (ZnPP) were used as the model chemotherapeutant and HO-1 inhibitor, respectively. In vitro and in vivo evaluation demonstrated that the intracellular redox-regulation mediated synergistic advantages of both two types of drugs translated into improved therapeutic outcomes. Consequently, such a thermosensitive micellar hydrogel formulation, which enabled precise control over the dosage and ratio of combination therapeutic agents to obtain the desired therapeutic effect with a single drug administration, holds great potential for spatiotemporal delivery of multiple bioactive agents for sustained combination therapy.
1 Introduction
Cancer is increasingly viewed as an eco-system, a community in which tumor cells cooperate with other tumor cells and host cells in their microenvironment.1 As conditions change, the eco-system evolves and adapts to ensure the survival and growth of cancer. The improved understanding of the intricate relationships in this ecosystem has led to revolutionary treatments, which have evolved from relatively nonspecific cytotoxic agents to selective, mechanism-based therapeutics, such as inhibition of molecular pathways for tumor growth and maintenance.2 Therefore, targeting unique biochemical alterations in cancer cells might be a feasible approach to achieve therapeutic activity and selectivity.
Increased generation of reactive oxygen species (ROS) and correspondingly induced redox adaptive mechanisms have long been observed in cancer cells.3–5 At advanced disease stage, cancer cells usually exhibit genetic instability and show a significant increase in oxidative stress during its development and progression. Due in part to “vicious cycle”, ROS induce gene mutations, especially in the mitochondrial genome, leading to further metabolic malfunction and ROS generation, which correlates with the aggressiveness of tumors and poor prognosis. Excessive level of ROS stress can be toxic to the cells, however, cancer cells become well-adapted to such intrinsic oxidative stress and develop enhanced endogenous antioxidant adaptive mechanisms, which not only activate ROS-scavenging systems to cope with the stress but also inhibit apoptosis. Thereinto, high expression of inducible isoform of heme oxygenase-1 (HO-1) is confirmed in many different types of cancer cells, whose expression can be potently induced in response to radio-, chemo-, or photodynamic therapies.6–8 HO-1 is considered to play a vital role in conferring drug resistance, which protects cells from oxidative stress by multiple mechanisms, including decreasing the prooxidant level (heme), increasing the antioxidant level (bilirubin), producing the antiapoptotic molecule (CO), and inducing ferritin to remove and detoxify free ferric ion.9–11 Moreover, mounting evidences suggest that HO-1 exerts pivotal regulatory role in tumor angiogenesis, metastasis and anti-apoptotic.12–14 Hence, from the view of dynamic equilibrium, synergistic intracellular redox-regulation through application of combinational therapy of ROS-generating agents and ROS-elimination inhibitors should be a feasible therapeutic approach to effectively kill cancer cells and overcome redox adaptation associated drug resistance.
Systemic administration of drugs or drug combinations via nanocarriers should be the preferred approach, which has enabled effective and targeting drug delivery through appropriate design.15 However, recent evidence points to less than 1% tumor accumulation of systemically administered nanoparticles, which limits the potential clinical transformation.16,17 In addition, although systemic treatment is necessary to eliminate metastasis, this approach is suboptimal for treating primary tumors. Eliminating the primary tumor that serves as the “source” for metastasis using chemotherapy drugs, as well as preventing metastasis before its spread by genetically modifying the tumor can revolutionize patients' point of care.18–22 Therefore, it is imperative to re-examine the available delivery platforms and determine the optimal administration route on a case-by-case basis, which will open up new vistas for effective combinational therapy, for the treatment of unresectable tumors, and for washout procedure following tumor resection to prevent recurrence. Hydrogels have proven to be highly biocompatible materials that allow for local delivery of therapeutic cargos, whose chemical and physical versatility can be exploited to attain disease-triggered in situ assembly, programmed degradation, and consequent drug release at set ratio.23–26
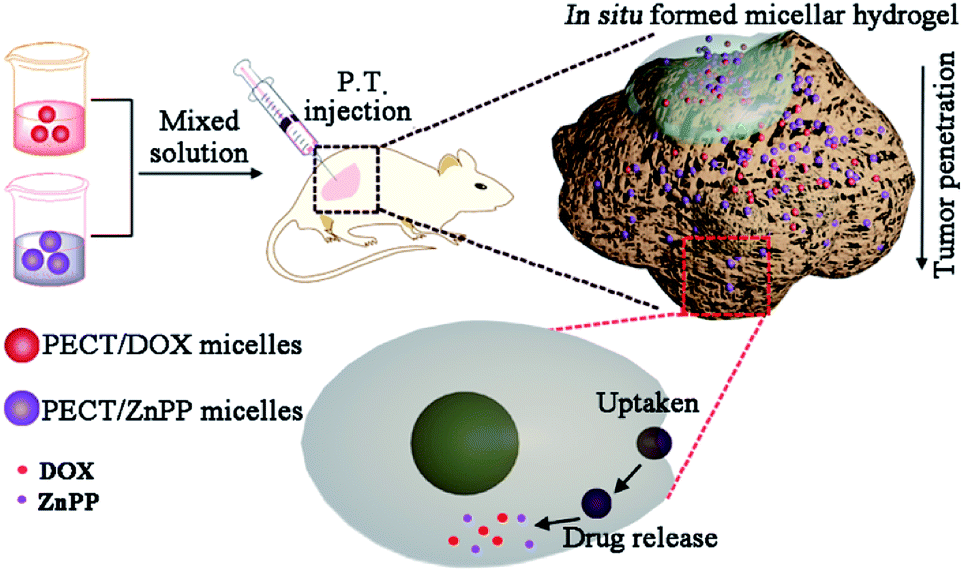
Herein, thermosensitive micellar hydrogel was prepared for co-delivery of nanomedicines in situ, which was capable of encapsulating and delivering multiple drugs with diverse therapeutic properties while maintaining the controlled synergistic ratio. It was expected to obtain enhanced anticancer effect through redox modulation mediated synergistic effects. The overall design and action pathway were shown in Fig. 1. Doxorubicin (DOX) and Zn(II) protoporphyrin IX (ZnPP) were used as the model chemotherapeutant and HO-1 inhibitor, respectively, which were encapsulated within poly(ε-caprolactone-co-1,4,8-trioxa[4.6]spiro-9-undecanone)–poly(ethylene glycol)–poly(ε-caprolactone-co-1,4,8-trioxa[4.6]spiro-9-undecanone) (PECT) micelles (Ms). Encapsulation and delivery multiple drugs with diverse therapeutic properties while maintaining the controlled synergistic ratio prove to be very difficult. In this work, the physicochemical properties of the two model drugs were different. It was difficult to load them in one micelle with needed ratio. From an overall perspective, DOX and ZnPP were encapsulated within PECT micelles individually, which would be more feasible for the control of drug ratio. After local injection, parent hydrogel depot was formed by thermosensitive self-aggregation of drug-loaded Ms, where continuously dissociated drug-loaded Ms were supposed to be spatially delivered within tumors with controlled synergetic ratio. In detail, endocytosis and synergetic treatment effect in vitro were tested against HepG2 cells. Subsequent evaluation of local antitumor activity was carried out using xenograft model of human hepatoma.
