
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA real-time decoding sequencing technology—new possibility for high throughput sequencing†
Dan Pu ab and
Pengfeng Xiao*b
ab and
Pengfeng Xiao*b
aSchool of Bioinformatics, Chongqing University of Posts and Telecommunications, Chongqing, 400065, China
bState Key Laboratory of Bioelectronics, School of Biological Science and Medical Engineering, Southeast University, Nanjing, 210096, China. E-mail: xiaopf@seu.edu.cn; Fax: +86-25-83793310; Tel: +86-25-83793310
First published on 16th August 2017
Abstract
A real-time decoding sequencing developed by our group offers long read length, high sequencing accuracy and high compatibility, making it have great potential in high throughput sequencing (HTS) platforms. Here, we first discuss its potential advantages in HTS in terms of read length, sequencing accuracy, and turnaround time. We then discuss its disadvantages including homopolymers and chain decoding mistakes. How to handle these two major disadvantages is also discussed with respect to resequencing and de novo sequencing. We also provide the characteristics of this technology for HTS in terms of error-correcting and discriminating SNP/deletion/insertion. Finally, the existing sequencing platforms with which this technology is compatible are discussed. This technology is not only compatible with the first-generation sequencing platform, but also the second-generation and even the third-generation sequencing platforms. It will further improve the advantages of existing sequencing platforms (read length of PGM and 454 system) and compensate some disadvantages of other next generation sequencing (NGS) platforms (sequencing accuracy of PGM sequencer). We fully hope it will provide a new promising technology for researchers and customers to extend applications of the current and upcoming platforms in almost every area in life and biomedical sciences.
Introduction
Over the past ten years, the cost of human genome sequencing has dramatically reduced from $100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 to $1000 because of extraordinary advancements in DNA sequencing technologies.1,2 The road to this milestone involved many sequencing technologies and these sequencing technologies have profoundly altered our understanding of biology, human diversity, and disease. Emerging sequencing technologies can be grouped into first-, second-, and third-generation sequencing. The automated Sanger method is considered as a ‘first-generation’ technology.3 Although the first human genome sequence was interrogated by this technology, limitations of low throughput and high cost of this technology showed a need for new and improved technologies to sequence large numbers of human genomes in parallel at lower cost.
000000 to $1000 because of extraordinary advancements in DNA sequencing technologies.1,2 The road to this milestone involved many sequencing technologies and these sequencing technologies have profoundly altered our understanding of biology, human diversity, and disease. Emerging sequencing technologies can be grouped into first-, second-, and third-generation sequencing. The automated Sanger method is considered as a ‘first-generation’ technology.3 Although the first human genome sequence was interrogated by this technology, limitations of low throughput and high cost of this technology showed a need for new and improved technologies to sequence large numbers of human genomes in parallel at lower cost.
To ameliorate these limitations, the second-generation sequencing (SGS) was developed. It might be categorized as neither sequencing-by-synthesis (SBS) based nor sequencing-by-ligation (SBL) based.3,4 SBS-based strategies, which are achieved by using DNA polymerase to extend many DNA strands in parallel, may be real-time or synchronous-controlled.5 The real-time SBS strategy identifies newly incorporated nucleotide ‘on the fly’ without interrupting the synthesis process (e.g., Roche/454,6 Ion PGM7). Synchronous-controlled approaches are achieved by using nucleotide substrates that are reversibly blocked or by simply adding only one kind of nucleotide at a time. These strategies include Illumina8 and Fluorogenic DNA sequencing in PDMS microreactors.9 SBL-based strategies use ligase enzymes to extend DNA strands instead of DNA polymerases. These strategies contain AB SOLiD10 and Polonator.11 Currently, due to longer read length and higher throughput of SBS-based platforms, the sequencing market is dominated by SBS-based platforms (e.g., Illumina HiSeq 2000 (Illumina), 454 GLS FLEX (Roche), and Ion PGM™ sequencer (Ion Torrent)) instead of SBL-based platforms (e.g., SOLiD 2.0 (Applied Biosystem)). One of the hallmark features of SGS technologies is their massive throughput at a modest cost, with hundreds of gigabases of sequencing now possible in a single run for several thousand dollars.3 However, the complexity associated with DNA library preparation and the biases introduced by polymerase chain reaction (PCR) amplification may limit broad applications to human genome resequencing.
Single-molecule sequencing (SMS) approaches, which are referred as the third-generation sequencing (TGS), were then proposed to ameliorate these limitations.12 These technologies can roughly be binned into three different categories: (i) SBS technologies in which single molecules of DNA polymerase are observed as they synthesize a single molecule of DNA (SMRT sequencing (Pacific Biosciences) and HeliScope (Helicos BioSciences)); (ii) nanopore-sequencing technologies (MinION (Oxford Nanopore Technologies)); and (iii) direct imaging of individual DNA molecules using advanced microscopy techniques (sequencing using fluorescence resonance energy transfer (FRET) (VisiGen Biotechnologies)).13 These technologies have advantages over current SGS in terms of throughput, turnaround time, and read length. However, while cost and time have been greatly reduced, the error rates of TGS reach up to 4–11%13–15 and are relatively higher than SGS. Furthermore, signal acquisition is still a challenge in TGS.
In addition to the improvements of current technologies, other promising technologies which can offer higher throughput, longer read length, and lower error rate are eager to be proposed. Not long ago, our group proposed a real-time SBS-based decoding sequencing technology in which a template is determined without directly measuring base sequence, but by decoding two sets of encodings obtained from two parallel sequencing runs. Although both this decoding sequencing and the SOLiD system are achieved by decoding the codes (or encodings in the decoding sequencing) into base sequence instead of by directly obtaining base sequence, this decoding sequencing is different from the SOLiD system in terms of sequencing chemistries and decoding procedures. As for the SOLiD system, it is based on sequencing-by-ligation (SBL), and 8-mer probe which contains ligation site (the first base), cleavage site (the fifth base), and 4 different fluorescent dyes (linked to the last base) which are applied to interrogate the libraries. The SOLiD cycle of hybridization and ligation, imaging, and probe cleavage is repeated ten times to yield ten color calls spaced in five-base intervals. The extended primer is then stripped from the immobilized templates and another ligation round is performed with a ‘n − 1’ primer. After 5 rounds of sequencing, color codes from the five ligation rounds are aligned to a reference genome to decode the inquired sequence.3 Decoding is achieved by prepending the leading base to result in k-mer color codes from which the base sequence can be reconstructed from the first to the last.
