
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtic salt-based nitrogen-doped mesoporous carbon for simultaneous electrochemical detection of Cd(II) and Pb(II)
Danfeng Qin†
ae,
Ruiyu Xu†ae,
Hangjia Shenae,
Xamxikamar Mamata,
Le Wangae,
Shanshuang Gaoa,
Ying Wangae,
Nuerbiya Yalikunae,
Thomas Wagberg*b,
Shiguo Zhangc,
Qunhui yuan*d,
Yongtao Lia and
Guangzhi Hu *ae
*ae
aKey Laboratory of Chemistry of Plant Resources in Arid Regions, State Key Laboratory Basis of Xinjiang Indigenous Medicinal Plants Resource Utilization, Xinjiang Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Urumqi 830011, Xinjiang, China. E-mail: guangzhihu@ms.xjb.ac.cn
bDepartment of Physics, Umea University, 90187 Umea, Sweden. E-mail: thomas.wagberg@umu.se
cDepartment of Chemistry and Biotechnology, Yokohama National University, 79-5 Tokiwadai, Hodogaya-ku, Yokohama 240-8501, Japan
dHarbin Institute of Technology, Shenzhen 518055, China. E-mail: yuanqunhui@hitsz.edu.cn
eUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 25th July 2017
Abstract
Nitrogen-doped mesoporous carbon (NMC) derived from a single small-molecule protic salt (p-phenylenediamine bisulfate) is used for sensing toxic heavy metal ions. Using Nafion, bismuth and NMC to anchor the glassy carbon electrode surface, the fabricate electrode shows high sensitivity for detecting Cd(II) and Pb(II). The limits of detection (S/N = 3) are estimated to be 0.3 μg L−1 for Cd(II) and 0.4 μg L−1 for Pb(II), respectively, which are 10 and 25 times lower than the maximum acceptable content for drinking water recommended by the WHO. Furthermore, the sensor is successfully used to analyze Cd(II) and Pb(II) in tap-water with high anti-interference capability and good recovery.
1 Introduction
Toxic heavy metals pose a serious threat to both the environment and human health, because of their strong toxicity, ability to accumulate in biological systems and long metabolic cycles.1 Among the heavy metals, Cd(II) and Pb(II) are two common pollutants, which are harmful to mental health, the central nervous system, deleterious blood composition as well as other tissues and organs.2 Hence, it is urgent to dynamically monitor Pb(II) and Cd(II) at trace levels. With the aim of realizing accurate and reliable determination of heavy metals in environmental samples, different techniques such as fluorescence3 and inductively coupled plasma mass spectroscopy (ICP-MS)4 have been applied. However, these methods normally need professional operation and they are costly and/or require complex instruments. On the contrary electrochemical detection methods are time efficient, sensitive and use low cost equipment. In particular, anodic stripping voltammetry (ASV) represents a powerful tool for analyzing trace metals due to the effective accumulation of the target ions,5 which should be noted relies strongly on the sensing material deposited on the working electrode surface.6Nitrogen-doped mesoporous carbon (NMC) has shown excellent physical and chemical properties, such as abundant pore structure, large surface area, and low density,7 making it frequently applied in lithium batteries,8 fuel cells,9 supercapacitors,10 catalyst supports,11 separation processes12 and sensing.13 To detect heavy metals, NMC is hypothesized to exhibit remarkable performance because the mesoporous structure not only provides efficient diffusion and mass transfer for heavy metal ions, but also contains a large amount of electron-donating nitrogen atoms which can improve the performance of sensor.14,15 For example, Cui et al. demonstrated a nitrogen-doped porous carbon was synthesized with Al-based organic gel template for detection of Cd(II) by ASV with good stability and excellent selectivity.16
Normally, mesoporous carbon is produced based on the sacrifice template method, but the method is multistep, and time-consuming.7,17–19 For example, Shen et al. recently reported a MgO/ordered mesoporous carbon composite that could be applied for determination of Pb(II).20 The casting template method used for the preparation of ordered mesoporous carbon is complex, including SBA-15 (as template) preparation, infiltration of the sucrose into the SBA-15, carbonization of the sucrose, and finally etching of the silica template.