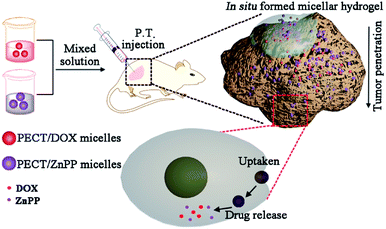 |
| | Fig. 1 Illustration of design and action pathway of PECT/DOX Ms and PECT/ZnPP Ms formed thermosensitive hydrogel. | |
2 Materials and methods
2.1 Materials
PECT copolymer was synthesized according to our previously reported methods, and the chemical structure was shown in ESI† file.27–29 Doxorubicin hydrochloride was purchased from Wuhan Hezhong Biochem Co. Ltd (Wuhan, China). Zn(II) protoporphyrin IX (ZnPP), 2,2,2-trifluoroethanol (TFE), 4,6-diamidino-2-phenylindole (DAPI), and cell counting kit (CCK-8) were purchased from Sigma-Aldrich (St. Louis MO, USA). The HepG2 cell line was purchased from American type culture collection (ATCC).
2.2 Preparation of drug-loaded PECT Ms
The drug loaded PECT Ms were prepared by nanoprecipitation method.27–29 Taking DOX as the example, DOX (2 mg) and PECT copolymer (100 mg) were co-dispersed in 4 mL TFE and then the mixture was added into 20 mL deionized water under constant stirring rate. After removal of TFE solvent, the micelle solution was lyophilized to obtain freeze-dried powder of PECT/DOX Ms. Blank PECT Ms were prepared with the same procedure without the addition of DOX. Correspondingly, ZnPP-loaded PECT Ms (PECT/ZnPP Ms) were prepared with similar method. The size and morphology of blank and drug-loaded Ms were measured by DLS and TEM, respectively. UV-Vis spectrophotometry was employed to determine the drug loading content (DLC) and drug loading efficiency (DLE).| | |
DLC (%) = (weight of loaded drug)/(weight of drug loaded NPs) × 100%
| (1) |
| | |
DLE (%) = (weight of loaded drug)/(weight of drug in feed) × 100%
| (2) |
2.3 Preparation and characterization of thermosensitive hydrogels
The freeze-dried powder of PECT Ms, PECT/DOX Ms, PECT/ZnPP Ms, mixture of PECT/DOX Ms and PECT/ZnPP Ms at different mass ratio were re-dispersed in saline at 25 °C with a mass concentration of 25% (w/w). The drug encapsulated in the formulation can be calculated by the following equation.| |
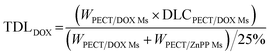 | (3) |
| |
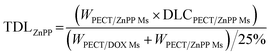 | (4) |
TDL: total drug loading amount; WPECT/DOX Ms: weight of PECT/DOX Ms; WPECT/ZnPP Ms: weight of PECT/ZnPP Ms. DLCPECT/DOX Ms: drug loading content of PECT/DOX Ms; DLCPECT/ZnPP Ms: drug loading content of PECT/ZnPP Ms.
In order to verify whether the drug loading and combination had influence on the gelation property, the gelation behavior was tracked using a Fluids Rheometer equipment (Stress Tech, Rheological Instruments AB).30
2.4 FRET-monitored dynamic drug encapsulation in the micellar hydrogel
2.4.1 Preparation of dye-encapsulated PECT Ms. Fluorescent dye DiO or DiI (1 mg) and PECT copolymer (100 mg) were co-dissolved in tetrahydrofuran (THF) and then the dispersion was added dropwised into 10 mL deionized water, during which dye molecules were loaded into core of PECT Ms owing to hydrophobic interactions. Then, THF solvent was removed by volatilization under RT in super clean bench for 24 h, which was maintained away from light. Finally, the solution was centrifuged (5000 rpm, 5 min) to remove the unpackaged dyes. The dye loading amount was determined by fluorescent quantitative analysis and the loading ratio for DiO and DiI was 0.81% and 0.84%, respectively.
2.4.2 FRET determination of dynamic dye encapsulation in the mixed Ms solution. After PECT/DiO Ms and PECT/DiI Ms solutions were prepared by the procedure described above, the two solutions were mixed in an equal volume. Then, the mixture was cultured at 37 °C and the FRET evolution was tracked by a Varian fluorescence spectrophotometer with excitation wavelength of 470 nm.
2.4.3 In vivo FRET determination. In vivo FRET evolution was conducted on BALB/c nude mice (6–7 weeks old, Vital River Laboratory Animal Technology Co. Ltd, China). After lyophilization of dye-loaded PECT Ms, the obtained powders were re-dispersed in water independently at a concentration of 25% (w/w). Prior to injection, the two solutions were mixed at an equivalent volume. 150 μL of blending solutions was subcutaneously administrated in the upper portion of the right flank. And then emission signals were monitored in real-time by a fluorescence imaging equipment (Kodak In Vivo FX Pro, New Haven, CT, USA) at 600 nm and 535 nm, respectively, with an excitation wavelength of 465 nm. The mean fluorescence intensities were also recorded to calculate the FRET efficiency and to characterize the drug loss rate from the hydrogel.
2.5 Cellular endocytosis evaluation
To observe cellular uptake of drug loaded Ms, 500 μL of HepG2 cell suspensions were seeded on a confocal microscopic dish at a density of 2 × 105 per dish. A certain amount of PECT/DOX Ms and PECT/ZnPP Ms solution was added and the drug concentrations of DOX and ZnPP were maintained at 5 μg mL−1. After further incubation for 4 h, cells were washed with PBS three times and then stained with 500 μL DAPI (1 μg mL−1 in PBS) for 5 min. After repeated wash with PBS, cellular endocytosis was examined using fluorescence microscopy (Leica AF 6500).
2.6 In vitro cell viability of individual drug and drug combinations on HepG2 cells
CCK-8 assay was conducted to evaluate the in vitro cell viability of DOX, ZnPP loaded Ms and their combinations. Briefly, HepG2 cells were seeded in 96-well plates at a density of 5000 cells per well 24 h prior to drug treatment. Subsequently, cells were treated with individual drug loaded Ms and drug combination with various molar ratios at a series of dilutions in full medium. After incubation, a 10% CCK-8 solution was added to wells, and the plate was incubated for another 2 h. The absorbance of each treated well was measured with multi-functional microplate reader at 490 nm. Each concentration was tested in four wells and data presented in means ± standard error means (SD).