However, decoding sequencing is based on real-time SBS instead of SBL. In addition, natural nucleotides instead of fluorescent dyes labeled probes are used; thus, there is no need to image and cleave probes, requiring many fewer steps in each cycle. This technology applies any two of the three sets of dual mononucleotide additions AG/CT, AC/GT, and AT/CG to interrogate a fragment in two parallel runs, and two sets of encodings, which contain information about the possible types and numbers of incorporated base(s) in each cycle, can be acquired. The template sequence can be reconstructed by decoding the two sets of encodings using a decoding scheme in which all the encodings and their degraded encodings from the two sets of encodings are compared with each other from the first to the last. The identical nucleotide between the two compared encodings is right the incorporated nucleotide. This strategy can increase read length and reduce error rate.16 Until now, this technology was successfully applied to simultaneously genotype several SNPs in a single run.17 Additionally, when it is applied to quantitatively analyze SNPs, it allows for the detection of alleles with frequencies as low as 3%, which is more sensitive than the ∼5% to ∼7% level detected by conventional pyrosequencing.18
Here, we first discuss the potential advantages of this technology for HTS in terms of read length, sequencing time, and accuracy. We then discuss two major disadvantages containing chain decoding mistakes and homopolymers. How to handle these disadvantages is also proposed in terms of resequencing and de novo sequencing in this technology. Finally, we discuss the existing platforms with which this technology is compatible. We fully hope it will provide a new promising technology for researchers and customers.
Results
Mechanism of the real-time decoding sequencing technology
In general, nucleotides A, G, C, and T, can form sixteen combinations of di-bases. These include AA, CC, GG, TT, AC, CA, AG, GA, AT, TA, CG, GC, CT, TC, TG, and GT, six of which are different di-base combinations (AG, CT, AC, GT, AT, and CG). These six combinations can form three sets of dual mononucleotide additions, AG/CT, AC/GT, and AT/CG. This methodology uses information about the type(s) and number of incorporated base(s) to encode for the sequencing signals in each cycle (a reaction is defined as a cycle) at a time. A template can be interrogated in three parallel runs by using the three sets of dual mononucleotide additions AG/CT, AC/GT, and AT/CG, and three sets of encodings which contain the information about the possible type(s) and number of incorporated base(s) in each cycle can be acquired. However, given any two sets of encodings, nucleic acid sequence is allowed to be sequentially reconstructed.16 Therefore, this technology is able to use any two of the three sets of dual mononucleotide additions AG/CT, AC/GT, and AT/CG to interrogate a template in two parallel runs. Fig. 1A shows a sequence that is interrogated by AG/CT and AC/GT in two sequencing runs, respectively. Take an example to demonstrate how dual mononucleotide addition AG/CT works. When dual mononucleotide addition AG/CT is used, di-base AG (a mixture of dATPaS (α-thio-dATP) and dGTP) is first added into the reaction in the 1st cycle, and then a mixture of dCTP and dTTP (di-base CT) is added in the 2nd cycle. The ratio between the two mixed nucleotides is 1:1. In the 3rd and the 4th cycles, a mixture of dATPaS and dGTP (di-base AG) and a mixture of dCTP and dTTP (di-base CT) are added. That is to say, sequencing procedures are carried out by sequentially adding a mixture of dATPaS and dGTP (di-base AG) and a mixture of dCTP and dTTP (di-base CT). After two sequencing runs, the strategy decodes the two sets of encodings using a decoding scheme which is achieved first by degrading all the encodings whose number of the incorporated bases is more than one, and then by comparing the two sets of encodings from the first to the last. The identical nucleotide between the two compared encodings is right the incorporated nucleotide. The decoding procedures are shown in Fig. 1B.
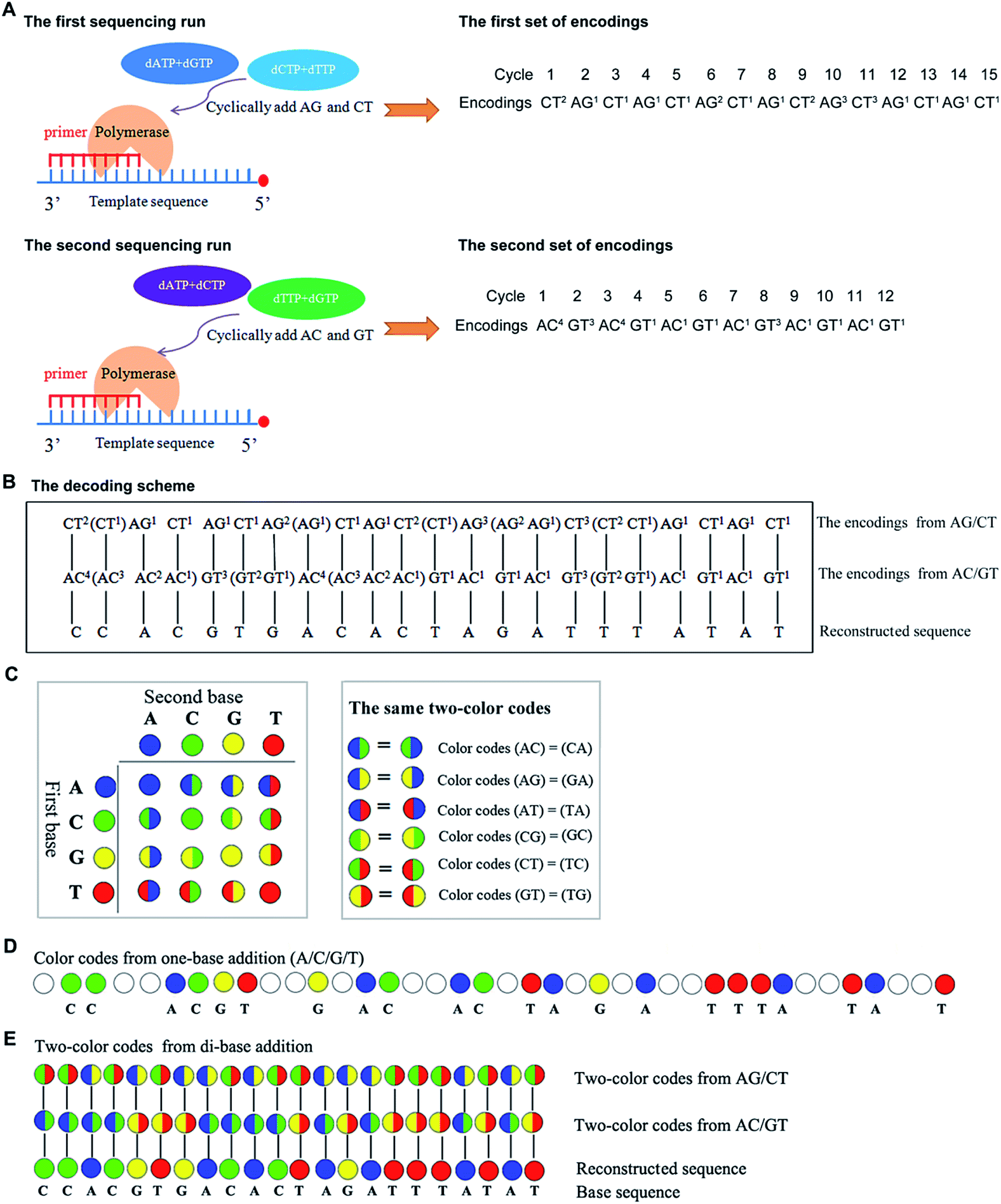 | ||
Fig. 1 The scheme of decoding sequencing system in terms of encodings and two-color codes. Sequence (5′-GGTGCACTGTGATCTAAATATA-3′) is interrogated. (A) Sequence is interrogated by using AG/CT and AC/GT in two parallel sequencing runs, and two sets of encodings are obtained. (B) The decoding scheme of this strategy. The encodings in the brackets behind each encoding are the possible degraded encodings of each encoding. (C) The color coding scheme of this technology. A two-color matrix is used to encode sequencing signals (left). The identical color-codes are shown in the right box. (D) Color codes from one-base addition. The nucleotide dispensation order is A/C/G/T. (E) Two-color codes from di-base addition and the decoding procedures. The color codes, blue, green, yellow, and red represent bases A, C, G and T, respectively. The white code represents there is no nucleotide to be incorporated in the cycle. The two-color code  represents di-base GT while represents di-base GT while  represents di-base AC. The two-color code represents di-base AC. The two-color code  represents di-base CT while represents di-base CT while  represents di-base AG. represents di-base AG. | ||