Nitrogen-doped porous carbon is commonly prepared in two ways: in situ synthesis or a post-treatment functionalization. The former method results in a homogeneous distribution of N,15 while with the latter it is difficult to control the nitrogen content and there is a risk of blocking the porous architecture.21 Xiao et al. successfully prepared nitrogen-doped microporous carbon with 25.0 at% nitrogen content for detection of Cd(II) and Pb(II) by in situ doping of nitrogen.22
In summary, traditional methods for the synthetic of NMC materials are typically tedious and time-consuming, therefore we describe a facile one-step template-free method to prepare NMC via carbonization of a protic salt ([pPDA][2HSO4]),19 which behaves as an carbon and nitrogen source during pyrolysis. We then use the prepared NMC in an electrochemical heavy metal sensor. The synthetic action of Nafion and bismuth on the NMC makes the sensor highly sensitive, and we show that it can be probed by simultaneous detection of Cd(II) and Pb(II).
2 Experimental section
2.1 Chemicals and apparatus
All chemicals used in this study were of analytical grade and used as received. p-Phenylenediamine and H2SO4 were obtained from Aladdin (Shanghai, China). 5.0 wt% Nafion, 1000 mg L−1 Cd(II) solution and 1000 mg L−1 Pb(II) solution were purchased from Sigma-Aldrich (Shanghai, China). 1000 mg L−1 atomic absorption standard Bi(III) solution was provided from Fluka (Shanghai, China).The morphology of NMC was characterized by a field emission scanning electron microscope (SEM, Carl Zeiss, Oberkochen, Germany). X-ray photoelectron spectra (XPS) were recorded with an Axis Ultra DLD electron spectrometer (Kratos Analytical Ltd., Manchester, UK). The structure analysis of NMC was operated on JEOL-2100F microscope (JEOL, Tokyo, Japan) at 200 kV. N2 adsorption–desorption isotherms was determined with an Autosorb-IQ-MP gas sorption analyzer (Quantachrome Instruments, Boynton Beach, Florida USA). The content of Cd(II) and Pb(II) was measured with Inductively Coupled Plasma-Mass Spectrometry (ICP-MS, ELAN DRC II, PerkinElmer, USA).
2.2 Synthesis of NMC and order mesoporous carbon
The NMC was prepared following a published method.19 Briefly, dilute aqueous sulfuric acid solution (20%, 25 mL) was slowly dropped into methanol (100 mL) containing p-phenylenediamine (5 g) in an ice bath. A precipitate was instantly formed, following which the mixture was stirred for 1 h. Afterwards the solvent was removed by using a rotary evaporator and the obtained mixture was kept at 80 °C for 24 h in a vacuum drying oven to get the protic salt. The as-obtained precursor was heated up to 900 °C at 10 °C min−1 in argon atmosphere and maintained at 900 °C for 2 h. After cooling down, black NMC was obtained. In order to compare the electrochemical performance of N doping, order mesoporous carbon (OMC) was prepared by using SBA-15 sacrifice template method.232.3 Preparation of NMC–Nafion/GCE, NMC–Nafion–Bi/GCE and DPASV test
The surface of GCE was sequentially polished with decreasing particle sizes of alumina power slurries. After polishing, the GCE was sonicated with absolute ethanol and deionized water for 3 min, respectively, and then dried by nitrogen. NMC was dispersed in DMF solution (containing 0.3 wt% Nafion) by sonication to get a 1.0 mg mL−1 suspension. A NMC–Nafion/GCE was obtained by casting 5 μL of suspension on GCE surface and drying at room temperature. Meanwhile, NMC–Nafion–Bi/GCE was prepared by an in situ plating of Bi film in accumulation step.The electrolyte (10 mL) included acetate buffer (0.1 M, pH = 5.0) and the target metal ions. If the modified electrodes need to deposit Bi film, 400 μg L−1 Bi(III) is added into the electrolyte. Under optimized conditions, differential pulse anodic stripping voltammetry (DPASV) was applied for heavy metals determination. The accumulation step was set at −1.1 V under stirring for 420 s. Following the preconcentration, the solution was kept quiet for 10 s. Then the DPASV curve was recorded at differential pulse mode from −1.0 V to −0.4 V.