2.7 Anti-tumor efficacy in xenograft human hepatoma model
The in vivo anti-tumor activity was conducted using xenograft tumor models. Female BALB/c nude mice (6–7 weeks) subcutaneously implanted with HepG2 tumors were obtained from Cancer Hospital, Chinese Academy of Medical Sciences. Animals developed tumors of approximately 100–150 mm3 were included in subsequent in vivo assessments. Mice were then randomly assigned to one of the following four groups: group 1 (n = 8), mice were peritumorally injected with PECT/DOX Ms hydrogel; group 2 (n = 8), mice were peritumorally injected with PECT/ZnPP Ms hydrogel; group 3 (n = 8), mice were peritumorally injected with PECT/DOX Ms + PECT/ZnPP Ms (at synergic ratio) hydrogel; group 4 (n = 8), mice were peritumorally injected with PBS as blank control. The total dose of DOX and ZnPP administrated was 20 mg kg−1 per animal and the injection volume for each peritumoral administration of hydrogel formulations was 200 μL. Tumor volumes were measured using a caliper at designated times and calculated according to the formula, tumor volume = a2 × b/2, where “a” is the shorter diameter and “b” is the longer one. Body weight of each animal was also recorded every two days.
The expression of HO-1 mRNA in tumor tissues was measured by quantitative real-time reverse transcription-PCR (qRT-PCR). Total RNA was harvested by using TRIzol according to the manufacturer's instructions. The concentrations were measured by spectrophotometer at 260 and 280 nm. cDNA was synthesized with the Taqman miRNA Reverse Transcription kit. qRT-PCR was performed using a commercial kit and an ABI7300 Real-time PCR system. The amplification condition was as follows: 2 min at 95 °C, followed 42 cycles of 30 s at 95 °C and 1 min at 60 °C. The primer sequences were as follows: HO-1 forward primer, 5′-GTGTAAGGACCCATCGGAGA-3′ reverse primer, 5′-GTGTAAGGACCCATCGGAGA-3.
All animal experiments were performed in accordance with the protocol approved by Chinese Academy of Medical Sciences and Peking Union Medical College Animal Care and Use Committee, which was based on the People's Republic of China national standard (GB/T 16886.6-1997).
2.8 Statistical analysis
All data are presented as mean ± standard deviations (SDs). The differences among groups were determined using student's t-test (GraphPad Prism 6.0) and p < 0.05 was considered to be statistically significant.
3 Results and discussion
3.1 Characterization of drug loaded Ms and hydrogels
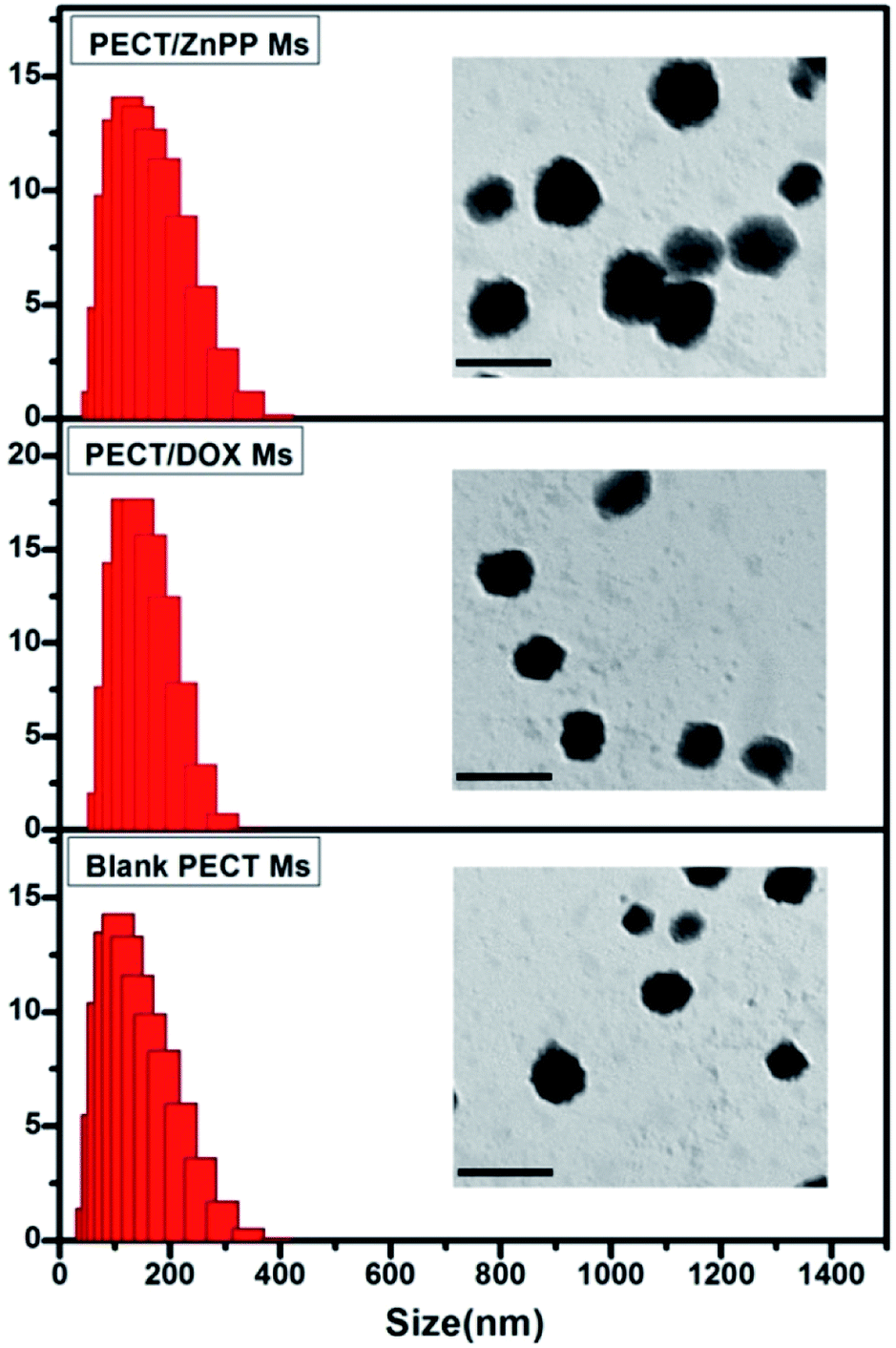
The formation of drug loaded Ms was mainly driven by the hydrophobic interaction between selected drugs (DOX and ZnPP) and the hydrophobic segment of amphiphilic PECT copolymers. As shown in Fig. 2, the particle sizes of obtained blank Ms were about 145 nm, which slightly increase about 10 nm due to the drug encapsulation. Importantly, the drug loading did not make obvious influence on the core–shell morphologies.
