
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile synthesis of Au/Pd nano-dogbones and their plasmon-enhanced visible-to-NIR light photocatalytic performance
Yueyue Yinab,
Yong Yang *a,
Liangzhu Zhangcd,
Yongsheng Lib,
Zhiyuan Lie,
Weiwei Leid,
Yunfeng Maa and
Zhengren Huanga
*a,
Liangzhu Zhangcd,
Yongsheng Lib,
Zhiyuan Lie,
Weiwei Leid,
Yunfeng Maa and
Zhengren Huanga
aState Key Laboratory of High Performance Ceramics and Superfine Microstructure, Shanghai Institute of Ceramics, Chinese Academy of Sciences, 1295 Dingxi Road, Shanghai 200050, China. E-mail: yangyong@mail.sic.ac.cn
bLab of Low-Dimensional Materials Chemistry, Key Laboratory for Ultrafine Materials of Ministry of Education, School of Materials Science and Engineering, East China University of Science and Technology, Shanghai 200237, China
cKey Laboratory of Inorganic Functional Materials and Devices, Shanghai Institute of Ceramics, Chinese Academy of Sciences, 215 Chengbei Road, Jiading, Shanghai 201800, China
dInstitute for Frontier Materials, Deakin University, Waurn Ponds Campus, Victoria 3216, Australia
eLaboratory of Optical Physics, Institute of Physics, Chinese Academy of Sciences, P. O. Box 603, Beijing 100190, China
First published on 25th July 2017
Abstract
Here, we report a facile synthesis of Pd/Au nano-dogbones (NDBs) by loading massive Pd nanoparticles on the two ends of Au nanorods (NRs) via a seed-mediated method. The as-prepared samples were investigated by using TEM, elemental analysis, UV-vis and FDTD simulation. The photocatalytic activity of Pd/Au NDBs was assessed by dehydrogenation of formic acid under visible-to-NIR light irradiation. The Pd/Au NDBs shows much higher enhanced reaction activity compared with samples with core–shell structure due to the increased electron and hole pair separation and total catalytic sites. More importantly, the introduction of Au NRs broadens the photoresponse range and improves the utilization of sunlight efficiently. This finding provides a rational approach to improve photocatalytic performance by forming dogbone structures via integrating plasmonic metals and conventional catalysts.
1. Introduction
The demanding energy crisis drives scientists to look for green and abundant fuels. H2 generated directly via photocatalysis has attracted significant notice, in consideration of solar energy sustainable supply and clean utilization.1 However, conventional catalysts are restricted in practical application because of their lower catalytic efficiency under visible light irradiation.2,3 Therefore, developing an efficient and stable visible-light response catalyst is undoubtedly meaningful. Plasmoic metal nanostructures greatly promote the visible light to NIR absorption due to surface plasmon resonances (SPR). More importantly, the separation of hot electrons and holes in plasmonic metals coupled with conventional catalysts has a synergy effect, leading to enhanced catalytic activity. Therefore, rational combining plasmonic metal with traditional catalytic particles to form a heterostructure holds promise in photocatalysis.4–6Au NRs have triggered scientist interests for the ability to harvest a wide range of visible light and even NIR light due to tunable SPR, which contributes to improve solar energy conversion efficiency. For example, Au NRs integrated with TiO2 or Pt showed enhanced catalytic activity in water splitting7,8 However, little effort was allocated to integrating Au NRs with catalytic catalyst for H2 production from formic acid under visible light irradiation. Generally, H2 is generated via a catalytically dehydrogenation pathway (HCOOH (l) → H2 (g) + CO2 (g), ΔG298 = −35.0 kJ mol−1), while, carbon monoxide (CO), a disastrous poison to catalysts, can also be generated through a dehydration route (HCOOH (l) → H2O (l) + CO (g), ΔG298 = −14.9 kJ mol−1).9–11 For the purpose of improving hydrogen production efficiency, it is obviously desirable to develop novel catalysts for directly H2 conversion. Inspiring, much progress has been made on heterogeneous catalysis for selective dehydrogenation of formic acid.12–17 Among different catalytic metals, Pd-based catalysts are most promising for practical hydrogen production from formic acid.18–22 However, the absence of strong SPR absorption over Pd inhibited light energy utilization.23–25 Recently, Pd single layer tipped Au NRs demonstrated efficient plasmon-enhanced catalytic formic acid dehydrogenation even when below room temperature (5 °C).26 However, there are not enough electron–hole separation channels in Pd single-crystal covered structure, which would hinder the further improvement on catalytic properties. If more Pd poly-crystal nano particles (NPs) covered on two ends of Au NRs, more Pd–Au catalytic active sites and more electron–hole separation channels will be generated due to enlarged surface area of more Pd NPs. Therefore, it is one promising route to improve the photocatalytic activity by smaller Pd polycrystalline NPs covered Au NRs. However, to synthesize such a well-defined spatial separation structure by all wet-chemistry routes is challenging.