3 Results and discussion
3.1 Characterization of NMC
The granular morphology of synthesized NMC nanoparticles was characterized by SEM (Fig. 1a), revealing a fluffy foamy NMC structure. The structure NMC was evaluated by TEM as that shown in Fig. 1b. As expected, NMC is pretty transparent due to its rich, disordered wormlike porous structure, as previously reported in the literature.19 | ||
| Fig. 1 (a) SEM and (b) TEM image of NMC, (c) the relative N2 sorption isotherms (inset: pore width distribution of NMC), and (d) XPS (inset: high-resolution N 1s spectrum). | ||
The pore structure of NMC was explored by nitrogen adsorption–desorption experiments. As presented in Fig. 1c, a hysteresis loop at pressures P/P0 > 0.5 is observed, which indicates the presence of mesopores. The BET surface area is calculated to be up to 596 m2 g−1 with a narrow pore-size distribution and an average pore size of 7.0 nm as shown as in the inset of Fig. 1c.
To investigate the chemical valence and elemental composition of the NCM, XPS measurement was performed in the region from 0 to 600 eV. The elemental content of NMC is determined to 84.2 at% for C, 10.0 at% for N, 5.0 at% for O, and 0.9 at% for S (Fig. 1d). The high resolution N 1s spectra of NMC (inset in Fig. 1d) could be deconvoluted into four peaks, at 397.80, 400.08, 402.16, and 405.05 eV, which correspond to pyridine-like, pyrrole-like nitrogen as well as oxidized species at higher binding energies (>402 eV),24–26 respectively. The XPS results clearly prove that N atoms have been successfully doped into the material. N introduction can accelerate the electron transfer and improves the sensitivity of the modified electrode.27
3.2 Electrochemical responses of the different electrodes
In order to study the electrochemical performance of bare GCE, OMC/GCE and NMC/GCE, cyclic voltammograms were investigated in Fe(CN)63−/4− and KCl solution. As shown in Fig. 2a, three pairs of well-defined redox peaks are observed in all electrodes. Compared with the bare GCE, the redox current of OMC/GCE is increased. It is believed to result from the good electrical conductivity of OMC. However, the largest peak current is appeared at NMC/GCE, which indicates that N accelerates electron transfer of [Fe(CN)6]3−/4− probe and shows better performance than OMC. | ||
| Fig. 2 (a) Comparison of cyclic voltammograms at bare GCE, OMC/GCE and NMC/GCE with a scan rate of 50 mV s−1 in 1 mM [Fe(CN)6]3−/4− and 0.1 M KCl. (b) DPASV curves at different electrodes after background subtracting in HAc–NaAc buffer (0.1 M, pH 5.0) with 50 μg L−1 Cd(II), 50 μg L−1 Pb(II). | ||
The electrochemical behaviors of Nafion–GCE (curve a), Bi/GCE (curve b), Nafion–Bi/GCE (curve c), OMC–Nafion/GCE (curve d), NMC–Nafion/GCE (curve e), and NMC–Nafion–Bi/GCE (curve f) were investigated by DAPSV in HAc–NaAc (0.1 M, pH 5.0), respectively. As presented in Fig. 2, there are two peaks around −0.8 V and −0.6 V, corresponding to the stripping peak of Cd and Pb at these six electrodes. The anodic peak currents of Cd and Pb at Bi/GCE are 5.09 μA and 6.31 μA, which is almost 45.5% and 64.3% at the NMC–Nafion–Bi/GCE. Compared to Nafion/GCE, the Nafion–Bi/GCE shows a clear increase in peak currents, ascribed to the ability of the Bi-film to form a fused alloy with Cd and Pb (Cd(II) + Bi(III) + 5e → Cd–Bi,Pb(II) + Bi(III) + 5e → Pb–Bi).28 Additionally, the stripping peak currents of Cd(II) and Pb(II) at NMC–Nafion/GCE are higher than OMC–Nafion/GCE, which suggests that NMC shows more outstanding analytical performance in determination heavy metals than OMC. Moreover, the peak current of NMC–Nafion–Bi/GCE improves about 47% for Cd(II) and 66% for Pb(II) than that of Nafion–Bi/GCE, respectively. These improvements in peak current can be attributed to the porous structure and large surface area of the NCM, which is beneficial for an effective accumulation of heavy metal ions.