A two-color code matrix is also used to encode sequencing signals. Two-color code(s) which contain(s) the information about the possible type(s) of incorporated base(s) also can be applied to represent sequencing signals in each cycle.17 Four color codes (blue, green, yellow, and red) and twelve two-color codes are applied to encode for the sixteen possible di-bases (Fig. 1C). The color codes, blue, green, yellow, and red, represent 4 monodibases AA, CC, GG, and TT, respectively. The remaining two-color codes are used to encode for the twelve di-bases. Additionally, the number of two-color code(s) represents the number of incorporated nucleotide(s) in each cycle. The two-color codes should satisfy the following requirements: first, two different di-bases that have the same first (or the same second) base get different two-color codes. For example, two-color codes (AC) ≠ (AT); two-color codes (AC) ≠ (TC). Second, monodibases get different color codes. For example, two-color codes (AA) ≠ (CC). Third, a di-base and its reverse get the same color code. For example, two-color codes (AC) = (CA). All the same two-color codes are shown in Fig. 1C. When a template is sequenced by both one-base addition and di-base addition, the corresponding color codes are shown in Fig. 1D and E. The system can reconstruct the base sequence based on a scheme that the same color code between the two compared two-color codes is right the incorporated nucleotide (see Fig. 1E). For example, the first two-color code which is half green and half red in the 1st run is compared with the first one which is half blue and half green in the 2nd run, and the same color between the two compared codes is green. Thus, the incorporated nucleotide is base C in this cycle. Continuing in this way, all bases including the last one can be decoded.
Advantages of this technology for HTS
In one-base addition technology, general average read length is about 0.5 bp in a sequencing reaction when homopolymers are taken into account.16 When the first base is measured, three sequencing reactions are acquired in order to ensure that at least one base is measured in each template. However, in this decoding sequencing, general average read length is about 1.5 bp in a sequencing reaction when homopolymers are considered.16 Since two nucleotides are simultaneously added into each reaction, two sequencing cycles in a single sequencing run can ensure that at least one base is determined in each template. Therefore, if we assume that the reaction efficiency and time are identical in both the two technologies, the shortest read length of the decoding technology would be three times longer than that of one-base addition technology. This can be corroborated in our previously published work.16 In Fig. S1,† when a sequence is interrogated by the decoding sequencing, the minimum read length per cycle is 1.583 bp (57/36 = 1.583) while a maximum read length per cycle is 2.111 bp (57/27 = 2.111). Both the maximum and the minimum read lengths are more than 1.5 bp per cycle. In contrast, in the one base addition technology, average read length is 0.474 bp (27/57 = 0.474) per cycle. When the given cycles are greater (e.g., 500 cycles) in both the technologies, read lengths measured in the decoding sequencing (750 bp) are greatly longer than that in the one base addition technology (250 bp). Thus, the decoding sequencing has significantly increased the potential read length.
First, we assume that the inquired sequence length is a constant N. Suppose that four nucleotides A, G, C, and T appear in a sequencing reaction at random; we compare the expectation value of the inquired sequencing cycles in one-base addition technology and in decoding sequencing, respectively. In the one-base addition technology, nucleotides A, G, C, and T are cyclically added. When the first base is measured and the downstream bases are independent, the test probability of the second base is 1/3 (without considering homopolymers). Thus, as for one-base addition technology, the expectation value of the required cycles is E1 = 1 + 3(N − 1). Here, N is segment length to be tested. For the decoding sequencing, two nucleotides are simultaneously added. When the first base is interrogated and the downstream bases are independent, the test probability of the second base is 1. Thus, the expectation value of the required cycles should be E2 = 1 + (N − 1) in a single run. Since the decoding sequencing technology contains two sequencing runs, the expectation value of the required cycles is E3 = (1 + (N − 1)) × 2 in the two sequencing runs. When the fragment length is N, the difference of required cycles between the two technologies is E1 − E3 = N − 2. Thus, the longer inquired sequence is, the fewer number of cycles the decoding sequencing needs. Therefore, time required for the decoding sequencing is much less than that for the one-base addition technology when a much longer sequence is interrogated. Moreover, the two sequencing runs in the decoding sequencing can be parallelly carried out; thus time required for two sequencing runs is the same as that for a single run. When the fragment length is constant, the decoding sequencing needs fewer cycles (and thus less operation time), compared to the one-base addition technology.
Second, we assume the number of cycles is constant in a single run. When the decoding sequencing is carried in two unparallel runs, the operation time which contains degeneration and primer extension is two times that of the one-base addition technology. However, the time-cost is undoubtedly worthwhile when the truth that read length per cycle is longer than that of the one-base addition technology is considered.