Herein, we developed an efficient method for synthesis of Au/Pd NDBs via the reduction of Pd2+ by L-ascorbic acid (AA) in the presence of CTAC-capped Au NRs and Ag+. As schematic illustration of the process is shown in Scheme 1. By controlling the CTAC bilayer confines the Au NR with only two ends accessible to Pd species. Ag was first deposited on nanorods, then was consumed by the Pd2+ ions in the solution via galvanic replacement reaction, leading to the formation of dogbones structure.27,28 The Au/Pd NDBs have a spatial separation structure, and Pd act as acceptor for hot electrons from Au NRs. This structure satisfies electron refilling requirement and exhibits plasmon-enhanced hydrogen production from formic decomposition under visible and NIR light irradiation.
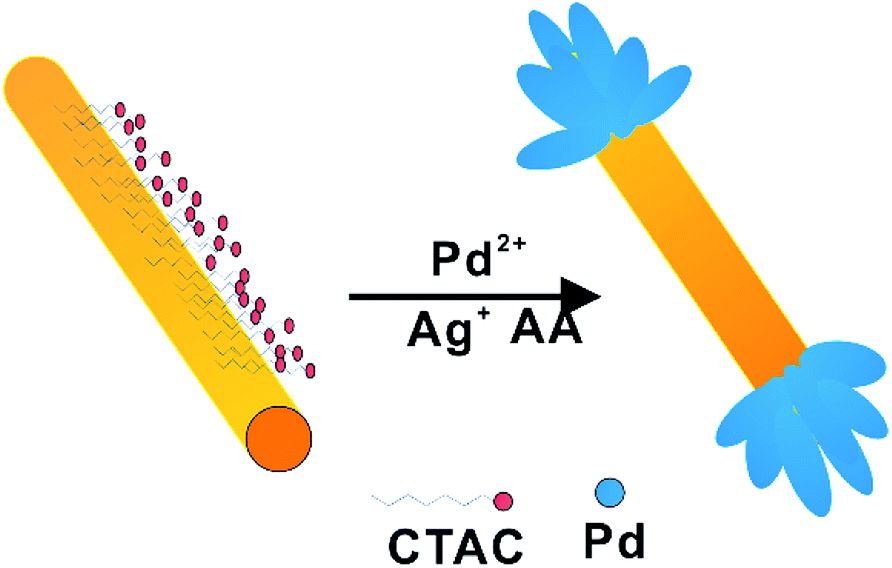 | ||
| Scheme 1 Synthetic procedure for Au/Pd NDBs. | ||
2. Experimental
2.1 Chemicals
All chemicals were purchased from commercial corporations. CTAC (96% powder), sodium oleate (NaOL, >97.0%) and sodium borohydride (NaBH4, 99%) were purchased from Aladdin. Hydrogen tetrachloraurate trihydrate (HAuCl4·3H2O), silver nitrate (AgNO3, 99.99%), CTAB (>98.0%), L-ascorbic acid (BioUltra, >99.5%) and palladium chloride (PdCl2, >99.9%) were purchased from Sigma-Aldrich. Ultrapure water (10 MΩ resistivity) was used for all experiments. 10 mM H2PdCl4 solution was obtained via mixing 44.5 mg of PdCl2 in 25 mL of 10 mM HCl. All glass ware were treated by freshly prepared aqua regia (HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3 in a 3:1 ratio by volume) and washed thoroughly with great amount water to remove chemical remains.
:HNO3 in a 3:1 ratio by volume) and washed thoroughly with great amount water to remove chemical remains.