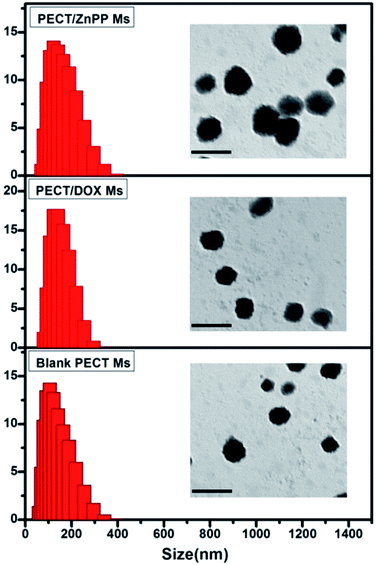 |
| | Fig. 2 The particle size and morphology of blank PECT Ms, PECT/DOX Ms, and PECT/ZnPP Ms prepared in saline at 25 °C. The scale bar was 0.2 μm. | |
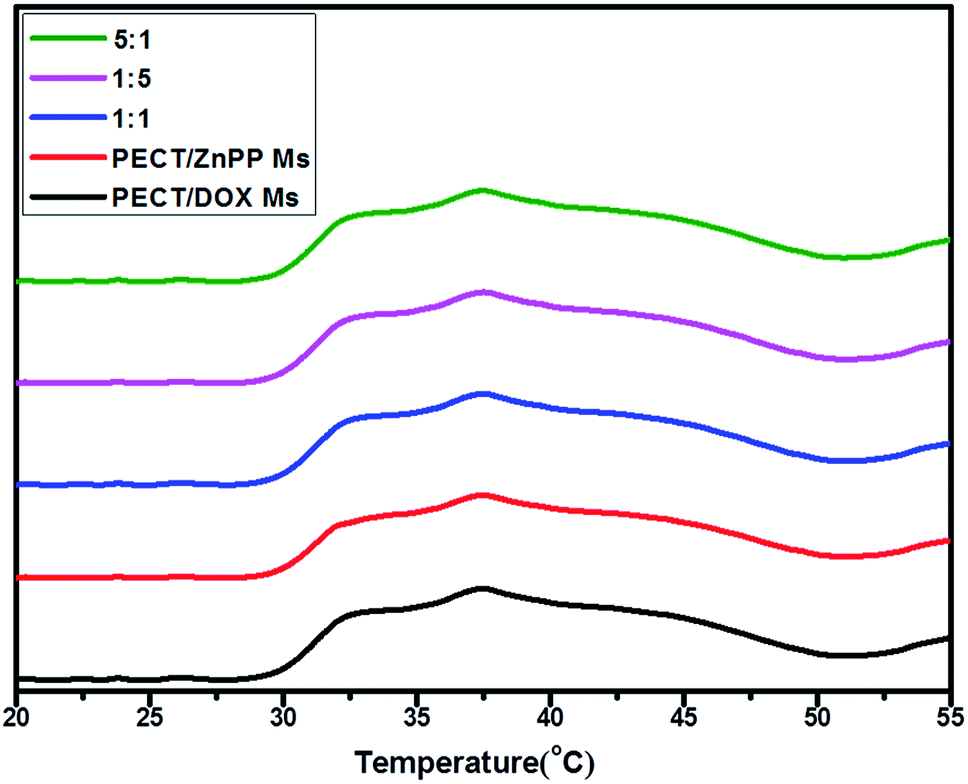
Furthermore, the influence of different drug loading on the gelation behavior was tracked by a rheometer. Drug-encapsulated hydrogel was constituted by the re-dispersion of lyophilized powders of Ms in saline at a mass concentration of 25% (w/w). Then, the thermosensitive aggregation behavior of the aqueous solution of Ms was investigated by rheological analysis. As shown in Fig. 3, both solutions of blank PECT Ms and drug loaded Ms exhibited sol-to-gel phase transition and the viscosity abruptly increased when the temperature was higher than 28 °C, indicating the formation of hydrogel. The encapsulation of drugs and drug combinations into hydrogel did not obviously affect the intrinsic thermosensitive aggregation profile of PECT Ms, as proven by the similar phase transition rheological properties. As studied in our another work, results of in vitro and in vivo evaluation confirmed that unique thermosensitive non-covalent Ms aggregation facilitated the hydrogel formation and sustained micelles shedding induced the hydrogel degradation, respectively, during which polymers were steadily incorporated into the Ms without any micelle disassembly or reassembly (unpublished work). Hence, this unique micellar hydrogel, consisting of nothing more than Ms could serve as a depot of nanomedicines and as a “combo” macroscale drug delivery platform for co-delivery of multi-modal therapeutics.
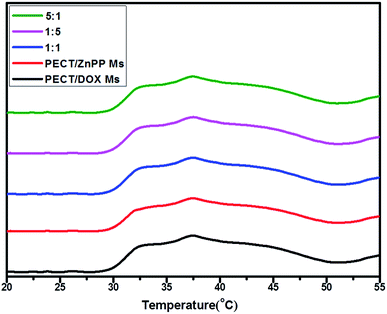 |
| | Fig. 3 The viscosity variation of PECT Ms, PECT/DOX Ms, PECT/ZnPP Ms, mixture of PECT/DOX Ms and PECT/ZnPP Ms at three different weight ratio as a function of temperature at a solution concentration of 25% (w/w). | |
3.2 FRET-monitored dynamic drug encapsulation in the micellar hydrogel
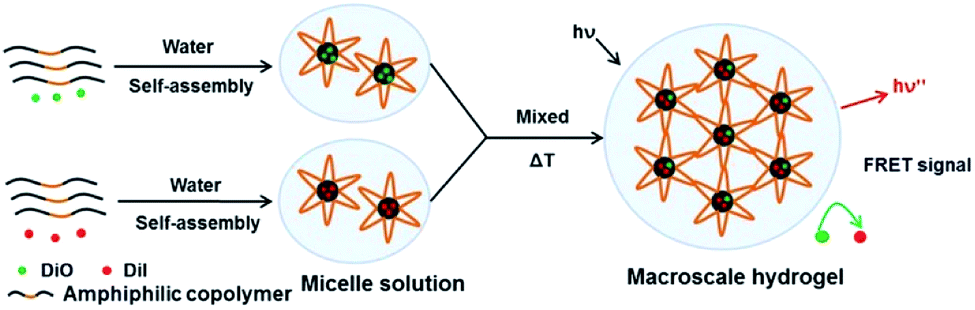
Macroscale hydrogel systems (MHS) have been engineered for spatiotemporally controlled delivery of various bioactive agents, including small drug molecules, genes, proteins and antibodies.31,32 Of many factors associated with the initial design of such drug delivery systems, drug encapsulation and release are fundamental concerns, which are directly related to the administration dose and drug bioavailability. Hence, understanding the real-time spatial pattern of drug presentation at a molecular level will greatly contribute to the rational design, construction and improvement of macroscale drug delivery systems. Herein, fluorescence resonance energy transfer (FRET) was used to track the drug encapsulation and release in this model thermosensitive micellar hydrogel.33,34 The design concept was illustrated in Fig. 4. 3,3′-dioctadecyloxacarbocyanine (DiO, donor) and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine (DiI, acceptor) were used as the FRET pair, which were independently and non-covalently encapsulated in the core of PECT Ms via the nanoprecipitation method to model the package of two hydrophobic dye molecules.35 The stokes shifts for DiI and DiO were less than 20 nm, thus, a high loading amount may result in aggregation induced quenching. Therefore, the loading amounts of DiO and DiI were 0.81% and 0.84%, respectively. After lyophilization, the obtained dye-loaded powders were re-dispersed at room temperature for 5 min in deionized water respectively to construct the hydrogel solution. Upon mixture, the temperature was increased to 37 °C and the flowing solution transform into a semi-solid hydrogel within 20 s. The assumption was as follows: (i) in gel state, if dye molecules exchanged between the hydrophobic core of micelles and in the bulk solvent, both kind of dye molecules could appeared simultaneously in a same micelle container, thus obvious FRET would be observed. Moreover, an equilibration state might appear for this dynamic exchange process in the hydrogel depot; (ii) otherwise, if dye molecules were stably sequestered in Ms individually and did not freely moved from one micelle to another, no FRET would be observed because the distance between the two dye molecules is much higher than their Förster radius (less than 10 nm). Hence, tracing the evolution of FRET in such a macroscale system would provide us insights into the dynamic drug encapsulation and the potential drug release kinetics. It should be mentioned that, DOX and ZnPP were not loaded during the FRET-monitored dynamic drug encapsulation in the micellar hydrogel. Hence, although DOX and ZnPP had fluorescent properties themselves, the tracking of DiO and DiI were not affected.