3.3 Optimization of experimental conditions
In order to obtain high sensitive of detection heavy metals at the NMC–Nafion–Bi/GCE, pH of solution, Bi(III) concentration, accumulation potential and preconcentration time were investigated, respectively.The effect of Bi(III) concentration was examined from 100 μg L−1 to 600 μg L−1. As shown in Fig. 3a, the stripping peak currents of Cd(II) is gradually decreased when the Bi(III) concentration was higher than 400 μg L−1 and Pb(II) concentration is higher than 500 μg L−1. Thus, we chose 400 μg L−1 Bi(III) as the optimum concentration.
 | ||
| Fig. 3 Influence of various experimental parameters on stripping responses of 50 μg L−1 Cd(II) and 50 μg L−1 Pb(II) at NMC–Nafion–Bi/GCE. (a) Bi(III) concentration; (b) preconcentration potential; (c) pH and (d) deposition time. | ||
The influence of deposition potentials on the DPASV signals of Pb(II) and Cd(II) was explored in the potential window from −0.9 to −1.3 V. The results displayed in Fig. 3b indicate that the stripping peak currents increase sharply when the accumulation potential is shifted negatively from −0.9 to −1.1 V, and the maximum current for these two ions are at −1.1 V and then shift further in the negative direction when the deposition potential is further decreased. This phenomenon is ascribed to the significant hydrogen evolution at more negative potentials.29 Here −1.1 V is selected as the optimized deposition potential.
Solution pH can also affect the DPASV peak current of the Cd(II) and Pb(II) oxidation. Fig. 3c presents the influence of buffer pH on the DPASV responses of Cd(II) and Pb(II). When the pH increases from 4.0 to 5.0, both stripping peak currents increase, while a further increase in pH leads to a decrease in stripping signal. This is ascribed to the formation of metal hydroxide complexes.1 Therefore, a pH 5.0 was applied in this work.
Accumulation time is a really important parameter to heavy metal detection by using DPASV method. It can be seen from Fig. 3d, the influence of the accumulation time on detection of Cd(II) and Pb(II) was investigated from 180 to 480 s. The stripping signals increase gradually to the deposition time from 180 up to 420 s. However, when the preconcentration time is beyond 420 s, the stripping peak currents almost remain constant due to the rapid surface saturation.30 In order to get high peak current, an accumulation time 420 s is chosen for further study.
3.4 Analytical performance of Cd(II) and Pb(II) analysis
The DPASV curve of Cd(II) and Pb(II) are showed in Fig. 4a. It can be seen that for Cd and Pb, the potential peak positions are positively shifted for higher concentrations, a phenomenon which is common and analogous to what is observed for metal film electrodes.13 This is caused by interaction between the specific composition/structural morphology on the electrode surface and the thin mercury or Bi film.31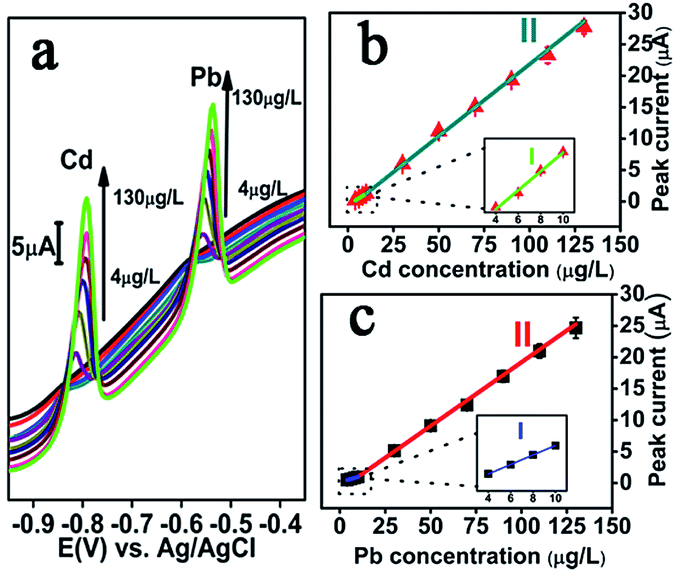 | ||
| Fig. 4 (a) DPASV records for various concentrations (4, 6, 8, 10, 30, 50, 70, 90, 110 and 130 μg L−1 from bottom to top) of Cd(II) and Pb(II) at NMC–Nafion–Bi/GCE. (b and c) The calibration curves of Cd(II) and Pb(II). | ||
Under the optimized experimental conditions, NMC–Nafion–Bi/GCE was applied in the successive analysis of Cd(II) and Pb(II) in the range from 4 μg L−1 to 130 μg L−1. DPASV response signal (vs. Ag/AgCl) for different concentrations of target metal ions and the relative calibration plots were illustrated in Fig. 4b and c. The analytical curves both for Cd(II) and Pb(II) cover two linear ranges from 4 to 10 μg L−1 and from 10 to 130 μg L−1, demonstrating the good linearity of the proposed sensing electrode for both Cd(II) and Pb(II). The regression equation of Cd(II) and Pb(II) are listed in Table 1. As seen from the Table 1, the slop of the linear in the low concentration range is much smaller than the one in the high concentration range. It could be due to the difference in rate determining step in these two concentration ranges. Moreover, the limit of detection (at S/N = 3) were valuated to be 0.3 μg L−1 for Cd(II) and 0.4 μg L−1 for Pb(II), which are 10 times for Cd(II) and 25 times for Pb(II) lower than the reference value of drinking water given by WHO (Cd: 3 μg L−1, Pb: 10 μg L−1),32 making the NMC–Nafion–Bi/GCE to a good candidate for the analysis of real aqueous samples.