2.2 Preparation of Au NRs
Au NRs were prepared by a modified seed-mediated growth method.29,30 Generally, the seed solution was obtained by reduction of HAuCl4·3H2O (0.5 mM, 5 mL) in CTAB (0.2 M, 5 mL) solution with ice-cold NaBH4 (6 mM, 1 mL). The seed solution was aged at room temperature for 30 min before use. The obtained seed solution (0.4 mL) was added into growth solution prepared by the sequential addition of AgNO3 (24 mL, 4 mM), HAuCl4·3H2O (250 mL, 1 mL), HCl (4.8 mL, 37 wt%) and AA (1.25 mL, 64 mM) into 250 mL CTAB-NaOL solution (7.0 g CTAB and 1.234 g NaOL dissolved in 250 mL purified water). The resulting Au NRs were divided into two parts, Au-NRs-1 were redispersed in 25 mL of 0.1 M CTAB, the as-prepared Au NRs were labeled as Au-NRs-1. The other redispersed in 0.1 M CTAC were labeled as Au-NRs-2.2.3 Preparation of Au/Pd NDBs
The Au-NRs-2 (2 mL) was injected to purified water (18 mL). H2PdCl4 (3 mL, 0.01 M), AgNO3 (4 mL, 1 mM) and various volumes AA (0.05 mM) were added sequentially in mixture. The solution were left undisturbed in a water bath at 30 °C for 12 h. The obtained nanostructures were centrifuged twice and redispersed in purified water (5 mL).2.4 Preparation of Au@Pd core–shell nanocuboids
The Au-NRs-1 (2 mL) was injected to purified water (18 mL). H2PdCl4 (3 mL, 0.01 M), and AA (300 μL, 0.05 mM) were added sequentially in mixture. The solution were left undisturbed in a water bath at 30 °C for 12 h. The obtained nanostructures were centrifuged twice and redispersed in purified water (5 mL).2.5 Preparation of Au@Pd core–shell nanostructure with irregular shapes
The Au-NRs-2 (2 mL) was injected to purified water (18 mL). H2PdCl4 (3 mL, 0.01 M), and AA (300 μL, 0.05 mM) were added sequentially in mixture. The solution were left undisturbed in a water bath at 30 °C for 12 h. The obtained nanostructures were centrifuged twice and redispersed in purified water (5 mL).2.6 Preparation of Au@Pd core–shell nanorods with serrated shapes
The Au-NRs-1 (2 mL) was injected to purified water (18 mL). H2PdCl4 (3 mL, 0.01 M), AgNO3 (4 mL, 1 mM) and AA (300 μL, 0.05 mM) were added sequentially in mixture. The solution were left undisturbed in a water bath at 30 °C for 12 h. The obtained nanostructures were centrifuged twice and redispersed in purified water (5 mL).2.7 Characterization of materials
Transmission electron microscopy (TEM) images, Scanning TEM (STEM) images and energy dispersive spectroscopy (EDS) mapping images were obtained on a JEOL JEM-2100F electron microscope operating at 200 KV. Absorption spectra was taken using quartz cuvettes of 1 cm path length on a PerkinElmer Lambda 950 instrument.2.8 FDTD calculation
The computational simulations were performed by using finite-difference-time-domain (FDTD) method with perfectly matched layers (PML) boundary conditions. The optical constants of Au were employed the results of tabulated values for bulk gold.31 The size of the NRs was adapted the average values. Particularly, the Au NR was modelled as a cylinder, with a length of 128 nm and width of 27 nm. For the model of Au/Pd NDBs, cylinder Pd nanorods with a length of 15 nm and width of 7 nm accumulated at two ends of the Au NR. For NRs dispersed in purified water, the refractive index of the medium was set to be 1.33.2.9 Hydrogen production from formic acid
The as-prepared catalysts were centrifuged and washed several times. The treated catalyst was added to 100 mL of formic acid (1 M) and poured into the reaction glassware. For visible-NIR light hydrogen evolution, the reaction mixtures were irradiated for 4 h with visible-NIR light (PerfectLight, PLS-SXE300, 300W) under magnetic stirring (300 rpm). UV light was removed by a 420 nm cutoff filter. A water bath was employed to control the reaction temperature at 15 °C. The amount of H2 in the gas phase was measured by Aosong Instrument GC-7890H gas chromatograph equipped with a MS-5A column and a thermal conductivity detector (TCD).3. Results and discussion
Fig. 1a and b show TEM and HRTEM images of the highly mono-dispersed single-crystalline Au NRs oriented growth along (200) plane by seed-mediated method. Fig. 1c and d show the prepared Au NRs presents good uniformity with lengths of 128 nm and width of 27 nm (aspect ratio of 4.80). Fig. 1e shows the Au NRs have a wide range of light absorption from 400 nm to 1200 nm with LSPR peak centred at about 998 nm, which indicates the Au NRs are capable harvest the light from UV to visible-NIR. | ||
| Fig. 1 (a) TEM images of Au NRs, (b) HRTEM of Au NRs, (c and d) size distributions (length and width) of gold NRs, (e) UV-vis-NIR absorption spectra of the Au NRs. | ||
Fig. 2a–c show TEM images of Au/Pd NDBs obtained in the presence of CTAC-capped Au NRs and Ag+. The products are composed of uniform NDBs structure with two ends of Au NRs coated with Pd nano-ball. HRTEM (Fig. 2d) image shows lots of single crystalline Pd nanoparticles with orientated growth along the (200) plane.