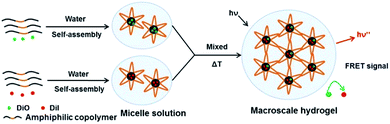 |
| | Fig. 4 The schematic dye encapsulation in a thermosensitive micellar hydrogel. DiO and DiI were independently encapsulated in the amphiphilic copolymer Ms and then the obtained micelle solution was mixed. Upon temperature increased to 37 °C, hydrogel was formed and then FRET was monitored. | |
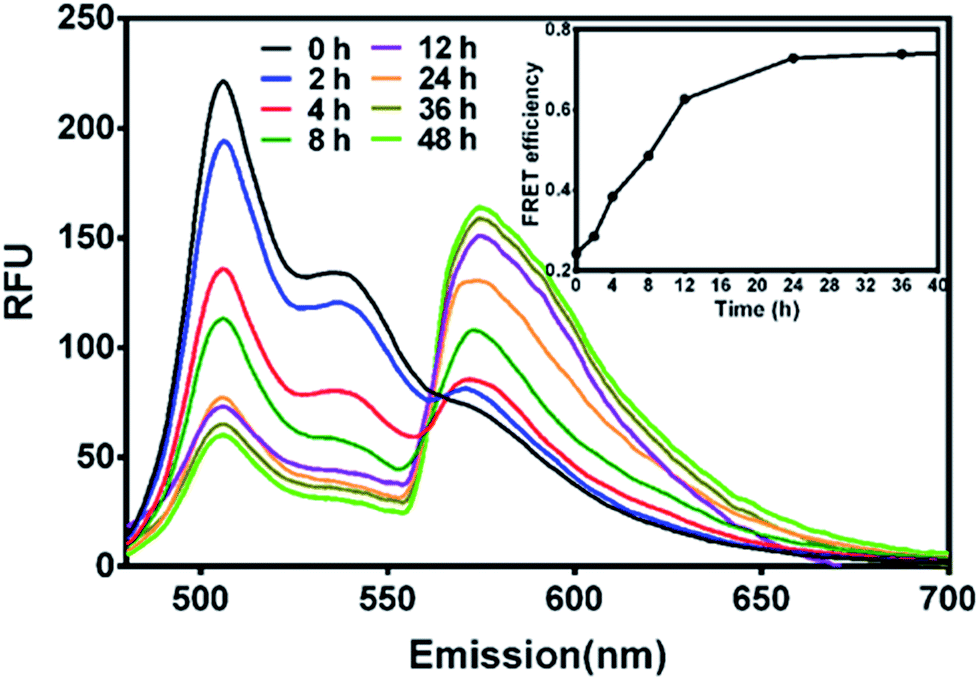
Firstly, to probe the drug encapsulation dynamics, solutions of PECT/DiO Ms and PECT/DiI Ms were mixed in vitro. Time-dependent emission fluorescence was monitored under the DiO excitation (470 nm). FRET was characterized by the donor (DiO) emission decrease and the concurrent acceptor emission (DiI) increase. As shown in Fig. 5, obvious enhancement of FRET was found over time, indicating the continuous thermodynamic dye distribution between micelles. The FRET efficiency calculated by IA/(IA + ID), where IA and ID are the emission fluorescence intensities of the acceptor and the donor, respectively, was plotted as a function of time. We define the FRET efficiency as the dynamic distribution ratio (DDR) of two kinds of dye molecules, which was found to gradually increase within the observation period until equilibrium, indicating that dye molecules persistently released from their native micelles and exchanged to the guest micelles within the observation period, instead of stably sealed in micellar cores. After incubation for 24 h, the variation of FRET reached balanced state. Results above suggested that drug encapsulation in a delivery vehicle was probably a dynamic equilibrium process, during which free drug movement, drug exchange or penetration between Ms might occur.