| Heavy metals | Regression equation (y: μA, x: μg L−1) | LOD (μg L−1) | Maximum acceptable content (μg L−1) | |
|---|---|---|---|---|
| Linear range I: 4–10 (μg L−1) | Linear range II: 10–130 (μg L−1) | |||
| Cd(II) | y = 0.179x − 0.550, R2 = 0.990 | y = 0.218x − 0.446, R2 = 0.997 | 0.3 | 3 |
| Pb(II) | y = 0.081x + 0.165, R2 = 0.999 | y = 0.198x − 0.934, R2 = 0.999 | 0.4 | 10 |
The sensing performance of NMC–Nafion–Bi/GCE towards Cd(II) and Pb(II) is compared with the other reported sensors and presented in Table 2. It is obvious that the proposed NMC–Nafion–Bi/GCE is comparable to the previously reported results.
| Modified electrode | Method | Range in μg L−1 | Limit of detection in μg L−1 | Deposition time in s | Ref. | ||
|---|---|---|---|---|---|---|---|
| Cd | Pb | Cd | Pb | ||||
| a Nitrogen doped microporous carbon derived from ZIF-8.b Multi-walled carbon nanotube.c Poly(pyrocatechol violet).d Graphene.e Mercury film electrode.f Dibenzo-24-crown-8.g Screen-printed carbon electrode.h Screen-printed electrode.i Electroreduced graphene oxide.j Thionine.k 2-Mercaptoethanesulfonate.l 2,5-Dimercapto-1,3,4-thiadiazole.m Polyaniline.n Bismuth-oxychloride.o Poly(2,5-dimercapto-1,3,4-thiadiazole). | |||||||
| aCarbon of ZIF-8/Nafion/Bi/GCE | DPASV | 2–100 | 0.5–100 | 1.5 | 0.05 | 180 | 22 |
| bMWCNT–cpoly(PVC)/GCE | DPASV | 1.0–300 | 1.0–200 | 0.2 | 0.4 | 420 | 33 |
| Nafion–dG–eHgFE/GCE | SWASV | 1–7 | 1–7 | 0.08 | 0.07 | 120 | 34 |
| Bi–fD24C8–Nafion/gSPCE | SWASV | 0.5–60 | 0.5–60 | 0.27 | 0.11 | 180 | 35 |
| MWCNTs–Nafion–Bi/hSPE | DPASV | 0.5–80 | 0.05–100 | 0.1 | 0.07 | 120 | 36 |
| iERGO–jTH/kMES/GCE | SWASV | 1–40 | 1–40 | 0.1 | 0.05 | 300 | 37 |
| Bi–Nafion–lDMcT–mPANI–MWCNT/GCE | DPASV | 0.02–20 | 0.1–30 | 0.01 | 0.04 | 360 | 38 |
| nBiOCl–MWCNT/GCE | SWASV | 5–10 | 5–10 | 0.57 | 1.2 | 120 | 39 |
| Nafion–oPDMcT–MWCNTs/GCE | DPASV | 0.05–20 | 0.1–22 | 0.03 | 0.05 | 120 | 40 |
| NMC–Nafion–Bi/GCE | DPASV | 4–130 | 4–130 | 0.3 | 0.4 | 420 | Our work |
3.5 Interference study
The co-exiting ions test was applied by adding different cations into 0.1 M HAc–NaAc solution containing both 50 μg L−1 Cd(II) and 50 μg L−1 Pb(II). Fig. 5 presents the DPASV peak current comparison of Cd(II) and Pb(II) in the absence (I0) and presence (I) of various interfering metal ions. As displayed, under ±10.0% tolerated ratios, 600-fold Ca(II), 200-fold Mg(II), 50-fold Zn(II), 10-fold Fe(II), 8-fold Al(III), 8-fold Cr(III), 5-fold Fe(III), and 5-fold Co(III) have a relatively small influence of the peak current of Cd(II) and Pb(II). However, it is found that Cu(II) will seriously affect the stripping current of Cd(II) and Pb(II), which was in line with ref. 41. When 1-fold of Cu(II) is added, the peak current of Cd(II) and Pb(II) decreased by 55.4% and 63.2%, respectively, owing to the formation of Cu–Pb and Cu–Cd alloy.33 | ||
| Fig. 5 Interferences caused by other cations on the stripping peak current of 50 μg L−1 Cd(II) (red) and 50 μg L−1 Pb(II) (blue) under the optimized conditions. | ||
3.6 Repeatability, reproducibility and stability of the sensor