 | ||
| Fig. 2 (a and b) TEM images of Au/Pd NDs, (c) TEM images of single Au/Pd NDs, (d) HRTEM images of NDs. | ||
The ball size of Au/Pd NDBs could be well-controlled by simply modifying the molar ratios of reduction agent AA and Pd atoms source H2PdCl4. As can be seen in Fig. 3a–d, the ball semidiameter progressively increased form 13 nm to 44 nm, with molar ratio of AA:H2PdCl4 (the total amount of H2PdCl4 was fixed) increasing from 0.1 to 1. The dissociated nanoshperes are also found. Fig. 4 shows the small variation of LSPR peak of four samples may result in the influence of the ball size on the aspect ratio.
 | ||
| Fig. 3 TEM images of Au/Pd NDBs with various AA:H2PdCl4 ratio: (a) 0.1, (b) 0.25, (c) 0.5, (d) 1. | ||
 | ||
| Fig. 4 UV-vis-NIR absorption spectra of the four samples. | ||
To further identify the element distribution Au/Pd NDBs, STEM and EDS analysis on Au/Pd NDBs were employed. EDS elemental maps of Au/Pd NDBs (Fig. 5) clearly show Pd atoms selectively locate at the ends of Au NRs while no Pd atoms are found in the side of Au NRs, which confirmed the dogbones nanostructure. The Ag mapping present the similar results, shows the ball of the nano-dogbones is PdAg alloy structure. As can be seen in Fig. 6, the dissociated nanoshperes are also PdAg alloy, which is further supported that Ag+ ions have great impact on forming polycrystalline Pd nanostructure.32
 | ||
| Fig. 5 STEM images and corresponding STEM-EDS elemental maps of Au, Pd, Ag of single Au/Pd NDBs, the scale bars represent 20 nm. | ||
 | ||
| Fig. 6 STEM images and corresponding STEM-EDS elemental maps of Au/Pd NDBs and PdAg nanoshperes, the scale bars represent 100 nm. | ||
In order to compare the catalytic performance between heterostructure and homogeneous structure of Pd modified Au NRs, core–shell structure was further prepared. Fig. 7a shows Au@Pd core–shell nanocuboids. The as-prepared CTAB-capped Au NRs played as core, which was coated with a thin Pd layer with a thickness of 9 nm. The homogeneously growth pattern of Pd layer belongs to Frank-van der Merwe mode, which was confirmed by HRTEM (Fig. 7b) that a single Pd layer was aligned along the same direction with Au NRs.33,34 The SEAD pattern in Fig. 7b indicates Au@Pd core–shell nanocuboids are single crystalline in nature with well defined face-centered cubic (fcc) structures.