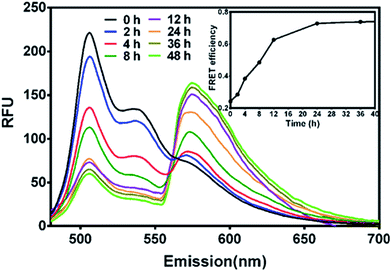 |
| | Fig. 5 The FRET evolution of mixed solution of PECT/DiO Ms and PECT/DiI Ms (the concentration of DiO and DiI encapsulated in PECT Ms was 0.1 mg mL−1; the concentration of PECT/DiO Ms and PECT/DiI Ms was 1 mg mL−1; excitation wavelength, 470 nm). | |
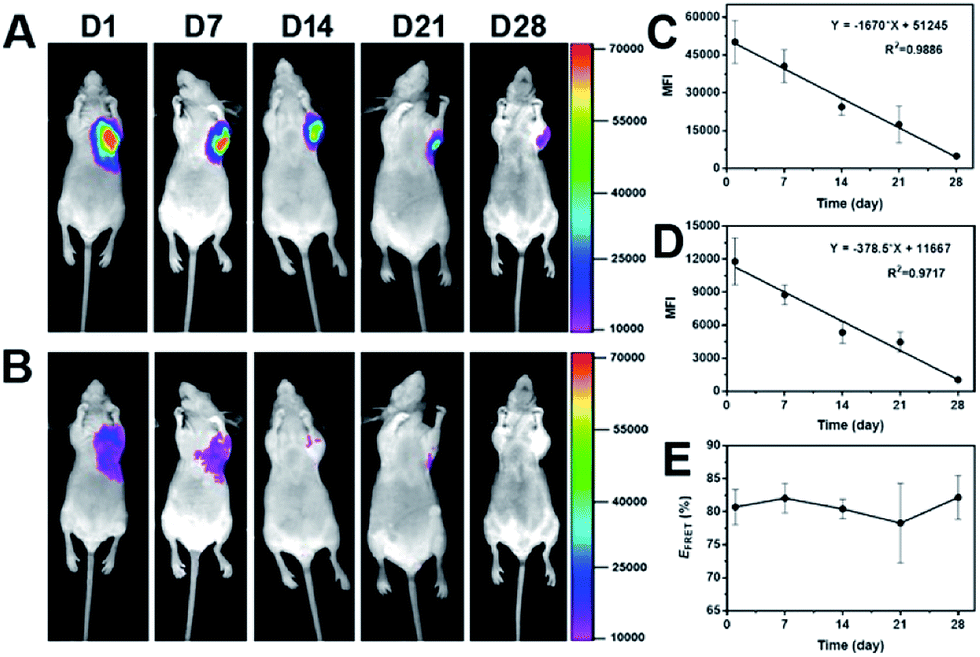
After in vitro FRET evaluation was implemented, PECT hydrogel packaged with DiO and DiI was subcutaneously injected in vivo. As shown in Fig. 6A and B, 24 h post-injection, strong FRET emission signals at 600 nm were detected while weak emission at 535 nm was observed. A similar phenomenon was found during the whole experiment period, demonstrating the dynamic distribution of dye molecules in the hydrogel depot. The mean fluorescence intensities (MFI) of both channels were recorded to quantify the real-time reduction rate of dye encapsulation and to calculate the FRET efficiency. As depicted in Fig. 6C and D, MFI decay versus time conformed to the linear regression, which demonstrated the rate of dye loss from the hydrogel was nearly constant. Our previous report on the PECT hydrogel degradation confirmed a linear attenuation of fluorescence signal.36 Thus, the carrier degradation, instead of dye diffusion, was responsible for the dye loss, because diffusion-controlled drug release usually shows an exponential profile. The FRET efficiency was in the range of 78.3–82.2% over time (Fig. 6E), which indicated that the loss of two dye molecules was in a similar rate. Collectively, it could be concluded that during the long-term drug encapsulation in the micellar hydrogel, an equilibrium state for dye distribution was first achieved and thereafter, drug molecules were released sustainedly without further dynamic exchange between Ms within the hydrogel. And it also reminded us to notice the drug–drug interaction when multiple drugs were concurrently encapsulated in one container for combination therapy, if lipophilic dyes were considered as model drugs. Significantly, we can propose a general mathematical equation for predicating the drug loss rate from the formulation with a linear regression property. The equation can be essentially written as Y = −aX + b, where Y is the residual drug amount in the formulation, a is the slope indicating the drug loss rate versus time, X is the time and b represents the total amount of drug encapsulated in the formulation.
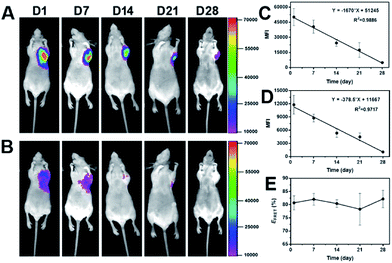 |
| | Fig. 6 (A) Representative photos of emission signals at 600 nm and (B) 535 nm over time. 300 μL of the mixture of PECT/DiO Ms and PECT/DiI Ms solutions at weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (total concentration was maintained at 25%, w/w) was subcutaneously injected and images were captured under the excitation of 465 nm by a fluorescence imaging equipment (Kodak In Vivo FX Pro). The linear fitting curves of mean fluorescence intensity (MFI) versus time for emission fluorescence at 600 nm (C) and 535 nm (D) and (E) the profile of FRET efficiency as a function of time. :1 (total concentration was maintained at 25%, w/w) was subcutaneously injected and images were captured under the excitation of 465 nm by a fluorescence imaging equipment (Kodak In Vivo FX Pro). The linear fitting curves of mean fluorescence intensity (MFI) versus time for emission fluorescence at 600 nm (C) and 535 nm (D) and (E) the profile of FRET efficiency as a function of time. | |
3.3 In vitro cell viability of individual drug and drug combinations
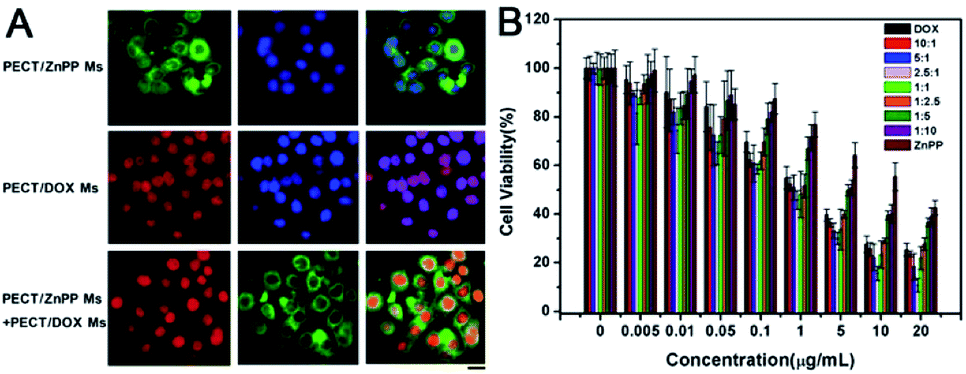
In order to achieve therapeutic effect, it was crucial for drug loaded Ms to be uptaken by tumor cells via endocytosis with rapidly intracellular drug release. As shown in Fig. 7A, the result of fluorescence microscopy observation demonstrated that both PECT/DOX Ms and PECT/ZnPP Ms could be internalized by HepG2 cells after incubation for 4 h. As can be seen clearly, intense DOX fluorescence covered all nuclear regions, which was consistent with its pharmacological properties. DOX, as a widely used antineoplastic agent in the treatment of various cancers, is known to exert drug effects via intercalation with DNA and inhibition of macromolecular biosynthesis. As to PECT/ZnPP Ms, bright green fluorescence could be observed in the cytoplasm. ZnPP is a potent inhibitor of HO-1, which is highly upregulated in many cancer tissues in vivo and confers an antioxidative function to cells. The combination of PECT/DOX Ms and PECT/ZnPP Ms facilitated the distribution of DOX in cell nucleus and ZnPP in cytoplasm. Subsequently, the in vitro cell viability of individual drug and drug combinations were evaluated by CCK-8 method. The cell viabilities against concentration of drugs were measured, where the molar proportion of each drug was maintained by controlling the mass ratio of PECT/DOX Ms and PECT/ZnPP Ms. As illustrated in Fig. 7B, the cytotoxicity of PECT/DOX Ms was obviously higher than that of PECT/ZnPP Ms, due to the strong inhibition of biosynthesis and duplication of DNA. In addition, certain combinational therapeutic effect was observed as the molar ratio of DOX and ZnPP in PECT Ms increased to 2.5:1. This combinational treatment effect could attribute to the inhibition of HO-1 through ZnPP to make tumor cells more vulnerable to oxystress of DOX. In addition, most of normal cells were not affected because HO-1 was expressed only at low level and insignificant in normal cells. Therefore, the rationale for this treatment was expected to obtain selective tumor regression.