Repeatability was evaluated by five measurements of 50 μg L−1 Cd(II) and 50 μg L−1 Pb(II) at the same NMC–Nafion–Bi/GCE. The relative standard deviations (RSD) based on five times measures were 3.7% for Cd(II) and 3.5% for Pb(II), indicating a satisfying durability of the developed electrode.Reproducibility of the proposed electrode was carefully examined by using five NMC–Nafion–Bi/GCE for simultaneous detection of 50 μg L−1 Cd(II) and 50 μg L−1 Pb(II). The RSD between electrodes was 3.3% for Cd(II) and 5.4% for Pb(II), respectively.
After storage at 4 °C for one day and half a month, the stripping signal stability of NMC–Nafion–Bi/GCE was evaluated in 50 μg L−1 Cd(II) and 50 μg L−1 Pb(II) by comparing the striping peak current between the 1st and 15th day. As illustrated in Fig. 6, the DPASV responses of Cd(II) and Pb(II) obtained at different storage time only slightly change, implying the good long-time stability of the proposed sensor.
 | ||
| Fig. 6 Interferences caused by storage time on the DPASV responses of 50 μg L−1 Cd(II) (red) and 50 μg L−1 Pb(II) (blue) under the optimized conditions. | ||
3.7 Analysis of real sample
To evaluate its accuracy in practical applications, the modified electrode was utilized to detect trace amounts of target metal ions in real water sample by standard addition method. The environmental samples of tap water were collected from municipal water supply system of Urumqi. The real samples also tested by inductively coupled plasma mass spectrometry (ICP-MS) as a reference method. As listed in Table 3, the amount of Cd(II) and Pb(II) recovered with the help of the sensor are in good agreement with reference method.| Added (μg L−1) | Found (μg L−1) | Recovery (%) | Found by ICP-MS (μg L−1) | |||
|---|---|---|---|---|---|---|
| Cd | Pb | Cd | Pb | Cd | Pb | |
| 0 | — | — | — | — | — | — |
| 6 | 6.5 ± 0.1 | 6.5 ± 0.1 | 108 | 108 | 6.0 | 5.8 |
| 8 | 7.7 ± 0.1 | 8.6 ± 0.1 | 96 | 108 | 7.9 | 8.1 |
| 30 | 28.6 ± 0.4 | 30.3 ± 0.4 | 95 | 101 | 29.7 | 29.9 |
4 Conclusions
A large specific surface area and good electrical conductivity NMC was obtained via direct carbonation of a single small-molecule protic salt [pPDA][2HSO4]. The NMC was used to make the sensitive sensor electrode for Cd(II) and Pb(II) detection by DPASV. In particular, the developed sensor allows the analysis of Cd(II) and Pb(II) at a more lower detection limit (Cd(II), 0.3 μg L−1; Pb(II), 0.4 μg L−1). In addition, we further show that the sensor successfully passes stringent interference tests for common interfering agents, with the exception for copper, and also displays good long term stability and reproducibility. Finally we show the sensor's applicability for real-life testing of tap water section should come in this section at the end of the article, before the acknowledgements.Acknowledgements
This work was supported by National Natural Science Foundation of China (21677171 and 21505154), Xinjiang International Science and Technology Cooperation Project (20166010), West Light Foundation of The Chinese Academy of Sciences (2016-YJRC-1), 1000-Talent Program (Recruitment Program of Global Expert, In Chinese: Qian-Ren-Ji-Hua), and Director Foundation of XTIPC, CAS (Grant No. 2015RC012). TW acknowledge support from the Artificial Leaf Project Umeå (K&A Wallenberg foundation) and by the Swedish Research Council (2013-5252).Notes and references
- M. Lv, X. Wang, J. Li, X. Yang, C. a. Zhang, J. Yang and H. Hu, Electrochim. Acta, 2013, 108, 412–420 CrossRef CAS.