 | ||
| Fig. 7 TEM and HRTEM images: (a and b) Au@Pd core–shell nanocuboids, (c and d) Au@Pd core–shell nanostructure with irregular shapes, (e and f) Au@Pd core–shell nanorods with serrated shapes. | ||
Fig. 7c shows Au@Pd core–shell nanorods with irregular shapes. The CTAC-capped Au NRs were coated by a irregular Pd thin layer. Fig. 7d shows Pd layer presents a epitaxial growth pattern, while polycrystalline Pd particles were found growth on the surface of Pd thin layer. The SAED patterns (Fig. 7d) identify the Au NRs coated by Pd single crystalline with fcc structure. Some small polycrystalline Pd nanoparticles grow on the surface of Pd layer. This may be caused by CTAC soft template that is not able to specifically stabilize a particular crystalline facet like CTAB, leading to the irregularly shaped morphology.35
Fig. 7e shows Au@Pd core–shell nanorods with serrated shapes. The CTAB-capped Au NRs were coated with disconnected Pd thin layer in the Ag ions. HRTEM (Fig. 7f) image shows that the mass Pd nanoparticles with orientated growth along the (200) plane. SAED (Fig. 7f) pattern shows a single crystalline pattern of Au and Pd. The formation of discontinuous Pd shell due to the existence of the lattice vacancies generated by the galvanic replacement between Ag and Pd2+ in the modified procedure.
The most exciting feature of the designed Au/Pd NDBs offer the possibilities to promote SPR-induced hot-electron generation under visible and NIR light irradiation. The samples prepared in the presence of CTAB, CTAB-Ag+, CTAC and CTAC-Ag+ were treated with perchloric acid (HClO4) to remove the coated surfactants (CTAB or CTAC) that hinder charge-transfer process before using in H2 production experiments. Fig. 8a shows different reaction rate of H2 evolution via formic acid dehydrogenation in the dark or under light irradiation at 15 °C. TCD test only detected the presence of H2 and CO2, which conforms with the results of Pd tipped Au NRs catalysts for formic acid dehydrogenation.26 With light irradiation, the H2 evolution rate over Au/Pd NDBs reached to 1.05 mmol g−1 h−1 while that in the dark do not shows catalytic performance. As can be seen from Fig. 8b, there is a difference in H2 production between Pd–Au heterogeneous structure and Pd–Au homogeneous structure. Au/Pd NDBs exhibited a high rate of H2 evolution while Au@Pd core–shell NP did not show catalytic ability. Fig. 8c and d shows the catalytic activity of Au/Pd NDBs under light irradiation. For Au/Pd NDBs, H2 linearly evolved from the suspensions with increasing irradiation time. The H2 evolution rate initially arises with the increasing ball size. However, the rate drops to zero at AA:Pd = 1.
 | ||
| Fig. 8 (a) The rate of H2 evolution under dark/light irradiation over Au/Pd NDBs (AA:Pd = 0.5), (b) the rate of H2 evolution under the light irradiation over the catalysts obtained in the presence of CTAC-Ag+, CTAB-Ag+, CTAC, CTAB, (c) effect of ball size of Au/Pd NDBs on the rate H2 evolution under the light irradiation over the catalysts obtained at AA:Pd ratio of 0.1, 0.25, 0.5 and 1, (d) the time-dependent profiles of H2 evolution over the Au/Pd NDBs obtained at AA:Pd ratio of 0.1, 0.25, 0.5 and 1. Light irradiation is λ > 420 nm, 100 mW cm−2 at 15 °C. | ||
Au NRs function as carrier and hot-electrons source for Pd nanoparticles. The dispersity of Pd particles and quantity of catalytic sites are two major factors that influence the H2 production in this experiment. Table 1 shows that the corresponding H2 evolution rate increases from 0.45 mmol g−1 h−1 to 1.05 mmol g−1 h−1 when the size of Pd particles increases from 13 nm to 30 nm. The enlarged Pd particles mean bigger surface area and more catalytic site for H2 production. TEM images in Fig. 3(a–c) show that Pd particles in the Au/Pd NDBs samples have good dispersity. The improved photocatalytic performance was caused by the increased catalytic sites in Pd particles. However, Au/Pd NDBs-1.00 do not have any catalytic performance. TEM image in Fig. 3d shows that the ball of Au/Pd NDBs-1.00 with large ball size are too dense, which impairs the dispersity of Pd particles and contributes to the worsen catalytic activity. Therefore, the optimal reaction rate can be obtained by tuning the ball size in a relatively high ratio of AA:H2PdCl4. The turnover frequency (TOF) over Au/Pd NDBs obtained at AA:Pd = 0.5 without any additive was measured to be 2.1 h−1 at 15 °C under the light irradiation.