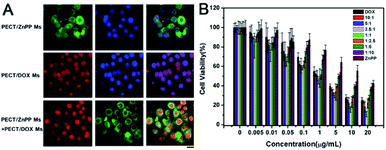 |
| | Fig. 7 (A) Endocytosis of PECT/ZnPP Ms, PECT/DOX Ms, PECT/DOX Ms and PECT/ZnPP Ms combination at a drug concentration of 5 μg mL−1: for each panel, DOX fluorescence signal in cells (red), ZnPP fluorescence signal in cells (green), cell nuclei stained by DAPI (blue), the scale bar was 25 μm; (B) viability of HepG2 cells after incubation with PECT/ZnPP Ms, PECT/DOX Ms, PECT/DOX Ms and PECT/ZnPP Ms combination at different drug molar ratio for 48 h. | |
3.4 In vivo antitumor activity
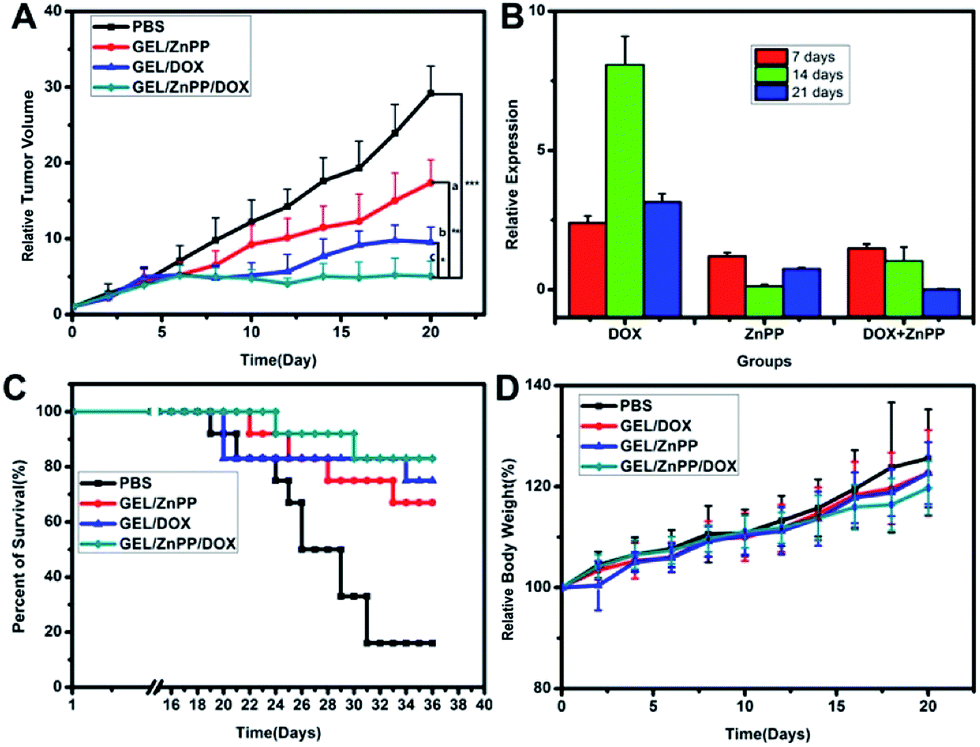
In order to verify if the merit of combinational therapeutic strategy could translate into improved therapeutic outcomes, we assessed the growth of xenografted HepG2 tumors following peritumoral administration of PECT hydrogel formulations. Human HepG2 cells (2 × 106 cells) were implanted subcutaneously in the dorsal skin of female BALB/c nude mice as described above. The antitumor activity was evaluated, when tumors had attained a size of approximately 100–150 mm3. As shown in Fig. 8A, administration of all the three therapeutic hydrogel formulations revealed significant tumor growth inhibition without remarkable fluctuation of body weight in comparison with blank control (p < 0.05). The PECT/DOX Ms and PECT/ZnPP Ms hydrogel (termed as GEL/DOX/ZnPP) combinational group led to statistically significant tumor growth delay compared with single treatment with PECT/DOX Ms hydrogel (termed as GEL/DOX) or PECT/ZnPP Ms hydrogel (termed as GEL/ZnPP), which could attribute to the inhibition of HO-1 leading to tumor cells more vulnerable to oxystress of DOX. To clarify whether the induction of tumor growth suppression caused by ZnPP mediated inhibition of HO-1, the expression of HO-1 mRNA in tumor tissues after treatment were detected by qRT-PCR method. As shown in Fig. 8B, the expression of HO-1 mRNA in GEL/DOX treated group was significantly higher than that of GEL/ZnPP group and GEL/DOX/ZnPP group, providing experimental evidence that HO-1 expressed in tumors holds promise as a potential molecular target for pharmacological intervention in cancer therapy. Inhibition of HO-1 activity by using GEL/ZnPP synergistically potentiated the antitumor activity of GEL/DOX. Therefore, these results demonstrated that the peritumoral injection of macroscale hydrogel enhanced and prolonged the local accumulation and retention of chemotherapeutic agents and HO-1 inhibitor, which resulted in synergetic treatment outcomes in vivo.
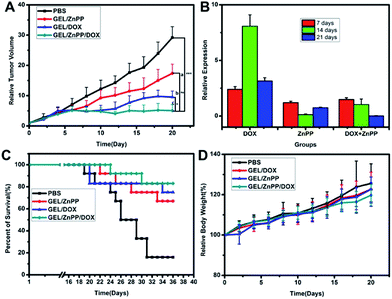 |
| | Fig. 8 (A) Relative tumor volumes of different formulations on the HepG2 tumor bearing BALB/c nude mice ((a) = in comparison with PBS, (b) = in comparison with GEL/DOX, (c) = in comparison with GEL/ZnPP, *p < 0.05, **p < 0.01, ***p < 0.001). (B) Relative HO-1 mRNA expression in tumor tissues. (C) The survival rate and (D) body weight of tumor bearing mice after various formulations were given. Data are presented as the mean ± standard deviation, n = 10. | |
4 Conclusions
In this work, a macroscale micellar hydrogel was used as a versatile platform to deliver chemotherapeutic agent and contemporaneously provide HO-1 inhibitor in the tumor tissue. In vitro and in vivo evaluation demonstrated that the synergistic advantages of both two types of drugs translated into improved therapeutic outcomes. Such an injectable thermosensitive micellar-hydrogel formulation, which not only enabled the precise control over the ratio of combinational drugs to obtain desired therapeutic effect, but also avoided repeated drug administrations, possesses great potential for spatio-temoral delivery of multiple bioactive agents for sustained combination therapy with improved patient compliance.