Challenges of the decoding sequencing technology for HTS
Although the technology may provide longer read length and more accurate sequencing data to both researchers and clinicians, two key limitations remain. First, chain decoding mistakes. After sequencing with di-base addition, signals can be encoded into two-color codes (or encodings) according to a two-color code matrix. These original two-color code data can be decoded by comparing the two sets of two-color codes to reconstruct the base sequence from the first to the last. Since the decoding procedures are achieved by comparing the two sets of two-color codes from the first to the last, the front two-color code will directly influence the decoding of its following bases. That is to say, a wrong two-color code will cause a chain of decoding mistakes. Second, another major limitation of this technology relates to homopolymers which include two types. One is consecutive instances of the same base, such as AAA or GGG. The other is consecutive instances of di-bases, such as ACACAC or ACCCAACAA. The two limitations will likely limit the ability of the technology to be applied on NGS platforms. Here, we provide their corresponding resolutions for application on NGS platforms.| The first set of encodings | The second set of encodings | Decoded bases | |
|---|---|---|---|
| a Underlined segments and the bases in the boxes indicate differences between the original data and the reference. | |||
| Reference | 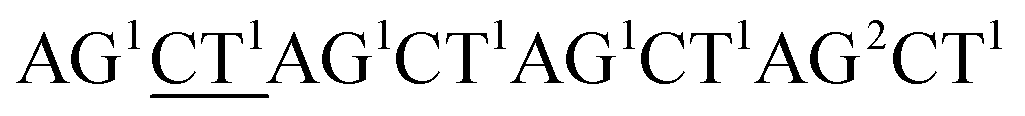 |
AC2GT1AC1GT1AC2GT1AC1 | 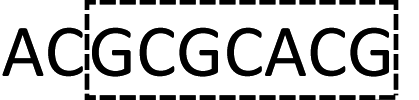 |
| Original data | 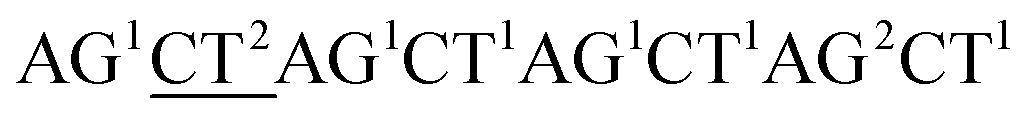 |
AC2GT1AC1GT1AC2GT1AC1 |  |
As with re-sequencing, data analysis depends on reference sequences for follow-up data analysis since reference sequences exist. In the decoding sequencing, each template is interrogated in two sequencing runs, and two sets of two-color codes are obtained. These two-color codes are compared with the two-color codes of the reference instead of the reference base sequence. Therefore, the procedures are as follows: (i) the reference base sequences are translated into two-color code sequences under different dual mononucleotide additions (AG/CT, AC/GT, and AT/CG) by software. (ii) The two-color codes of reference sequences are compared with those of the original data to get information for mapping with a newly developed mapping algorithm. Two situations exist when the two-color codes of original data do not completely align with the two-color codes of references. First, one set of two-color code sequences perfectly match to the corresponding two-color code sequence of reference, but the other has one mismatched two-color code. The mismatched two-color code is referred to as sequencing errors. Second, both the two sets of two-color code sequences have several mismatched two-color codes and a certain number of (such as 18 consecutive two-color codes) matched consecutive two-color code segments. In this case, mismatched fragments are removed, and a certain number of matched consecutive code segments are reserved and used for assembly.
As de novo sequencing, there is no reference available, and thus the original sequencing data cannot align to the reference sequence. In general, a template can be interrogated by any two sets of dual mononucleotide additions from AG/CT, AC/GT, or AT/CG, and the sequence is able to be reconstructed by any two sets of two-color codes (or encodings). However, chain decoding mistakes may occur in any sets of two-color codes. In such case, a template should be interrogated by three sets of di-base combinations AG/CT, AC/GT, and AT/CG in three sequencing runs, and three sets of two-color codes (or encodings) will be obtained. The three sets of two-color codes (or encodings) can provide more information to decode the original sequence, reducing the chain decoding errors.
When raw data from high-throughput DNA sequencing are used to assemble de novo genome sequence, a corresponding number of coverage is required. Since a template is interrogated by at least two parallel sequencing runs in this decoding technology, each base is independently detected at least twice. This may dramatically reduce or even eliminate the chain decoding errors, just as the error correction in SOLiD system.26,27
| Title | Sequence (5′–3′) |
|---|---|
| a Templates T1–T3 represent the templates used. SP represents sequencing primer. The underlined segments are the hybridization regions with the sequencing primer SP. The segments in the bracket are SNP/deletion/insertion. | |
| T1 | 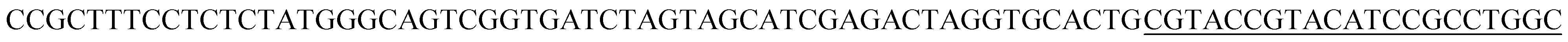 |
| T2 | 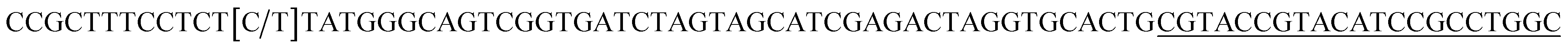 |
| T3 |  |
| T4 |  |
| SP | GCCAGGCGGATGTACGGTACG |
For re-sequencing, a reference sequence is available for assembly. The reference base sequence is first converted into two-color code sequence under each di-base addition (AG/CT, AC/GT, and AT/CG) by software. Given a set of two-color codes from any di-base addition, the two-color codes of references are compared with those of original sequencing reads. Between the two sets of two-color codes obtained in two sequencing runs, when one has an ambiguous number of homopolymers in a given fragment, the other must be clear encodings in this fragment. Thus, when the two sets of two-color codes are compared with the two sets of two-color codes of references, one will perfectly match, but the other cannot match only in a given fragment. The solution is to use ambiguous alignment to align the two-color codes of original sequences to those of references by previously designing a dynamic range of base number. Take template T1 as an example, when the template is sequenced by AC/GT and AG/CT, respectively, two sets of two-color codes (or encodings), S1 and S2, are obtained (Fig. 2A). S2 has an ambiguous number of homopolymers in a given fragment while S1 has clear encodings in this fragment. By using ‘ignore’ strategy, the homopolymer segments are discarded and the remaining parts are applied to decode the base sequence.