| Catalyst | Size of Pd nanoparticles (nm) | Rate of H2 evolution (mmol g−1 h−1) |
|---|---|---|
| Au/Pd NDBs-0.10 | 13 | 0.45 |
| Au/Pd NDBs-0.25 | 24 | 0.51 |
| Au/Pd NDBs-0.50 | 30 | 1.05 |
| Au/Pd NDBs-1.00 | 44 | 0 |
In order to examine function region of electric field amplification by SPR excitation and its potential contribution to the plasmon-induced enhancement of H2 production, the 3-D FDTD simulation were performed to present the spatial distribution of electric field intensity via varying incident polarization direction (Fig. 9a and b). Considering the random orientation of Au/Pd NDBs in suspensions, two patterns of stimulate light polarization direction (parallel and perpendicular) were employed to the Au/Pd NDBs. The results shows that the SPR enhancement of electric field mainly focus on the balls, regardless of the polarization direction. This indicates that dogbones structure are effective for the occurrence of electron–hole pairs separation. Therefore, combining experimental results and FDTD simulation, a reasonable reaction mechanism for formic acid dehydrogenation over Au/Pd NDBs under light irradiation is proposed, as illustrated in Fig. 9c. The NDBS structure are effective to harvest light to generate electron–hole pairs. Hot electrons accumulating in Pd surface improve the H2 catalytic performance. However, Fig. 9d shows the core–shell structure denied the opportunity for allowing longer-time electrons-hole separation due to the strongly charge recombination in homogeneous structure, leading to the absence of activity.36,37
 | ||
| Fig. 9 (a and b) Spatial distribution of the SPR-induced enhancement of electric field intensity for Au/Pd NDBs (AA:H2PdCl4 = 0.5), simulated by FDTD method, here E denotes the vector of the electric field and k denotes the wavevector of 900 nm excitation laser, (c) reaction mechanism for H2 production. | ||
4. Conclusion
In summary, an easy, bottom-up, wet-chemistry technique for the synthesis of Au/Pd NDBs has been successfully developed by loading massive Pd nanoparticles on two ends of Au nanorods (NRs) via seed-mediated method. The Pd/Au NDBs shows higher enhanced reaction activity compared with samples with core–shell structure, which was attributed to the increased electron and hole pair separation and total catalytic size. More importantly, the introduction of Au NRs broadens the photoresponse range and improves the utilization of sunlight efficiently, which directs us to extend this work to other dogbones like heterostructure system, e.g. Cu/Pd NDBs and therefore provides new opportunities in designing advanced photoactive materials with high stability and enhanced performance for solar energy conversion.Conflict of interest
There are no conflicts of interest to declare.Acknowledgements
This study was supported by a fund from the National Natural Science Foundation of China (No. 51471182) and Shanghai International Science & Technology Cooperation Program (No. 17520711700).Notes and references
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- C. Clavero, Nat. Photonics, 2014, 8, 95–103 CrossRef CAS.
- S. Mubeen, J. Lee, D. Liu, G. D. Stucky and M. Moskovits, Nano Lett., 2015, 15, 2132–2136 CrossRef CAS PubMed.
- X. Huang, Y. Li, Y. Chen, H. Zhou, X. Duan and Y. Huang, Angew. Chem., Int. Ed., 2013, 52, 6063–6067 CrossRef CAS PubMed.
- Z. Zheng, T. Tachikawa and T. Majima, J. Am. Chem. Soc., 2014, 136, 6870–6873 CrossRef CAS PubMed.
- B. Wu, D. Liu, S. Mubeen, T. T. Chuong, M. Moskovits and G. D. Stucky, J. Am. Chem. Soc., 2016, 138, 1114–1117 CrossRef CAS PubMed.
- M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 CAS.
- C. Hu, S. W. Ting and K. Y. Chan, Int. J. Hydrogen Energy, 2012, 37, 6372–6380 CrossRef CAS.
- S. Park, Y. Xie and M. J. Weaver, Langmuir, 2002, 18, 5792–5798 CrossRef CAS.
- K. Tedsree, T. Li, S. Jones, C. W. Chan, K. M. Yu, P. A. Bagot, E. A. Marquis, G. D. Smith and S. C. Tsang, Nat. Nanotechnol., 2011, 6, 302–307 CrossRef CAS PubMed.