- P. Hajeb, J. J. Sloth, S. Shakibazadeh, N. A. Mahyudin and L. Afsah Hejri, Compr. Rev. Food Sci. Food Saf., 2014, 13, 457–472 CrossRef CAS.
- A. H. El-Sheikh, Y. S. Al-Degs, J. A. Sweileh and A. J. Said, Talanta, 2013, 116, 482–487 CrossRef CAS PubMed.
- D. V. Biller and K. W. Bruland, Mar. Chem., 2012, 130–131, 12–20 CrossRef CAS.
- P. Zhang, G. Xu, J. Lv, J. Cui, Z. Zheng and Y. Wu, J. Electroanal. Chem., 2012, 685, 91–96 CrossRef CAS.
- B. Wang, B. Luo, M. Liang, A. Wang, J. Wang, Y. Fang, Y. Chang and L. Zhi, Nanoscale, 2011, 3, 5059–5066 RSC.
- C. Liang, Z. Li and S. Dai, Angew. Chem., 2008, 47, 3696–3717 CrossRef CAS PubMed.
- P. Kanchana, N. Sudhan, S. Anandhakumar, J. Mathiyarasu, P. Manisankar and C. Sekar, RSC Adv., 2015, 5, 68587–68594 RSC.
- R. Silva, D. Voiry, M. Chhowalla and T. Asefa, J. Am. Chem. Soc., 2013, 135, 7823–7826 CrossRef CAS PubMed.
- H. Zhu, J. Yin, X. Wang, H. Wang and X. Yang, Adv. Funct. Mater., 2013, 23, 1305–1312 CrossRef CAS.
- G. Hu, F. Nitze, E. Gracia-Espino, J. Ma, H. Barzegar, T. Sharifi, X. Jia, A. Shchukareev, L. Lu, C. Ma, G. Yang and T. Wagberg, Nat. Commun., 2014, 5, 5253–5261 CrossRef CAS PubMed.
- L. Zhang, K. Cai, F. Zhang and Q. Yue, Chem. Res. Chin. Univ., 2015, 31, 130–137 CrossRef CAS.
- L. Xiao, S. Zhou, G. Hu, H. Xu, Y. Wang and Q. Yuan, RSC Adv., 2015, 5, 77159–77167 RSC.
- S. van Dommele, K. P. de Jong and J. H. Bitter, Chem. Commun., 2006, 4859–4861 RSC.
- Y. Shao, J. Sui, G. Yin and Y. Gao, Appl. Catal., B, 2008, 79, 89–99 CrossRef CAS.
- L. Cui, J. Wu and H. Ju, ACS Appl. Mater. Interfaces, 2014, 6, 16210–16216 CAS.
- S. Tanaka, N. Nishiyama, Y. Egashira and K. Ueyama, Chem. Commun., 2005, 16, 2125–2127 RSC.
- T. Y. Ma, L. Liu and Z. Y. Yuan, Chem. Soc. Rev., 2013, 42, 3977–4003 RSC.
- S. Zhang, T. Mandai, K. Ueno, K. Dokko and M. Watanabe, Nano Energy, 2015, 13, 376–386 CrossRef CAS.