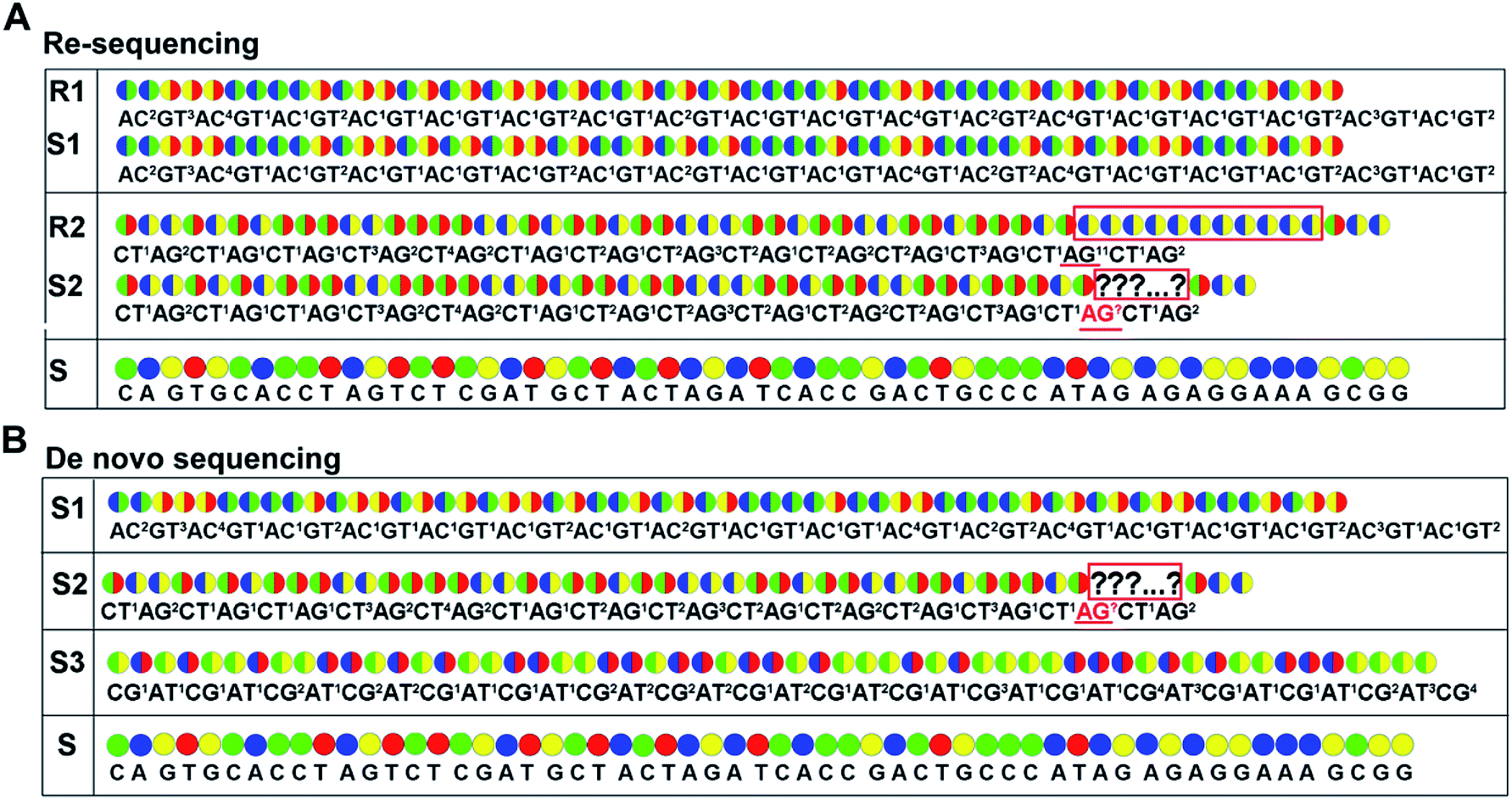 | ||
Fig. 2 Encodings and two-color codes from interrogating 57 bp of template T1 by using the decoding sequencing technology. Sequences S1–S3 indicate two-color codes (upper) and encodings (lower) obtained from AC/GT, AT/CG, and AG/CT while sequences R1–R3 indicate two-color codes (upper) and encodings (lower) obtained from reference by using AC/GT, AT/CG, and AG/CT. The two-color codes in the boxes are two-color codes with unknown length. Underlined segments are encodings with unknown length in homopolymer. The color codes, blue, green, yellow, and red, represent bases A, C, G, and T, respectively. The two-color code  represents di-base GT while represents di-base GT while  represents di-base AC. The two-color code represents di-base AC. The two-color code  represents the di-base CT while represents the di-base CT while  represents di-base AG. The two-color code represents di-base AG. The two-color code  represents di-base CG while represents di-base CG while  represents di-base AT. Two-color codes in the red boxes indicate differences between the original data and the reference. represents di-base AT. Two-color codes in the red boxes indicate differences between the original data and the reference. | ||
As for de novo sequencing, reference sequence is not available. Thus, the original encodings cannot align with the reference encoding sequences, making the unambiguous alignment of sequencing reads impossible. In such cases, the template needs to be sequenced using three different di-base additions (AG/CT, AC/GT, and AT/CG) in three sequencing runs, and three sets of encodings will be obtained. Among the three sets of encodings, if homopolymers exist in one set of encodings, the other two sets of encodings in this region must be without homopolymers. Therefore, correct sequence could be decoded by using the two sets of encodings without homopolymers. In Fig. 2B, three sets of encodings (S1, S2, and S3) are obtained. S2 contains homopolymers, but no homopolymer exists in S1 and S3. Thus, the correct sequence could be decoded by using S1 and S3.
Characteristic of the decoding sequencing technology for HTS
Except for an increase of read length, the decoding sequencing has potential in-error-correcting. That is because, for each fragment, two sets of encodings will be obtained in this technology. These encodings provide much more information for alignment and thus allow for error-correcting and discriminating of SNP/deletion/insertion just like the SOLiD system.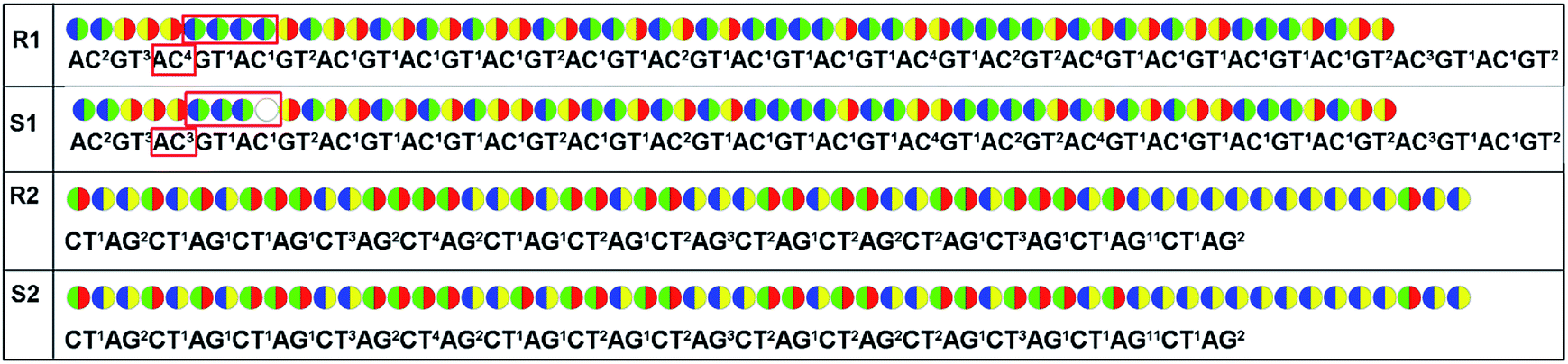 | ||
| Fig. 3 Sequencing error occurs in the sequencing data. Sequences S1 and S2 indicate two-color codes (upper) and encodings (lower) obtained from AC/GT and AG/CT when template T1 is interrogated. Sequences R1 and R2 indicate two-color codes (upper) and encodings (lower) obtained from reference by using AC/GT and AG/CT. Two-color codes in the red boxes indicate differences between the original data and the reference. | ||
 | ||
| Fig. 4 SNP, deletion or insertion occurs in original sequencing data. Sequences S1 and S2 indicate two-color codes (upper) and encodings (lower) obtained from AC/GT and AG/CT when templates T2–T4 are interrogated. Sequences R1 and R2 indicate two-color codes (upper) and encodings (lower) obtained from reference by using AC/GT and AG/CT. (A) A SNP, C/T is contained in template T2. (B) Deletion is contained in template T3. (C) Insertion is contained in template T4. Underlined segments and the two-color codes in the red boxes indicate differences between the original data and the reference. | ||
Possible applications to current sequencing platforms
So far, a number of SBS-based sequencing platforms have emerged in the market. These include Max-Seq (Qiagen), 454 GS FLX/FLX Junior (Roche), Ion PGM/proton (Ion torrent), HeliScope (Helicos biosciences), PacBio RS II (Pacific biosciences), and so on. Based on the principle of the decoding sequencing technology, they will be compatible with existing equipment, such as conventional pyrosequencing, 454 systems, Fluorogenic DNA sequencing in PDMS microreactors, Ion PGM/proton, and HeliScope.Conventional pyrosequencing is a real-time SBS technology which is based on adding a limiting amount of dNTP bases one at a time to control DNA synthesis and it yields detectable light by a cascade of enzymatic reactions.30 Since this technique is SBS-based and uses natural nucleotides during synthesis, it is compatible with decoding sequencing technology. Based on the conventional pyrosequencing platform (PSQ 96MA system), we validated this decoding sequencing technology in our previous works (we called it pyrosequencing with di-base addition).16 We found that it was perfectly compatible with a conventional pyrosequencing platform and that generated visible light was proportional to the number of incorporated nucleotides when the number of identical nucleotides was less than 7 bp.16 Moreover, the read length of pyrosequencing with di-base was nearly 1.5 times that of conventional pyrosequencing, therefore further improving its read length. Increasing read length will reduce the cost of DNA analysis and extend its applications. For example, several SNPs were simultaneously examined in a single run which were unable to be examined by conventional pyrosequencing.17
Roche/454 system uses emulsion PCR31 and pyrosequencing technology.29,32 Therefore, the decoding sequencing is compatible with Roche 454. The key advantage of the Roche/454 system is its longer sequence reads. If it is combined with the di-base addition, the read length will be further improved and thus reduce its cost. It will be more suited for de novo sequencing of new genome. However, similar to conventional pyrosequencing, a major limitation of 454 system relates to homopolymers owing to the lack of a terminating moiety. If the decoding sequencing is used on this platform, the limitation can be remedied by solutions we have proposed.