- S. Fukuzumi, T. Kobayashi and T. Suenobu, J. Am. Chem. Soc., 2010, 132, 1496–1497 CrossRef CAS PubMed.
- X. Zhou, Y. Huang, W. Xing, C. Liu, J. Liao and T. Lu, Chem. Commun., 2008, 30, 3540–3542 RSC.
- M. Ojeda and E. Iglesia, Angew. Chem., Int. Ed., 2009, 48, 4800–4803 CrossRef CAS PubMed.
- Z. L. Wang, J. M. Yan, Y. Ping, H. L. Wang, W. T. Zheng and Q. Jiang, Angew. Chem., Int. Ed., 2013, 52, 4406–4409 CrossRef CAS PubMed.
- Q. Y. Bi, X. L. Du, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, J. Am. Chem. Soc., 2012, 134, 8926–8933 CrossRef CAS PubMed.
- Y. Cao, Y. Yang, Y. Shan and Z. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 5998–6003 CAS.
- N. V. Long, Y. Yang, C. M. Thi, N. V. Minh, Y. Cao and M. Nogami, Nano Energy, 2013, 2, 636–676 CrossRef.
- A. Cuesta, G. Cabello, M. Osawa and C. Gutierrez, ACS Catal., 2012, 2, 728–738 CrossRef CAS.
- M. Ojeda and E. Iglesia, Angew. Chem., Int. Ed., 2009, 121, 4894–4897 CrossRef.
- S. Jones, J. Qu, K. Tedsree, X. Q. Gong and S. C. E. Tsang, Angew. Chem., Int. Ed., 2012, 51, 11275 CrossRef CAS PubMed.
- Y. Huang, X. Zhou, M. Yin, C. Liu and W. Xing, Chem. Mater., 2010, 22, 5122–5128 CrossRef CAS.
- X. Gu, Z. Lu, H. Jiang, T. Akita and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11822–11825 CrossRef CAS PubMed.
- S. Zhang, O. Metin, D. Su and S. Sun, Angew. Chem., Int. Ed., 2013, 52, 3681–3684 CrossRef CAS PubMed.
- Z. Zheng, T. Tachikawa and T. Majima, J. Am. Chem. Soc., 2015, 137, 948–957 CrossRef CAS PubMed.
- D. Y. Kim, K. W. Choi, X. L. Zhong, Z. Y. Li, S. H. Im and O. O. Park, CrystEngComm, 2013, 15, 3385–3391 RSC.
- F. R. Fan, D. Y. Liu, Y. F. Wu, S. Duan, Z. X. Xie, Z. Y. Jiang and Z. Q. Tian, J. Am. Chem. Soc., 2008, 130, 6949–6950 CrossRef CAS PubMed.
- X. Ye, C. Zheng, J. Chen, Y. Gao and C. B. Murray, Nano Lett., 2013, 13, 765–771 CrossRef CAS PubMed.
- H. Jing and H. Wang, CrystEngComm, 2014, 16, 9469–9477 RSC.
- P. B. Johnson and R. W. Christy, Phys. Rev. B: Solid State, 1972, 197(6), 4370–4379 CrossRef.
- L. Zhang, J. Zhang, Z. Jiang, S. Xie, M. Jin, X. Han, Q. Kuang, Z. Xie and L. Zheng, J. Mater. Chem., 2011, 21, 9620–9625 RSC.
- X. Xia, Y. Wang, A. Ruditskiy and Y. Xia, Adv. Mater., 2013, 25, 6313–6333 CrossRef CAS PubMed.
- H. Jing and H. Wang, CrystEngComm, 2014, 16, 9469–9477 RSC.
- X. Guo, Q. Zhang, Y. H. Sun, Q. Zhao and J. Yang, ACS Nano, 2012, 6, 1165–1175 CrossRef CAS PubMed.
- S. K. Cushing, A. D. Bristow and N. Wu, Phys. Chem. Chem. Phys., 2015, 17, 30013–30022 RSC.
- S. K. Cushing, J. Li, J. Bright, B. T. Yost, P. Zheng, A. D. Bristow and N. Wu, J. Phys. Chem. C, 2015, 119, 16239–16244 CAS.
| This journal is © The Royal Society of Chemistry 2017 |