Acknowledgements
This project was financially supported by the National Natural Science Foundation of China (81471727, 51603231), CAMS Initiative for Innovative Medicine (2016-I2M-3-022), Tianjin Health Bureau Key Research Project (15KG138), Tianjin Natural Science Foundation (16ZXXYSY00040, 16JCQNJC02500, 16JCZDJC33000, 17JCQNJC13800) and PUMC Youth Fund and the Fundamental Research Funds for the Central Universities (3332016098).
Notes and references
- S. J. Horning, Science, 2017, 355, 1103 CrossRef CAS PubMed.
- M. Vanneman and G. Dranoff, Nat. Rev. Cancer, 2012, 12, 237–251 CrossRef CAS PubMed.
- R. R. Allison and K. Moghissi, Clinical Endoscopy, 2013, 46, 24–29 CrossRef PubMed.
- D. Trachootham, J. Alexandre and P. Huang, Nat. Rev. Drug Discovery, 2009, 8, 579–591 CrossRef CAS PubMed.
- B. Schenk and S. Fulda, Oncogene, 2015, 34, 5796–5806 CrossRef CAS PubMed.
- N. G. Abraham and A. Kappas, Pharmacol. Rev., 2008, 60, 79–127 CrossRef CAS PubMed.
- A. Jozkowicz, H. Was and J. Dulak, Antioxid. Redox Signaling, 2007, 9, 2099–2118 CrossRef CAS PubMed.
- H. F. Bai, N. N. Qi, X. C. Wu and Z. Y. Nie, J. Gastroenterol. Hepatol., 2013, 2, 353–358 Search PubMed.
- D. Nowis, M. Legat, T. Grzela, J. Niderla, E. Wilczek, G. Wilczynski, E. Głodkowska, P. Mrowka, T. Issat and J. Dulak, Oncogene, 2006, 25, 3365–3374 CrossRef CAS PubMed.
- M. Minetti, C. Mallozzi, A. M. M. Di Stasi and D. Pietraforte, Arch. Biochem. Biophys., 1998, 352, 165–174 CrossRef CAS PubMed.
- I. Petrache, L. E. Otterbein, J. Alam, G. W. Wiegand and A. M. Choi, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2000, 278, L312–L319 CAS.
- P. O. Berberat, Z. Dambrauskas, A. Gulbinas, T. Giese, N. Giese, B. Künzli, F. Autschbach, S. Meuer, M. W. Büchler and H. Friess, Clin. Cancer Res., 2005, 11, 3790–3798 CrossRef CAS PubMed.
- A. Nishie, M. Ono, T. Shono, J. Fukushi, M. Otsubo, H. Onoue, Y. Ito, T. Inamura, K. Ikezaki, M. Fukui, T. Iwaki and M. Kuwano, Clin. Cancer Res., 1999, 5, 1107–1113 CAS.
- H. Was, J. Dulak and A. Jozkowicz, Curr. Drug Targets, 2010, 11, 1551–1570 CrossRef CAS PubMed.
- Z. Cheng, A. Al Zaki, J. Z. Hui, V. R. Muzykantov and A. Tsourkas, Science, 2012, 338, 903–910 CrossRef CAS PubMed.
- J. W. Nichols and Y. H. Bae, Nano Today, 2012, 7, 606–618 CrossRef CAS PubMed.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS.
- N. Rao, G. Agmon, M. T. Tierney, J. L. Ungerleider, R. L. Braden, A. Sacco and K. L. Christman, ACS Nano, 2017, 11, 3851–3859 CrossRef CAS PubMed.
- J. Conde, N. Oliva, Y. Zhang and N. Artzi, Nat. Mater., 2016, 15, 1128–1138 CrossRef CAS PubMed.
- A. Gilam, J. Conde, D. Weissglas-Volkov, N. Oliva, E. Friedman, N. Artzi and N. Shomron, Nat. Commun., 2016, 7, 12868 CrossRef PubMed.
- J. Conde, N. Oliva, M. Atilano, H. S. Song and N. Artzi, Nat. Mater., 2015, 15, 353–363 CrossRef PubMed.
- J. Conde, N. Oliva and N. Artzi, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E1278–E1287 CrossRef CAS PubMed.
- J. S. Kahn, Y. Hu and I. Willner, Acc. Chem. Res., 2017, 50, 680–690 CrossRef CAS PubMed.
- C. Löwenberg, M. Balk, C. Wischke, M. Behl and A. Lendlein, Acc. Chem. Res., 2017, 50, 723–732 CrossRef PubMed.
- Y. Shao, H. Jia, T. Cao and D. Liu, Acc. Chem. Res., 2017, 50, 659–668 CrossRef CAS PubMed.
- H. Shigemitsu and I. Hamachi, Acc. Chem. Res., 2017, 50, 740–750 CrossRef CAS PubMed.
- W. Wang, L. Deng, S. Liu, X. Li, X. Zhao, R. Hu, J. Zhang, H. Han and A. Dong, Acta Biomater., 2012, 8, 3963–3973 CrossRef CAS PubMed.
- W. Wang, L. Deng, S. Xu, X. Zhao, N. Lv, G. Zhang, N. Gu, R. Hu, J. Zhang, J. Liu and A. Dong, J. Mater. Chem. B, 2013, 1, 552–563 RSC.
- W. Wang, L. Chang, X. Li, Y. Wu, J. Xing, L. Deng and A. Dong, Soft Matter, 2012, 8, 1575–1583 RSC.
- P. Huang, Y. Zhang, W. Wang, J. Zhou, Y. Sun, J. Liu, D. Kong, J. Liu and A. Dong, J. Controlled Release, 2015, 220, 456–464 CrossRef CAS PubMed.
- N. Oliva, J. Conde, K. Wang and N. Artzi, Acc. Chem. Res., 2017, 50, 669–679 CrossRef CAS PubMed.
- S. W. Morton, X. Zhao, M. A. Quadir and P. T. Hammond, Biomaterials, 2014, 35, 3489–3496 CrossRef CAS PubMed.
- E. A. Jares-Erijman and T. M. Jovin, Nat. Biotechnol., 2003, 21, 1387–1395 CrossRef CAS PubMed.
- S. Jiwpanich, J. H. Ryu, S. Bickerton and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 10683–10685 CrossRef CAS PubMed.
- J. H. Ryu, R. T. Chacko, S. Jiwpanich, S. Bickerton, R. P. Babu and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 17227–17235 CrossRef CAS PubMed.
- W. Wang, J. Liu, C. Li, J. Zhang, J. Liu, A. Dong and D. Kong, J. Mater. Chem. B, 2014, 2, 4185–4192 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06357a |
| ‡ These authors contributed equally in the present work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *c
*c