Ion Personal Genome Machine (PGM) uses semiconductor sequencing technology. When a nucleotide is incorporated into the DNA molecules by the polymerase, a pyrophosphate is released and hydrolyzed to yield a proton. Instead of detecting the light change in pyrosequencing technology, this technology detects the change in pH to recognize whether the nucleotide is added or not. Similar to pyrosequencing technology, the amount of voltage detected is proportional to the number of incorporated nucleotides.7,33 Both the decoding technology and the semiconductor sequencing technology use unlabeled nucleotides and are based on real-time SBS technology. Thus, the decoding sequencing is compatible with PGM. When the dispensation orders of AG/CT, AC/GT, and AT/CG are applied, each time the chip is flooded with one di-base after another, and if it is not the correct nucleotide, no voltage will be found; if there are 2 nucleotides incorporated, then double voltage is detected. Based on the amount of the detected voltage, the encoding(s) in each reaction cycle can be obtained. Since the decoding sequencing has potential of increasing read length, the combination of PGM and the decoding sequencing will further improve the read length, making it much more useful for clinical applications and small labs.
Pyrosequencing and semiconductor sequencing use natural nucleotides and do not need chemical cleavage. However, they use instantaneous light emission or detection of the electrochemical signals which need constant monitoring, and each individual reaction requires a separate detector, thus affecting their throughput. In 2011, Sims et al. developed Fluorogenic DNA sequencing in PDMS microreactors.9 This technology combined the advantages of pyrosequencing and semiconductor sequencing with fluorescent labeled nucleotide monomers (TPLFNs). When a TPLFN is incorporated into the template immobilized in the micro-reactor by a DNA polymerase, a recessive fluorescently labeled pyrophosphate will be released. The pyrophosphate is degraded to yield fluorescent light. Based on the principle of the decoding sequencing, this technology is compatible with the decoding sequencing. It was confirmed that the original accuracy rate of this technology was about 99% and it not only provided the benefits of pyrosequencing, such as fast turnaround and one-color detection, but also had high throughput. When it is combined with this decoding sequencing, accuracy rate and throughput will be further improved.
The HeliScope from Helicos Biosciences is based on true single molecule sequencing technology and relies on the cyclic interrogation of a dense array of sequencing features.34 It applies a highly sensitive fluorescence detection system to directly interrogate single DNA molecules via sequencing by synthesis. Besides, no terminating moiety is present on the labeled nucleotides. Thus, our technology is compatible with this platform. If it is combined with the decoding sequencing, the read length will be further improved and thus reduce the cost of this platform. In addition, sequencing accuracy is the major problem in TGS. A two-pass strategy was developed to improve raw sequencing accuracy.35 However, if it is combined with the decoding sequencing, at least two sequencing runs are carried out, and the sequencing accuracy will be improved without performing two-pass strategy.
Discussion
Though truly novel core techniques emerge from time to time, it is clear that most new sequencing applications have resulted from efforts to combine the building blocks of existing designs and protocols in different ways.36 In 2005, ABI SOLiD sequencing was developed, which did not directly measure the base sequence, but measured DNA bases in pairs and in an encoded form (two-base encoding). In order to compare such data to a reference sequence, the encodings must be decoded into base sequence.26 Owing to a two-base decoding system, SOLiD could reach a high accuracy of 99.85% after filtering.29 However, read length is limited for fluorescently-labeled nucleotides and for large numbers of steps to read one base pair. By combining real-time SBS technology with another kind of two-base encoding, novel decoding sequencing emerges. This technology enables not only ability to increase the read length but also to maintain high accuracy. The longer read length is, the easier genome reassembly is.37 With longer reads, researchers are able to sequence through extended repetitive regions and detect mutations, many of which are associated with diseases.Another attractive advantage of decoding sequencing is its compatibility. According to its principle, it is not only compatible with the first-generation sequencing platform, but also the NGS and even the TGS platforms. Due to limited conditions in our laboratory, we only validated its feasibility on a conventional pyrosequencing instead of the NGS or TGS platforms. Decoding technology has the potential of further improving the advantages of sequencing platforms, such as read length of PGM and 454 systems. Moreover, it also enables one to compensate some disadvantages of other NGS platforms. For example, HeliScope Genetic Analysis System will compensate the disadvantage of low sequencing accuracy when it is used. Although this technology has been compatible with only a few platforms so far, it will also be compatible with others as new techniques emerge from time-to-time. We expect it will broadly expend applications of current and upcoming sequencing platforms.
Decoding sequencing has shown advantages in some unique niches. However, two major disadvantages including the chain decoding mistakes and homopolymers exist. Similar to other SBS-based technologies, homopolymers have previously prevented it from being applied more broadly since repetitive DNA sequences are abundant in bacteria and mammal human genomes.38 These regions are difficult to sequence even in relatively small genomes. Here, corresponding solutions have been provided to remedy limitations in terms of re-sequencing and do novo sequencing.
Current HTS platforms provide a huge variety of sequencing applications to many researchers and projects, and they make it possible for research groups to generate longer read lengths very rapidly at substantially lower costs. When decoding sequencing is applied with compatible platforms, its applications may be further expended. We fully hope it will increase the applicability of current and upcoming platforms in almost every arena in life and biomedical sciences.