- H. Shen, D. Qin, Y. Li, S. Li, C. Yang, Q. Yuan, T. Wagberg and G. Hu, Electroanalysis, 2016, 12, 2939–2946 CrossRef.
- Y. Zheng, Y. Jiao, M. Jaroniec, Y. Jin and S. Z. Qiao, Small, 2012, 8, 3550–3566 CrossRef CAS PubMed.
- L. Xiao, H. Xu, S. Zhou, T. Song, H. Wang, S. Li, W. Gan and Q. Yuan, Electrochim. Acta, 2014, 143, 143–151 CrossRef CAS.
- M. Armandi, B. Bonelli, E. I. Karaindrou, C. O. Areán and E. Garrone, Catal. Today, 2008, 138, 244–248 CrossRef CAS.
- W. Zhu, W. Liu, T. Li, X. Yue, T. Liu, W. Zhang, S. Yu, D. Zhang and J. Wang, Electrochim. Acta, 2014, 146, 419–428 CrossRef CAS.
- T. Sharifi, F. Nitze, H. R. Barzegar, C.-W. Tai, M. Mazurkiewicz, A. Malolepszy, L. Stobinski and T. Wågberg, Carbon, 2012, 50, 3535–3541 CrossRef CAS.
- J. P. Paraknowitsch, A. Thomas and M. Antonietti, J. Mater. Chem., 2010, 20, 6746 RSC.
- S. Deng, G. Jian, J. Lei, Z. Hu and H. Ju, Biosens. Bioelectron., 2009, 25, 373–377 CrossRef CAS PubMed.
- S. Lee, S. Bong, J. Ha, M. Kwak, S.-K. Park and Y. Piao, Sens. Actuators, B, 2015, 215, 62–69 CrossRef CAS.
- A. Afkhami, H. Bagheri, H. Khoshsafar, M. Saber-Tehrani, M. Tabatabaee and A. Shirzadmehr, Anal. Chim. Acta, 2012, 746, 98–106 CrossRef CAS PubMed.
- J. Wang, Ü. A. Kirgöz and J. Lu, Electrochem. Commun., 2001, 3, 703–706 CrossRef CAS.
- Y. Wang, Y. Wu, J. Xie and X. Hu, Sens. Actuators, B, 2013, 177, 1161–1166 CrossRef CAS.
- World Health Organization, Guidelines for Drinking-water Quality, WHO chronicle, 4th edn, 2011, pp. 104–108 Search PubMed.
- M. A. Chamjangali, H. Kouhestani, F. Masdarolomoor and H. Daneshinejad, Sens. Actuators, B, 2015, 216, 384–393 CrossRef CAS.
- C. M. Willemse, K. Tlhomelang, N. Jahed, P. G. Baker and E. I. Iwuoha, Sensors, 2011, 11, 3970–3987 CrossRef CAS PubMed.
- K. Keawkim, S. Chuanuwatanakul, O. Chailapakul and S. Motomizu, Food Control, 2013, 31, 14–21 CrossRef CAS.
- L. Fu, X. Li, J. Yu and J. Ye, Electroanalysis, 2013, 25, 567–572 CrossRef CAS.
- Z. Li, L. Chen, F. He, L. Bu, X. Qin, Q. Xie, S. Yao, X. Tu, X. Luo and S. Luo, Talanta, 2014, 122, 285–292 CrossRef CAS PubMed.
- Z. Su, Y. Liu, Y. Zhang, Q. Xie, L. Chen, Y. Huang, Y. Fu, Y. Meng, X. Li, M. Ma and S. Yao, Analyst, 2013, 138, 1180–1186 RSC.
- S. Cerovac, V. Guzsvany, Z. Konya, A. M. Ashrafi, I. Svancara, S. Roncevic, A. Kukovecz, B. Dalmacija and K. Vytras, Talanta, 2015, 134, 640–649 CrossRef CAS PubMed.
- X. He, Z. Su, Q. Xie, C. Chen, Y. Fu, L. Chen, Y. Liu, M. Ma, L. Deng, D. Qin, Y. Luo and S. Yao, Microchim. Acta, 2011, 173, 95–102 CrossRef CAS.
- Z. Wang, H. Wang, Z. Zhang, X. Yang and G. Liu, Electrochim. Acta, 2014, 120, 140–146 CrossRef CAS.
Footnote |
| † This authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2017 |