Conclusions
Overall, SBS-based decoding sequencing, using di-base decoding theory, provides long reads with a high error rate and is highly compatible. This decoding sequencing has great potential in HTS in terms of read length, sequencing accuracy, and turnaround time. Moreover, it is highly compatible with most other SBS-based platforms, and also has the ability to compensate limitations of some SBS-based platforms, thus extending its application to researchers and customers. When it is applied to a compatible SBS-based sequencing platform, it will provide longer average read length than the SBS-based SGS platforms, and the sequencing accuracy will be higher than TGS platforms. Two major disadvantages are homopolymers, including chain decoding mistakes and homopolymers. However, corresponding solutions have been provided to remedy these limitations. Decoding sequencing has the potential of providing a new promising technology compatible with existing sequencing platforms thus enabling researchers and customers to extend application of HTS technologies in the fields of biology and biomedicine.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The work was funded by the National Natural Science Foundation of China (61571114), and the Science and Technology Research Program of Chongqing Municipal Education Commission [KJ1704092, KJ1400410].Notes and references
- I. H. G. S. Consortium, Nature, 2004, 431, 931–945 CrossRef PubMed.
- E. L. van Dijk, H. Auger, Y. Jaszczyszyn and C. Thermes, Trends Genet., 2014, 30, 418–426 CrossRef CAS PubMed.
- M. L. Metzker, Nat. Rev. Genet., 2010, 11, 31–46 CrossRef CAS PubMed.
- M. Kircher and J. Kelso, BioEssays, 2010, 32, 524–536 CrossRef CAS PubMed.
- C. W. Fuller, L. R. Middendorf, S. A. Benner, G. M. Church, T. Harris, X. Huang, S. B. Jovanovich, J. R. Nelson, J. A. Schloss, D. C. Schwartz and D. V. Vezenov, Nat. Biotechnol., 2009, 27, 1013–1023 CrossRef CAS PubMed.
- M. Margulies, M. Egholm, W. E. Altman, S. Attiya, J. S. Bader, L. A. Bemben, J. Berka, M. S. Braverman, Y. J. Chen, Z. Chen, S. B. Dewell, L. Du, J. M. Fierro, X. V. Gomes, B. C. Godwin, W. He, S. Helgesen, C. H. Ho, G. P. Irzyk, S. C. Jando, M. L. Alenquer, T. P. Jarvie, K. B. Jirage, J. B. Kim, J. R. Knight, J. R. Lanza, J. H. Leamon, S. M. Lefkowitz, M. Lei, J. Li, K. L. Lohman, H. Lu, V. B. Makhijani, K. E. McDade, M. P. McKenna, E. W. Myers, E. Nickerson, J. R. Nobile, R. Plant, B. P. Puc, M. T. Ronan, G. T. Roth, G. J. Sarkis, J. F. Simons, J. W. Simpson, M. Srinivasan, K. R. Tartaro, A. Tomasz, K. A. Vogt, G. A. Volkmer, S. H. Wang, Y. Wang, M. P. Weiner, P. Yu, R. F. Begley and J. M. Rothberg, Nature, 2005, 437, 376–380 CAS.
- J. M. Rothberg, W. Hinz, T. M. Rearick, J. Schultz, W. Mileski, M. Davey, J. H. Leamon, K. Johnson, M. J. Milgrew, M. Edwards, J. Hoon, J. F. Simons, D. Marran, J. W. Myers, J. F. Davidson, A. Branting, J. R. Nobile, B. P. Puc, D. Light, T. A. Clark, M. Huber, J. T. Branciforte, I. B. Stoner, S. E. Cawley, M. Lyons, Y. Fu, N. Homer, M. Sedova, X. Miao, B. Reed, J. Sabina, E. Feierstein, M. Schorn, M. Alanjary, E. Dimalanta, D. Dressman, R. Kasinskas, T. Sokolsky, J. A. Fidanza, E. Namsaraev, K. J. McKernan, A. Williams, G. T. Roth and J. Bustillo, Nature, 2011, 475, 348–352 CrossRef CAS PubMed.
- D. R. Bentley, S. Balasubramanian, H. P. Swerdlow, G. P. Smith, J. Milton, C. G. Brown, K. P. Hall, D. J. Evers, C. L. Barnes, H. R. Bignell, J. M. Boutell, J. Bryant, R. J. Carter, R. Keira Cheetham, A. J. Cox, D. J. Ellis, M. R. Flatbush, N. A. Gormley, S. J. Humphray, L. J. Irving, M. S. Karbelashvili, S. M. Kirk, H. Li, X. Liu, K. S. Maisinger, L. J. Murray, B. Obradovic, T. Ost, M. L. Parkinson, M. R. Pratt, I. M. Rasolonjatovo, M. T. Reed, R. Rigatti, C. Rodighiero, M. T. Ross, A. Sabot, S. V. Sankar, A. Scally, G. P. Schroth, M. E. Smith, V. P. Smith, A. Spiridou, P. E. Torrance, S. S. Tzonev, E. H. Vermaas, K. Walter, X. Wu, L. Zhang, M. D. Alam, C. Anastasi, I. C. Aniebo, D. M. Bailey, I. R. Bancarz, S. Banerjee, S. G. Barbour, P. A. Baybayan, V. A. Benoit, K. F. Benson, C. Bevis, P. J. Black, A. Boodhun, J. S. Brennan, J. A. Bridgham, R. C. Brown, A. A. Brown, D. H. Buermann, A. A. Bundu, J. C. Burrows, N. P. Carter, N. Castillo, E. C. M. Chiara, S. Chang, R. Neil Cooley, N. R. Crake, O. O. Dada, K. D. Diakoumakos, B. Dominguez-Fernandez, D. J. Earnshaw, U. C. Egbujor, D. W. Elmore, S. S. Etchin, M. R. Ewan, M. Fedurco, L. J. Fraser, K. V. Fuentes Fajardo, W. Scott Furey, D. George, K. J. Gietzen, C. P. Goddard, G. S. Golda, P. A. Granieri, D. E. Green, D. L. Gustafson, N. F. Hansen, K. Harnish, C. D. Haudenschild, N. I. Heyer, M. M. Hims, J. T. Ho, A. M. Horgan, K. Hoschler, S. Hurwitz, D. V. Ivanov, M. Q. Johnson, T. James, T. A. Huw Jones, G. D. Kang, T. H. Kerelska, A. D. Kersey, I. Khrebtukova, A. P. Kindwall, Z. Kingsbury, P. I. Kokko-Gonzales, A. Kumar, M. A. Laurent, C. T. Lawley, S. E. Lee, X. Lee, A. K. Liao, J. A. Loch, M. Lok, S. Luo, R. M. Mammen, J. W. Martin, P. G. McCauley, P. McNitt, P. Mehta, K. W. Moon, J. W. Mullens, T. Newington, Z. Ning, B. Ling Ng, S. M. Novo, M. J. O'Neill, M. A. Osborne, A. Osnowski, O. Ostadan, L. L. Paraschos, L. Pickering, A. C. Pike, D. Chris Pinkard, D. P. Pliskin, J. Podhasky, V. J. Quijano, C. Raczy, V. H. Rae, S. R. Rawlings, A. Chiva Rodriguez, P. M. Roe, J. Rogers, M. C. Rogert Bacigalupo, N. Romanov, A. Romieu, R. K. Roth, N. J. Rourke, S. T. Ruediger, E. Rusman, R. M. Sanches-Kuiper, M. R. Schenker, J. M. Seoane, R. J. Shaw, M. K. Shiver, S. W. Short, N. L. Sizto, J. P. Sluis, M. A. Smith, J. Ernest Sohna Sohna, E. J. Spence, K. Stevens, N. Sutton, L. Szajkowski, C. L. Tregidgo, G. Turcatti, S. Vandevondele, Y. Verhovsky, S. M. Virk, S. Wakelin, G. C. Walcott, J. Wang, G. J. Worsley, J. Yan, L. Yau, M. Zuerlein, J. C. Mullikin, M. E. Hurles, N. J. McCooke, J. S. West, F. L. Oaks, P. L. Lundberg, D. Klenerman, R. Durbin and A. J. Smith, Nature, 2008, 456, 53–59 CrossRef CAS PubMed.
- P. A. Sims, W. J. Greenleaf, H. Duan and X. S. Xie, Nat. Methods, 2011, 8, 575–580 CrossRef CAS PubMed.
- K. McKernan, A. Blanchard, L. Kotler and G. Costa, US Pat., application 20080003571, 2006.
- J. Shendure, G. J. Porreca, N. B. Reppas, X. Lin, J. P. McCutcheon, A. M. Rosenbaum, M. D. Wang, K. Zhang, R. D. Mitra and G. M. Church, Science, 2005, 309, 1728–1732 CrossRef CAS PubMed.
- T. D. Harris, P. R. Buzby, H. Babcock, E. Beer, J. Bowers, I. Braslavsky, M. Causey, J. Colonell, J. Dimeo, J. W. Efcavitch, E. Giladi, J. Gill, J. Healy, M. Jarosz, D. Lapen, K. Moulton, S. R. Quake, K. Steinmann, E. Thayer, A. Tyurina, R. Ward, H. Weiss and Z. Xie, Science, 2008, 320, 106–109 CrossRef CAS PubMed.
- E. E. Schadt, S. Turner and A. Kasarskis, Hum. Mol. Genet., 2010, 19, R227–R240 CrossRef CAS PubMed.
- J. A. Reuter, D. V. Spacek and M. P. Snyder, Mol. Cell, 2015, 58, 586–597 CrossRef CAS PubMed.
- J. M. Di Bella, Y. Bao, G. B. Gloor, J. P. Burton and G. Reid, J. Microbiol. Methods, 2013, 95, 401–414 CrossRef CAS PubMed.
- D. Pu, Y. Qi, L. Cui, P. Xiao and Z. Lu, Anal. Chim. Acta, 2014, 852, 274–283 CrossRef CAS PubMed.
- D. Pu, C. Mao, L. Cui, Z. Shi and P. Xiao, Anal. Bioanal. Chem., 2016, 408, 3113–3123 CrossRef CAS PubMed.
- D. Pu, R. Pan, W. Liu and P. Xiao, Electrophoresis, 2017, 38, 876–885 CrossRef CAS PubMed.
- J. Bowers, J. Mitchell, E. Beer, P. R. Buzby, M. Causey, J. W. Efcavitch, M. Jarosz, E. Krzymanska-Olejnik, L. Kung, D. Lipson, G. M. Lowman, S. Marappan, P. McInerney, A. Platt, A. Roy, S. M. Siddiqi, K. Steinmann and J. F. Thompson, Nat. Methods, 2009, 6, 593–595 CrossRef CAS PubMed.
- J. Korlach, P. J. Marks, R. L. Cicero, J. J. Gray, D. L. Murphy, D. B. Roitman, T. T. Pham, G. A. Otto, M. Foquet and S. W. Turner, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1176–1181 CrossRef CAS PubMed.
- N. Whiteford, N. Haslam, G. Weber, A. Prugel-Bennett, J. W. Essex, P. L. Roach, M. Bradley and C. Neylon, Nucleic Acids Res., 2005, 33, e171 CrossRef PubMed.
- M. J. Chaisson and P. A. Pevzner, Genome Res., 2008, 18, 324–330 CrossRef CAS PubMed.
- R. Böttcher, R. Amberg, F. P. Ruzius, V. Guryev, W. Verhaegh, P. Beyerlein and P. J. van-der-Zaag, Nucleic Acids Res., 2012, 40, e125 CrossRef PubMed.
- D. R. Kelley, M. C. Schatz and S. L. Salzberg, Genome Biol., 2010, 11, R116 CrossRef CAS PubMed.
- M. J. Chaisson, D. Brinza and P. A. Pevzner, Genome Res., 2009, 19, 336–346 CrossRef CAS PubMed.
- H. Breu, White paper, Life Technologies, 2010 Search PubMed.
- H. Breu and D. Guo, US Pat., 9342651B2, 2016.
- M. Ronaghi, M. Uhlen and P. Nyren, Science, 1998, 281, 363–365 CrossRef CAS PubMed.
- L. Liu, Y. Li, S. Li, N. Hu, Y. He, R. Pong, D. Lin, L. Lu and M. Law, J. Biomed. Biotechnol., 2012, 2012, 251364 Search PubMed.
- A. Ahmadian, M. Ehn and S. Hober, Clin. Chim. Acta, 2006, 363, 83–94 CrossRef CAS PubMed.
- D. S. Tawfik and A. D. Griffiths, Nat. Biotechnol., 1998, 16, 652–656 CrossRef CAS PubMed.
- P. Nyren, B. Pettersson and M. Uhlen, Anal. Biochem., 1993, 208, 171–175 CrossRef CAS PubMed.
- B. A. Flusberg, D. R. Webster, J. H. Lee, K. J. Travers, E. C. Olivares, T. A. Clark, J. Korlach and S. W. Turner, Nat. Methods, 2010, 7, 461–465 CrossRef CAS PubMed.
- Z. Su, B. Ning, H. Fang, H. Hong, R. Perkins, W. Tong and L. Shi, Expert Rev. Mol. Diagn., 2011, 11, 333–343 Search PubMed.
- X. Zhou, L. Ren, Y. Li, M. Zhang, Y. Yu and J. Yu, Sci. China: Life Sci., 2010, 53, 44–57 CrossRef CAS PubMed.
- J. Shendure and E. Lieberman-Aiden, Nat. Biotechnol., 2012, 30, 1084–1094 CrossRef CAS PubMed.
- M. C. Schatz, A. L. Delcher and S. L. Salzberg, Genome Res., 2010, 20, 1165–1173 CrossRef CAS PubMed.
- T. J. Treangen and S. L. Salzberg, Nat. Rev. Genet., 2012, 13, 36–46 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra06202h |
| This journal is © The Royal Society of Chemistry 2017 |